
Abstract
CCN3 is a member of the CCN family of cell growth and differentiation regulators that play key roles during embryonic development, and are associated with severe human pathologies. The level of CCN genes’ expression is of prognostic value in several types of tumors. In the present manuscript, we report the isolation and characterization of a new set of antibodies targeted against each individual module of the human CCN3 protein. The need for module-specific antibodies stemmed from recent reports indicating that the expression of truncated CCN variant proteins was associated with development of cancers. Each of the four CCN3 modules were expressed as GST fusion proteins and used for rabbits immunization. Polyclonal IgGs purified by two rounds of affinity–chromatography specifically detected both the individual CCN3 domains and the full length CCN3 protein expressed in mammalian cell lines and tissues, as well as recombinant full length and truncated CCN3 proteins. The purified module-specific antibodies were successfully used for Western blotting, immunoprecipitation, immunofluorescence and immunocytochemistry. These antibodies permitted the detection of CCN3 proteins under native and denaturing conditions, and confirmed the sublocalisation of CCN3 proteins in the extracellular compartment, at the cell membrane, in the cytoplasm and in the nucleus of positive cells. Immunocytochemistry and Western blotting studies performed with the module-specific antibodies identified truncated CCN3 proteins in kidney tumor samples. The detection of these rearranged variants provides clues for their involvement in tumorigenesis. Therefore, these antibodies constitute unique tools for the identification of truncated CCN3 proteins in human tissues and may be of great interest in molecular medicine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The CCN3 protein is one of the three founders of the CCN family, which contains six members in human. The CCN proteins are key regulators of embryonic development (reviewed in Planque and Perbal 2003a; Perbal 2004; Rachfal and Brigstock 2005; Perbal and Takigawa 2005). They are involved in various cellular processes including proliferation, differentiation, survival, adhesion and migration. In the adult, the CCN proteins participate in the repair of injured tissues. Deregulations of expression of the CCN proteins have been described in fibrotic and cancer pathologies (reviewed in Planque and Perbal 2003b; Perbal 2004; Rachfal and Brigstock 2005). The CCN proteins are secreted factors of 35–48 kDa that contain four structural modules: (a) an insulin-like growth factor binding protein (IGFBP)-type motif, (b) a Von Willebrand type C (VWC) domain likely responsible for oligomerization, (c) a thrombospondin type 1 (TSP1) repeat, responsible for the interaction with extracellular matrix proteins, and (d) a carboxy-terminal module (CT) that contains a cystine-knot motif.
CCN3 exerts antiproliferative functions on both normal and tumor cells of various origins (Planque et al. 2005; Fukunaga-Kalabis et al. 2006; McCallum et al. 2006; Van Roeyen et al. 2007). CCN3 also modulates cell adhesion and migration (Benini et al. 2005), and CCN3 expression in primary Ewing’s tumors was associated with higher risk of developing metastasis (Manara et al. 2002). These results suggest a critical role for CCN3 in cancer progression. In agreement with these data, CCN3 expression was correlated with good or bad prognosis of survival in various types of cancers (reviewed in Planque et al. 2005). During embryonic development in mammalians, preferential sites of CCN3 expression included central nervous system, urogenital tract, and musculo-skeletal apparatus (reviewed in Planque et al. 2005 and Perbal and Planque 2006). In the adult, CCN3 expression persists in some of these sites (Perbal and Planque 2006).
In addition to the 54 kDa full-length secreted CCN3 protein, an amino-truncated form of about 30 kDa was observed in various embryonic and adult mammalian tissues (Ellis et al. 2000; Kocialkowski et al. 2001; Su et al. 2001), in diverse biological fluids (Burren et al. 1999), as well as in culture supernatants and cell extracts of normal and tumor cells (Chevalier et al. 1998; Perbal 1999; Perbal et al. 1999; Thomopoulos et al. 2001; Fu et al. 2004; Kyurkchiev et al. 2004). Sequencing of the truncated CCN3 form produce by baculovirus-infected cells indicated that it contained the TSP1 and CT modules (Perbal et al. 1999). It was postulated that this variant might be generated by proteolytic cleavage of the full-length secreted form (Perbal 2001). The immunological detection of CCN3 proteins used polyclonal antibodies targeted against the last 19 C-terminal amino acids.
Because truncated CCN protein variants lacking internal modules were associated with tumor development (Joliot et al. 1992; Tanaka et al. 2001), we have prepared polyclonal antibodies targeted against each of the four modules of the CCN3 protein in order to study whether deletion variant CCN3 proteins are expressed in human tumor tissues. For this purpose, each exon of the human ccn3 gene was cloned into a pGEX vector and the corresponding GST fusion proteins were used as antigens for rabbit immunization after they were purified by affinity chromatography. The module-specific IgGs reacted with each CCN3 module, either individually or in the context of the full-length protein. The four IgGs preparations were successfully validated for Western blot analysis, immunofluorescent staining and immunocytochemistry, therefore providing unique tools for immunological identification of truncated CCN3 proteins in normal and pathological samples.
Material and methods
Preparation of the immunogenic proteins and purification of the immune sera
The cDNAs of exons encoding each of the four human CCN3 modules were cloned into the pGEX-4T1 vector (Amersham – GE Healthcare) and the corresponding recombinant plasmids were further used to transform E. coli BL21.
GST-NH fusion proteins (nomenclature depicted in Fig. 1a) were purified by affinity chromatography on Sepharose beads as previously described (Bleau et al. 2007). GST fusion proteins were dialyzed against 10 mM NH4HCO3 and quantified by SDS-PAGE. Proteins were revealed by Coomassie Blue and silver staining. Protein concentrations were estimated by comparison with a Bovine Serum Albumin standard loaded onto the same gel. Of each GST-NH fusion protein preparation, 1.5 mg were used for rabbit immunization (Agro-Bio, La Ferté St Aubin, France). Rabbits were injected five times with 250 μg of purified GST-NH proteins. Three bleedings of 20–25 ml were then performed, and the rabbits were boosted with another 250 μg of antigens. IgG were purified from the immune sera by affinity chromatography on Protein A Sepharose beads. Briefly, nProtein A Sepharose 4 Fast Flow (Amersham – GE Healthcare) was equilibrated with 20 mM Tris pH 8.0. Anti-NH sera were loaded on the Protein A column and then washed with 20 mM Tris pH 8.0 and 20 mM glycine pH 5.0. IgG were eluted using 100 mM glycine pH 2.5 and neutralized with 1 M Tris pH 8.0. Fractions were pooled, and concentrated on Vivaspin concentrator (Vivascience).

