
Abstract
The gap junctions (GJs), which form intercellular communicating channels between two apposing cells or form hemichannel with extracellular environment, perform crucial functions to maintain small molecule homeostasis. The central nervous system (CNS) GJs are important for maintenance of myelin sheath and neuronal activity. Connexin (Cx) proteins are building blocks of GJs. Recent cell-biological investigations show that amongst the CNS specific Cxs, the most abundant Cx protein, Cx43 and its oligodendrocytic coupling partner Cx47 primarily important for maintenance of CNS myelin. Recent investigations elucidate that the expression of Cx43 and Cx47 is very important to maintain K+ buffering and nutrient homeostasis in oligodendrocytes, CNS myelin and oligodendrocyte function. The investigations on Multiple Sclerosis (MS) patient samples and EAE hypothesized that the functional loss of Cx43/Cx47 could be associated with spread of chronic MS lesions. Exploring the mechanism of initial GJ alteration and its effect on demyelination in this model