Preparation of the GST fusion proteins used as immunogens. a Schematic diagram depicting modular organization of the CCN3 protein and the nomenclature used in this manuscript. cDNAs of the human CCN3 exons (NH2, NH3, NH4 and NH5) were inserted in the pGEX4T1 vector downstream to the GST gene. b Control of purity and quantification of the immunogen proteins. Coomassie Blue staining of the SDS-PAGE gel. GST-NH proteins were extracted and purified from E. coli BL21 bacteria using glutathione affinity chromatography. MW: protein molecular weight marker (kDa)
Removal of anti-GST antibodies was performed by affinity chromatography. Purified GST protein was coupled to CNBr-activated Sepharose 4B PD-10 columns according to the protocol recommended by the manufacturer (Amersham – GE Healthcare). The IgG preparations were loaded on these columns, and absorbance of the collected fractions was measured at 280 nm. Anti-NH IgG were in the flow-through fractions while anti-GST IgG were adsorbed on the beads. Flow-through fractions were pooled and concentrated with Vivaspin concentrators (Vivascience).
Construction of expression vectors, production and purification of various recombinant proteins
pTEF-MF yeast expression vectors (Dualsystems Biotech) were used to produce each individual modules of the human CCN3 protein in a tag-free version or with a 6-Histidine tag at their amino-terminus (Bleau et al. 2007). This system allows secretion of the recombinant proteins in yeast culture medium. The non-tagged recombinant NH proteins were purified by immuno-affinity chromatography (Bleau et al. 2007). 6HIS-tagged constructs were purified using Ni-NTA purification system according to the manufacturer’s instructions (Invitrogen). Proteins were eluted with 500 mM imidazole. Recombinant Y-NH proteins were dialyzed overnight at 4°C against 10 mM NH4HCO3, lyophilized, and frozen until use. For biochemical assays, the proteins were usually dissolved in PBS. Quantification was performed after SDS-PAGE followed by silver staining of the gels.
The construction of pcDNA vectors expressing non-secreted Myc-tagged CCN3 variants was previously described (Planque et al. 2006). To generate secreted Myc-tagged variants (sMNH, Fig. 2a), the CCN3 signal peptide was PCR-amplified and inserted at the HindIII/BamHI sites of pMNH25, -NH24, -NH35 and -NH45. These constructs allow secretion of the sMNH proteins in cell culture medium when introduced into mammalian cells.

Western blot analysis of recombinant secreted CCN3 variants. a Schematic diagram depicting the CCN3 constructs and the nomenclature used in this experiment. The table on the right summarizes the reactivity observed with each anti-NH IgG in b (positive sign, anti-NH IgG recognizes the corresponding protein; negative sign, no detection). b Supernatants were collected from human gliobastoma G59 cell cultures expressing either non-tagged full-length CCN3 protein (sNH25 from G540 cells; lane 1), or tagged full-length CCN3 (sMNH25; lane 2), or variants lacking one or several modules: sMNH24 (lane 3), sMNH35 (lane 4), sMNH45 (lane 5). Of each individual CCN3 module purified from yeast culture supernatants, 100 ng were also loaded onto the gels (Y-NH2, Y-NH3, Y-NH4, Y-NH5; lane 6). Secreted proteins were analyzed by Western blotting with each of the four anti-NH IgG at the concentration of 25 μg/ml for anti-NH2, 8.7 μg/ml for anti-NH3, 60 μg/ml for anti-NH4, and 3.8 μg/ml for anti-NH5. c Detection of intracellular CCN3 forms. Cell lysates of G540 cells and G59 parental cells were immunoprecipitated with each purified anti-NH IgG coupled to protein A-magnetic beads. Eluted proteins were electrophoresed under denaturating conditions and immunoblotted with each corresponding antibodies
Culture and transfection of mammalian cell lines
The G59 glioblastoma cell line was originally isolated and provided by Dr M. Westphal, and has been described previously (Li et al. 1996). G59 were grown in Dulbecco’s Modified Eagle Medium (DMEM; Sigma) supplemented with 10% fetal calf serum (FCS) Sigma) and l-Glutamin–Penicillin–Streptomycin mix (Sigma; complete medium). The G540 cells isolated after transfection with the pCMV-CCN3 plasmid (pCMV82) express the human CCN3 protein (Gupta et al. 2001; Bleau et al. 2007). G59 cells were also transfected with psMNH25, psMNH35, psMNH24, psMNH45 (nomenclature depicted Fig. 2a) or pcDNA DNAs. Cell transfections were performed in 3.5 cm dishes in OPTIMEM medium with Lipofectamin 2000 (Invitrogen) according to the manufacturer’s recommendations. For each dish, 2 μg of DNA were mixed with 5 μl of Lipofectamin 2000 in a final volume of 1 ml of OPTIMEM, and incubated for 20 min at room temperature before being applied to cell cultures. Seventy two h after transfection, cell lysates and culture supernatants were tested for the expression of the recombinant proteins by immunoblotting with mouse monoclonal anti-Myc antibodies (Roche; 1:2000).
Human adenocortical carcinoma NCI H295R (American Type Cell Collection) cell line was cultured in DMEM-F12 (Sigma) supplemented with 2.5% of Nu serum (Invitrogen) and ITS+ supplement (Sigma). These cells express high levels of CCN3 (Thomopoulos et al. 2001).
For immunofluorescence detection, G59 and Baby Hamster Kidney 21 (BHK21) cells were seeded on coverslips in 24-well plates. For transfection, 600 ng of DNAs were incubated with 2 μl of EXGEN500 (Euromedex) in a final volume of 30 μl of 150 mM NaCl according to the manufacturer’s instructions.
Western blotting
Western blotting analyses were performed both with cell culture supernatants and cell lysates.
Laemmli buffer was directly added to 25 μl of conditioned media from G59 cells secreting the various CCN3 proteins (sNH25, sMNH25, sMNH24, sMNH35 and sMNH45, see above). In addition, cell culture supernatants from G540 and NCI-H295R cells were directly incubated with heparin Sepharose beads (Amersham – GE Healthcare) overnight at 4°C on a rotating wheel. Beads were then extensively washed with PBS, resuspended in Laemmli buffer and boiled for 10 min.
Human glioblastoma G540 and G59 parental cells were lysed in 50 mM Tris hydrochloride, pH 8.0, 150 mM NaCl, 5 mM EDTA and 2% NP40, containing protease inhibitors (Complete mini tablets, Roche) and phosphatase inhibitors (50 mM sodium fluoride, 2 mM sodium orthovanadate) for 30 min at 4°C. Cell debris were removed by centrifugation at 15,000×g. The protein content of the cell lysates was measured using the BCA Protein Assay kit (Pierce). Lysates were stored at −80°C until use. The CCN3 proteins contained in the various lysates were immuno-precipitated using protein A coupled to magnetic beads. Briefly, Bio-Adembeads Protein A (Ademtech France) magnetic beads were coated and chemically cross-linked with each anti-NH IgG according to the manufacturer’s instructions. Of the G540 or G59 cell lysates, 10–20 μg were incubated with the Bio-Adembeads Protein A-antibodies for 30 min, and subsequently washed with the lysis buffer. Immunoprecipitated proteins from cell extracts were eluted from magnetic beads with 20 μl of 50 mM glycine pH 2.7 containing 0.65% Tween 20. After neutralization with Tris 1 M pH 8, proteins were denaturated by boiling for 10 min in Laemmli buffer containing 5% 2-mercaptoethanol before gel analysis.
Proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes (Hybond-C extra, Amersham – GE Healthcare). Membranes were blocked with 5% non-fat milk in PBS-0.2% Tween 20 (Sigma; PBST; blocking solution) and incubated under gentle agitation for 1 h at room temperature in the presence of the anti-NH IgGs diluted in blocking solution. Optimal concentrations were 25 μg/ml for anti-NH2, 8.7 μg/ml for anti-NH3, 60 μg/ml for anti-NH4, 3.8 μg/ml for anti-NH5. The blots were extensively washed with PBST and incubated under gentle agitation with goat anti-rabbit IgG conjugated with horseradish peroxidase (1/20,000; Sigma) in blocking solution for 45 min at room temperature. After extensive washes with PBST, the membranes were developed using the Uptima chemoluminescent kit from Interchim.
Immunofluorescence staining
G59 and BHK21 cells were plated on 12 mm Ø coverglasses in 24-well dishes, and were transiently transfected with either pCMV47, pcDNA, pNH25 or pNH35DsRed as described above. pCMV47 allows the expression of full-length CCN3 protein (Bleau et al. 2007). pNH25 and pNH35DsRed govern the expression of intracellular forms of CCN3 (Planque et al. 2006). Forty eight hours after transfection, the coverslips were fixed with PBS containing 4% paraformaldehyde for 30 min at 4°C, and permeabilized for 5 min with PBS containing 0.1% Triton X-100. Coverslips were sequentially incubated with PBS containing 5% FCS for 1 h at room temperature, incubated with primary antibodies (anti-NH IgG, dilution 1:1,000) for 1 h in PBS containing 5% FCS, rinsed four times with PBS, incubated with secondary antibodies (AlexaFluor 488 goat anti-rabbit, Molecular Probe, dilution 1:2,000) for 30 min, rinsed four times with PBS, and finally mounted in FluoroGard antifade mounting medium (Biorad). Pictures were captured with a Leica DMR microscope.
OS7 and OS9 osteosarcoma cell lines which express various levels of CCN3 (Lazar et al. manuscript in preparation) were seeded in Iscove’s Modified Dulbecco’s Medium (IMDM) 10% FCS and grown on coverslips for 48 h before being fixed in 4% para-formaldehyde 30 min at 4°C and permeabilized with 0.15% Triton X-100 in PBS. All preparations were incubated with PBS containing 4% BSA to saturate non-specific binding. Immunofluorescence staining for CCN3 was performed with the following primary antibodies: -K19M, -NH2, -NH3, -NH4, -NH5 all diluted 1:200 with 4% BSA/PBS at 4°C overnight. Slides were incubated with secondary antibody (CY3-conjugated anti-rabbit IgG, Sigma; 1:5,000 dilution) for 1 h at room temperature. Nuclei were stained with Hoechst 33258 (0.5 μg/ml PBS/glycerol, Sigma). Digital images were taken in identical conditions, by using the image analysis software (Quips-XL genetic workstation, Abbot Visys).
Immunohistochemistry
Samples of normal embryonic (16 and 28 weeks gestation), fetal, and adult kidneys, were included in a Tissue Micro Array (TMA). TMA sections of 5 μm were deparafinized and antigen retrieval was performed by incubating the tissue sections with citrate buffer (pH 6.0) in a pressure cooker at 1.5 ATM for 3 min. The endogenous peroxidase was blocked using 3% hydrogen peroxidase for 5 min at room temperature. For immunohistochemistry the purified antibodies were used at the following dilutions: anti-NH2, anti-NH3, and anti-NH4 (1:400), anti-NH5 (1:600), anti-K19M (1:800). The immunohistochemical procedure included incubation of primary antibodies for 1 hour, immunodetection using ChemMate™ DAKO ENVISION™ /HRP Rabbit/Mouse (ENV) system followed by revelation of the reaction product by exposure to 3,3’-diaminobenzidine chromogen. Normal fetal kidneys specimens not treated by the primary antibodies were used as control. The levels of CCN3 immunostaining were evaluated independently by three investigators (MMS, SN and ALB).
Results
Production and purification of anti CCN3 module antibodies
E. coli BL21 cells, transformed with the various pGEX-NH plasmids, produced GST fusion proteins of about 38 kDa molecular weight (corresponding to 26-kDa of GST plus 9, 10, 11, and 12 kDa of NH2, NH3, NH4, and NH5, respectively; Fig. 1a). Fusion proteins were purified from bacterial lysates by a single-step affinity chromatography on glutathione-Sepharose beads and quantified as described under “Material and methods.” The purity of each GST fusion preparation was determined by Coomassie blue staining of the proteins separated by SDS-PAGE (Fig. 1b).
Polyclonal anti NH IgGs were produced in rabbits after injection. For each construct, rabbits were injected with a total of 1.5 mg of fusion protein. All sera were treated separately. The concentrations of anti-module specific IgGs obtained after two successive rounds of affinity chromatography (see “Material and methods”) were as follows: anti-NH2: 10 mg/ml; anti-NH3: 7 mg/ml; anti-NH4: 12 mg/ml; anti-NH5: 3.8 mg/ml.
These preparations were tested for their ability to detect various full-length and truncated forms of the human CCN3 protein under both denaturating and native conditions.
Western blot analysis of eukaryotic cell supernatants and cell extracts
Recombinant CCN3 proteins that were purified from yeast culture media and from G540-conditioned cell culture medium were used to assess the sensitivity and specificity of the module-specific antibodies. The G540 conditioned medium contains both the full-length 54 kDa (Gupta et al. 2001; Bleau et al. 2007) and the 32 kDa amino truncated human CCN3 proteins (Kyurkchiev et al. 2004). Each of the CCN3 modules was produced in yeast, as described under Materials and Methods. The proteins contained in the yeast culture media were purified either by immuno-affinity chromatography, or by imidazol-affinity chromatography.
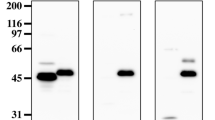
Western blot analysis revealed that each anti-NH antibody detected the individual CCN3 modules produced in yeast (Y-NH; Fig. 2b, lane 6), as well as the full-length protein (sNH25) secreted by the G540 cells (Fig. 2b, lane 1). The band of high molecular weight that is detected in the Y-NH5 preparation with the anti-NH5 antibodies (Fig. 2b, lower right panel, lane 6) is thought to result from oligomerization driven by the CT module (Perbal and Planque 2006). Similar high molecular CCN3 species were observed in patients with AML (McCallum et al. 2006) and were shown to be directly related to the level of expression of CCN3 in these patients. The additional band detected by the anti-NH5 in the conditioned medium of G540 cells (Fig. 2b, lower right panel, lane 1) corresponds to the 32 kDa CCN3 variant that was originally identified with the K19M antibody (Kyurkchiev et al. 2004). All the bands detected by the anti-NH5 antibody were also detected, though less strongly, by the K19M antibody (data not shown). Also, the anti-NH antibodies raised against one particular module did not react with the other three modules produced in yeast (data not shown).
Anti-NH antibodies detected CCN3 modules within the recombinant proteins secreted by G59 stable transfectants expressing truncated CCN3 variants (sMNH, Fig. 2a). For example the anti-NH2 antibodies stained sMNH25 and sMNH24 (Fig. 2b, upper panel left, lane 2 and 3), and did not react with the sMNH35 and sMNH45 proteins that do not contain the IGFBP module (Fig. 2b, upper panel left, lane 4 and 5). Anti-NH3 recognized sMNH25, sMNH24, sMNH35, but not sMNH45 (Fig. 2b, upper panel right) and anti-NH4 permitted the detection of all the variants (Fig. 2b, lower panel left). As expected, anti-NH5 reacted with all the variant proteins but sMNH24 that does not contain the C-terminal module of CCN3 (Fig. 2b, lower panel right).
Inasmuch as the NH antibodies showed different sensitivities, the IgG concentrations used for Western blotting needed to be adjusted for optimal staining of the various CCN3 variants in conditioned medium (see Fig. 2b). In spite of these adjustments, staining of sMNH24 and sMNH35 with anti-NH3 was repeatedly weak, therefore suggesting that the VWC module is not a powerful immunogen.
Previous experiments performed with the K19M antibody identified CCN3 proteins inside normal and tumor cells.
As a first step in examining whether intracellular CCN3 proteins were also detected by the NH antibodies, G59 and G540 cell lysates were first immunoprecipitated with anti-NH IgG cross-linked to Protein A coupled to magnetic beads and further treated for Western blotting (Fig. 2c). The results obtained indicated that the four NH antibodies detected CCN3 proteins in the immunoprecipitate, therefore establishing that they readily reacted with native CCN3 protein.
In a second step, we assessed the ability of the NH antibodies to recognize the human endogenous CCN3 protein. For that purpose, conditioned medium from NCI-H295R adrenocortical carcinoma cells was used. We had previously established that these cells naturally secrete high levels of CCN3 proteins (Thomopoulos et al. 2001). The 54 kDa full-length CCN3 protein, was detected with the four NH antibodies (Fig. 3a). An amino-truncated 32 kDa CCN3 protein was also identified in the conditioned medium of various human cell lines including NCI-H295R (Thomopoulos et al. 2001, Perbal 2004; Kyurkchiev et al. 2004). When straight conditioned medium was used, only the NH5 antibody detected the 32 kDa CCN3 protein. However, when the concentration of CCN3 proteins contained in the medium was increased by chromatography through heparin columns, the 32 kDa CCN3 protein was detected by both NH4 and NH5 antibodies. This protein was neither detected by the NH2 nor NH3 antibodies in the heparin binding fractions (Fig. 3a) therefore suggesting that it corresponded to a CCN3 variant lacking both the IGFBP and VWC modules.

Western blot of human endogenous CCN3 proteins in cancer cells. a Culture supernatants from NCI-H295R cells were concentrated with heparin Sepharose beads. Equal amounts of supernatants were loaded onto the gel in quintuplet. Membranes were incubated with each of the four anti-NH antibodies at their optimal concentration (see above). Anti-K19M antibodies were used as positive control. b Comparison of the relative sensitivity of the anti-NH antibodies. Equal amounts of supernatant from NCI-H295R cells culture concentrated with heparin Sepharose beads were loaded onto the gel in quintuplet. Membranes were incubated with each of the four anti-NH IgG and K19M antibodies at the same concentration (60 μg/ml)
A similar amino-truncated CCN3 protein has previously been identified in the culture medium of insect cells secreting recombinant full-length CCN3 protein (Perbal et al. 1999). Purification and sequencing revealed that this amino-truncated form lacked both IGFBP and VWC modules (similar to the NH45 recombinant protein used in this work). Until now, the lack of domain-specific antibodies, did not permit confirmation that the 32 kDa-protein detected in various cell supernatants was indeed composed of the TSP1 and CT modules.
When identical amounts of conditioned medium were used with the same concentration of antibodies preparations, the anti-NH5 antibody was found to be the most reactive and anti-NH2 the least reactive (Fig. 3b). Similar conclusions were drawn from results obtained with purified GST-NH25 protein (not shown).
Immunofluorescence detection of CCN3 in cultured cells
In order to determine whether the NH antibodies were able to detect cellular CCN3 by immunofluorescence, human glioblastoma G59 cells that express full-length CCN3 were used. The four IgG preparations (anti-NH2, -NH3, -NH4 and -NH5) detected the human CCN3 protein in transfected cells (Fig. 4). Positive staining was considered confirmatory since no fluorescence signal was observed in control pcDNA-transfected cells (Fig. 4, lower panel left and not shown). K19M antibodies were used as positive control (Fig. 4, lower panel right).

Detection of native secreted CCN3 protein in mammalian cells by immunofluorescent staining. G59 cells were transiently transfected with the pCMV47 vector expressing a full-length secreted form of the human CCN3 protein (sNH25). Pictures of two independent fields are shown for each anti-NH IgG. The K19M antibodies were used as positive control (lower panel right). As negative control, G59 cells were transfected with pcDNA. The background generated with the anti-NH5 IgG is shown lower panel left
Intracellular CCN3 variants have been detected in the cytoplasm and in the nucleus of cancer cells (Perbal 1999; Gellhaus et al. 2004; Fu et al. 2004). We previously reported that CCN3 proteins deprived of signal peptide were detected in the cell cytoplasm and routed to the nucleus, provided that they contained the carboxy-terminal module (Planque et al. 2006). To check whether the NH antibodies could detect such CCN3 protein variant, BKH21 cells were transiently transfected with pNH35DsRed, that is driving the expression of an amino-truncated form of CCN3 (NH35) fused to a carboxy-terminal Dictosoma striata Red (DsRed) tag. The NH35 protein showed both cytoplasmic and nuclear localization when assayed with the K19 antibodies (Planque et al. 2006). This construct also allowed direct detection of the recombinant proteins by auto-fluorescence of the DsRed protein.
The NH35DsRed protein was readily detected both in the cytoplasm and the nucleus of transfected cells with the anti-NH3, -NH4, and -NH5 antibodies (Fig. 5, left column). It is worth noting that the anti-NH2 IgG did not recognize the NH35DsRed protein, which does not contain the IGFBP module, therefore reinforcing the conclusion that labeling obtained with NH antibodies were specific (Fig. 5, upper panel left). None of the four anti-NH IgGs cross-reacted with the DsRed protein (not shown). Both cytoplasmic and nuclear untagged CCN3 protein expressed in pcDNA-NH25-transfected BHK21 cells (Planque et al. 2006) were also detected by the four NH antibodies. The staining obtained with the anti-NH and -K19M antibodies in these samples were identical (Fig. 5, lower panel left) therefore indicating that the NH antibodies could be used for the detection of CCN3 proteins in cells (Fig. 5, right column).

Detection of native non-secreted CCN3 proteins in mammalian cells by immunofluorescent staining. BHK21 cells were transiently transfected with either pNH35DsRed (left column) or pcDNA-NH25 (right column). Both nuclear and cytoplasmic staining were detectable in transfected cells (for the subcellular localization of the NH35DsRed and NH25 proteins, see Planque et al. 2006)
In order to establish that the module-specific antibodies also detected endogenous CCN3 proteins expressed in human tumor cells, osteosarcoma cell lines expressing various levels of CCN3 were used. As expected from the results obtained from Western blotting, positive, staining for CCN3 was detected in the cytoplasm of cells (Fig. 6). The results obtained with anti-K19M antibody support the specific reactivity of the NH antibodies in immunofluorescence (Fig. 6). Large variations in staining that were observed with the various antibodies might result from a different accessibility of the CCN3 epitopes in the various cell lines, as already suggested in previous work (Kyurkchiev et al. 2004).

Detection of the endogenous CCN3 protein in human osteosarcoma cells by immunofluorescent labeling. CCN3 expression in human osteosarcoma (OS7, and OS9) cell lines was assayed by indirect immunofluorescence using anti-NHs antibodies. Nuclei were counterstained with DAPI
In situ detection of the endogenous CCN3 protein in kidney tissues
Because we ultimately aimed at using the anti-NH antibodies to detect the various CCN3 proteins expressed in tumor samples, the expression of CCN3 was also analyzed by immunohistochemistry on paraffin-embedded sections of normal human embryonic, fetal and adult kidney tissues for which the pattern of CCN3 has already been characterized (Chevalier et al. 1998; Kocialkowski et al. 2001).
The results that were obtained are summarized in Table 1. In embryonic kidney the anti-NH2 and anti-NH3 antibodies stained only some tubular structures while the embryonic glomeruli and the blastemal interstitial cells were negative. Anti-NH4 also stained the tubular component of the developing kidney (Fig. 7) while anti-NH5 demonstrated intense positivity in almost all components of the kidney: blastemal cells, nephrogenic vesicles, embryonic glomeruli and most of the tubular epithelia (Fig. 7). In the fetal kidney anti-NH4 stained the tubules while anti-NH5 displayed the most intense positivity in all areas of the nephron (glomeruli and tubules). Staining was both cytoplasmic and nuclear (Fig. 7). In the adult kidneys, anti-NH2 reacted faintly with some epithelial cells of the proximal and distal tubules, with a granular cytoplasm distribution. The glomeruli and the stroma were negative. The anti-NH3 stained almost all tubular structures, with variable intensity in the cytoplasm of the epithelial lining while the glomeruli did not show any reactivity. The staining patterns for anti-NH4 and anti-NH3 were similar while a strong positivity was observed with the anti-NH5 in the tubules and in isolated cells within the glomerular tuft, with diffuse or granular decoration of the epithelial cell cytoplasm (Fig. 7).

Detection of the endogenous CCN3 protein in human kidney tissues by immunohistolabeling. Embryonic kidney (16 weeks gestation), fetal kidney (28 weeks gestation) and normal adult kidney stained with anti-NH2, -NH3, -NH4, -NH5 and -K19M polyclonal antibodies (×40). Fetal kidney stained with anti-K19M. Glomerular cells, epithelial tubular cells and interstitial cells appear intensively labeled. Fetal kidney stained with anti-NH5, displaying intense positivity in the cytoplasms and nuclei of the cells within the tubules as well in the glomeruli and interstitial blastema. Embryonic kidney stained with anti-K19M presenting intense positivity in developing glomeruli and in tubules. Embryonic kidney showing positivity for anti-NH4 in tubules and blastemal cells. Embryonic kidney showing intense positivity for NH5 in all tissue components. Normal adult kidney stained with anti-K19M. Strong positivity in tubules with a granular cytoplasmic distribution. Normal adult kidney stained with anti-NH5. Intense positivity of almost all cellular components
The various staining patterns obtained with the anti-NH antibodies matched the staining that was obtained with the anti-K19M and were in agreement with the previous identification of CCN3 sites of expression in the developing and adult kidneys. In addition they also suggested that either some CCN3 epitopes are masked in specific structures or that CCN3 isoforms, lacking specific modules are expressed in a developmentally regulated way in the various kidney structures. This particular aspect is under current investigation.
Discussion
In the past decade, several studies have reported the existence of truncated CCN proteins in normal cells and tumor samples (see reviews in Perbal and Takigawa 2005). The first evidence for the expression of a truncated CCN protein being associated with tumorigenic potential has been provided with the discovery of CCN3 in avian nephroblastomas (Joliot et al. 1992). Since that time, compelling observations have accumulated suggesting a role for amino-truncated CCN3 in tumorigenicity as the result of nuclear localization (Perbal 1999, 2004; Planque and Perbal 2003b; Planque et al. 2005, 2006).
The discovery of amino-truncated CCN3 highly expressed in particular subtypes of human melanomas (Vallacchi et al. 2007, submitted for publication) has confirmed the biological importance of these CCN protein variants and have raised the possibility that tumors of other tissues might also express rearranged CCN3 proteins, as previously shown in the case of CCN4 (Yanagita et al. 2007; Cervello et al. 2004; Tanaka et al. 2001, 2003). Because tumor samples are generally small and precious, it is not always possible to perform Western blotting on protein lysates and protein analysis is often performed by immunocytochemistry on tumor sections or tissue arrays.
Based on the variety of biological functions that depend upon the highly conserved multimodular structure of CCN proteins, we assumed that alteration of any single module would have quite important biological consequences. Since published data established that CCN3 variant species can be generated through post translational processing (Perbal et al. 1999; Bleau et al. 2007) we reasoned that wide screening of tumor samples for the expression of truncated CCN3 proteins would be eased by the use of module-specific antibodies. In our previous studies we have successfully used a polyclonal antibody (Anti-K19M) that was raised against the 19 terminal amino acids of CCN3 (Chevalier et al. 1998). Even though this type of antibody proved very powerful in identifying truncated CCN3 proteins by Western blotting, it did not permit discrimination of full-length and truncated CCN3 proteins by immunocytochemical methods performed on tissues.
In this work we have generated and characterized a set of four polyclonal rabbit antibodies, namely anti-NH2, -NH3, -NH4, and -NH5, raised against each of the IGFBP, VWC, TSP1, and CT modules of CCN3, respectively. We show that these antibodies are unique tools to detect normal and variant CCN3 proteins by Western blotting, immunofluorescence, and immunocytochemistry. Since these antibodies also permit immunoprecipitations we assume that they recognize both native and denatured CCN3 proteins.
Western blotting performed with conditioned medium and lysates from cell lines expressing various recombinant CCN3 proteins of different sizes, established that the module-specific antibodies recognized the CCN3 modules both as single species and when they are contained within recombinant proteins.
Use of this new panel of antibodies established that the 32 kDa protein that was detected in conditioned medium of CCN3 producing cells, is composed of the TSP1 and CT domains of CCN3. As compared to the full-length protein which is also expressed in the medium, the 32 kDa protein is lacking the two amino-proximal modules of CCN3. A truncated CCN3 protein with apparent molecular mass of 30–32 kDa was detected with the anti-K19M antibody in conditioned medium from cells such as NCI H295R (Thomopoulos et al. 2001; Kyurkchiev et al. 2004), in different human biological fluids such as urine, amniotic fluid, and cerebrospinal fluid (Burren et al. 1999), and in tissue extracts (Su et al. 2001). Based on the observation that an amino-truncated CCN3 of the same size and composition was also detected in the conditioned medium of SF9-infected insect cells producing recombinant CCN3 (Perbal et al. 1999) the 32 kDa detected in the conditioned medium of higher eukaryotic cells might result from proteolytic cleavage of the full-length 54 kDa CCN3 protein. A CCN3 protein with an apparent molecular weight of 32 kDa is also detected in cell lysates (Kyurkchiev et al. 2004) therefore raising the possibility that other biological mechanisms might be involved in the production of the amino-truncated CCN3 variant.
In spite of their different affinities for the modules they specifically recognize, anti-NH5 being by far the most sensitive, all module-specific antibodies also recognized the corresponding modules within recombinant CCN3 proteins containing various combinations of modules. These observations indicated that the detection of each CCN3 module was not hampered by the intrinsic composition of the rearranged CCN3 protein and confirmed that CCN3 proteins lacking various modules were not degraded.
Most interestingly, the anti-NH5 antibody readily detected the CT domain of the native CCN3 whereas the anti-K19M which is directed against the carboxy terminal amino acids of the CT module did not recognize efficiently the native protein in solution (Kyurkchiev et al. 2004). These observations also suggest that the C terminus of the native CCN3 protein is not accessible to antibodies either because of structural constraints, or because it is engaged in interaction with other partners (Perbal 2001).
The ability of the anti-NH antibodies to react with CCN3 in cells and tissues processed for immunofluorescence and immunocytochemistry, is a very important point towards our goals. Because the osteosarcoma cell lines expressed high quantity of CCN3 protein, we selected them for testing the capacity of the NH-antibodies to assess the detection of endogenous CCN3 protein by immunofluorescence in tumor cells. The results that we obtained establish this methodology as a unique tool for CCN3 evaluation in tumor samples from patients. Only previous results indicated that CCN3 proteins were detected by immunofluorescence using the anti-K19M antibody in several human cancer cell lines, including HeLa, osteosarcoma 143 and choriocarcinoma Jeg3, in rat C6 glioma cells, as well as in some cases of osteosarcomas and Ewing’s sarcomas (Perbal 1999; Gellhaus et al. 2004; Fu et al. 2004; Manara et al. 2002). The immunoreactivity demonstrated by the NH-antibodies was comparable to that of the polyclonal antibodies specific to K19M.
The detection of CCN3 by immunohistochemistry on paraffin-embedded sections also provided the demonstration that the anti-NH antibodies constituted a unique tool to screen human tumors for the expression of full-length or truncated CCN3 proteins.
In conclusion, we report here the characterization of a new set of module-specific antibodies that permit the structural composition of CCN3 proteins expressed in normal and pathological conditions. The new antibodies specifically react with the CCN3 modules contained in the full-length and truncated CCN3 proteins, under various experimental conditions including Western blot, immunofluorescence, and immunohistochemistry. Therefore, this set of antibodies represent unique tools to undertake wide screening of CCN3 expression in tumor arrays and should prove to be useful for proteomic analysis.
References
Benini S, Perbal B, Zambelli D, Colombo MP, Manara MC, Serra M, Parenza M, Martinez V, Picci P, Scotlandi K (2005) In Ewing’s sarcoma CCN3(NOV) inhibits proliferation while promoting migration and invasion of the same cell type. Oncogene 24:4349–4361
Bleau AM, Planque N, Lazar N, Zambelli D, Ori A, Quan T, Fisher G, Scotlandi K, Perbal B (2007) Antiproliferative activity of CCN3: involvement of the C-terminal module and post-translational regulation. J Cell Biochem (in press)
Burren CP, Wilson EM, Hwa V, Oh Y, Rosenfeld RG (1999) Binding properties and distribution of insulin-like growth factor binding protein-related protein 3 (IGFBP-rP3/NovH), an additional member of the IGFBP Superfamily. J Clin Endocrinol Metab 84:1096–1103
Cervello M, Giannitrapani L, Labbozzetta M, Notarbartolo M, D’Alessandro N, Lampiasi N, Azzolina A, Montalto G (2004) Expression of WISPs and of their novel alternative variants in human hepatocellular carcinoma cells. Ann N Y Acad Sci 1028:432–439
Chevalier G, Yeger H, Martinerie C, Laurent M, Alami J, Schofield PN, Perbal B (1998) novH: differential expression in developing kidney and Wilm’s tumors. Am J Pathol 152:1563–1575
Ellis PD, Chen Q, Barker PJ, Metcalfe JC, Kemp PR (2000) Nov gene encodes adhesion factor for vascular smooth muscle cells and is dynamically regulated in response to vascular injury. Arterioscler Thromb Vasc Biol 20:1912–1919
Fu CT, Bechberger JF, Ozog MA, Perbal B, Naus CC (2004) CCN3 (NOV) interacts with connexin43 in C6 glioma cells: possible mechanism of connexin-mediated growth suppression. J Biol Chem 279:36943–36950
Fukunaga-Kalabis M, Martinez G, Liu ZJ, Kalabis J, Mrass P, Weninger W, Firth SM, Planque N, Perbal B, Herlyn M (2006) CCN3 controls 3D spatial localization of melanocytes in the human skin through DDR1. J Cell Biol 175:563–569
Gellhaus A, Dong X, Propson S, Maass K, Klein-Hitpass L, Kibschull M, Traub O, Willecke K, Perbal B, Lye SJ, Winterhager E (2004) Connexin43 interacts with NOV: a possible mechanism for negative regulation of cell growth in choriocarcinoma cells. J Biol Chem 279:36931–36942
Gupta N, Wang H, McLeod TL, Naus CC, Kyurkchiev S, Advani S, Yu J, Perbal B, Weichselbaum RR (2001) Inhibition of glioma cell growth and tumorigenic potential by CCN3 (NOV). Mol Pathol 54:293–299
Joliot V, Martinerie C, Dambrine G, Plassiart G, Brisac M, Crochet J, Perbal B (1992) Proviral rearrangements and overexpression of a new cellular gene (nov) in myeloblastosis-associated virus type 1-induced nephroblastomas. Mol Cell Biol 12:10–21
Kocialkowski S, Yeger H, Kingdom J, Perbal B, Schofield PN (2001) Expression of the human NOV gene in first trimester fetal tissues. Anat Embryol (Berl) 203(6):417–427
Kyurkchiev S, Yeger H, Bleau AM, Perbal B (2004) Potential cellular conformations of the CCN3(NOV) protein. Cell Commun Signal 2:9
Li WX, Martinerie C, Zumkeller W, Westphal M, Perbal B (1996) Differential expression of novH and CTGF in human glioma cell lines. Mol Pathol 49:91–97
Manara MC, Perbal B, Benini S, Strammiello R, Cerisano V, Perdichizzi S, Serra M, Astolfi A, Bertoni F, Alami J, Yeger H, Picci P, Scotlandi K (2002) The expression of ccn3(nov) gene in musculoskeletal tumors. Am J Pathol 160:849–859
McCallum L, Price S, Planque N, Perbal B, Pierce A, Whetton AD, Irvine AE (2006) A novel mechanism for BCR-ABL action: stimulated secretion of CCN3 is involved in growth and differentiation regulation. Blood 108:1716–1723
Perbal B (1999) Nuclear localisation of NOVH protein: a potential role for NOV in the regulation of gene expression. Mol Pathol 52:84–91
Perbal B (2001) NOV (nephroblastoma overexpressed) and the CCN family of genes: structural and functional issues. Mol Pathol 54:57–79
Perbal B (2004) CCN proteins: multifunctional signalling regulators. Lancet 363:62–64
Perbal B, Planque N (2006) CCN3. AfCS-Nature Molecule Pages (DOI:10.1038/mp.a003943.01). The AfCS-Nature Signaling Gateway. Available at: http://www.signaling-gateway.org/
Perbal B, Takigawa M (2005) The CCN family of proteins: an overview. In: Perbal B, Takigawa M. (eds) The CCN proteins: a new family of cell growth and differentiation regulators. Imperial College Press, London, pp 1–18
Perbal B, Martinerie C, Sainson R, Werner M, He B, Roizman B (1999) The C-terminal domain of the regulatory protein NOVH is sufficient to promote interaction with fibulin 1C: a clue for a role of NOVH in cell-adhesion signalling. Proc Natl Acad Sci USA 96:869–874
Planque N (2006) Nuclear trafficking of secreted factors and cell-surface receptors: new pathways to regulate cell proliferation and differentiation, and involvement in cancers. Cell Commun Signal 4:7
Planque N, Perbal B (2003a) The CCN Proteins, a new family of cell growth regulators. In: Pandalai SG (ed) Recent research developments in human pathology (Vol. 1, Part II). Transworld Research Network, Trivandrum, pp 333–369
Planque N, Perbal B (2003b) A structural approach to the role of CCN (CYR61/CTGF/NOV) proteins in tumourigenesis. Cancer Cell Int 3:15
Planque N, Bleau AM, Perbal B (2005) The CCN3 protein: a multifunctional cell growth regulator. In: Perbal B, Takigawa M (eds) The CCN proteins: a new family of cell growth and differentiation regulators. Imperial College Press, London, pp 239–257
Planque N, Li CL, Saule S, Bleau AM, Perbal B (2006) Nuclear addressing provides a clue for the transforming activity of amino-truncated CCN3 proteins. J Cell Biochem 99:105–116
Rachfal AW, Brigstock DR (2005) Structural and functional properties of CCN proteins. Vitam Horm 70:69–103
Su BY, Cai WQ, Zhang CG, Martinez V, Lombet A, Perbal B (2001) The expression of ccn3 (nov) RNA and protein in the rat central nervous system is developmentally regulated. Mol Pathol 54:184–191
Tanaka S, Sugimachi K, Saeki H, Kinoshita J, Ohga T, Shimada M, Maehara Y, Sugimachi K (2001) A novel variant of WISP1 lacking a Von Willebrand type C module overexpressed in scirrhous gastric carcinoma. Oncogene 20(39):5525–5532
Tanaka S, Sugimachi K, Kameyama T, Maehara S, Shirabe K, Shimada M, Wands JR, Maehara Y (2003) Human WISP1v, a member of the CCN family, is associated with invasive cholangiocarcinoma. Hepatology 37(5):1122–1129
Thomopoulos GN, Kyurkchiev S, Perbal B (2001) Immunocytochemical localization of NOVH protein and ultrastructural characteristics of NCI-H295R cells. J Submicrosc Cytol Pathol 33:251–256
Van Roeyen C, Eitner F, Scholl T, Boor P, Kunter U, Planque N, Groene HJ, Bleau AM, Perbal B, Ostendorf T, Floege J (2007) CCN3 is a novel, endogenous, and PDGF-regulated inhibitor of glomerular cell proliferation. Kidney Int (in press)
Yanagita T, Kubota S, Kawaki H, Kawata K, Kondo S, Takano-Yamamoto T, Tanaka S, Takigawa M (2007) Expression and physiological role of CCN4/Wnt-induced secreted protein 1 mRNA splicing variants in chondrocytes. FEBS J 274(7):1655–1665
Acknowledgements
We are grateful to Pr. P. Picci for his support and encouragement. This work was supported by the French Ministry of Education and Research and by European Union (Prothets grant; contract number LSHC-CT-2004-5030306). Dr N. Lazar is supported by the European Prothets grant. Dr A.M. Bleau was supported by the Fonds de la Recherche en Santé du Québec. Dr N. Planque and Prof. B. Perbal are supported by the French Ministry of Education and Research. M. Boudebous, K. Massaba, S. Ramelet and V. Broquerie are acknowledged for their technical support. Thanks are due to A. Perbal for her help and to Dr H. Yeger and S. Gabaron for critical reading and correction of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lazar, N., Manara, C., Navarro, S. et al. Domain-specific CCN3 antibodies as unique tools for structural and functional studies. J. Cell Commun. Signal. 1, 91–102 (2007). https://doi.org/10.1007/s12079-007-0009-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-007-0009-8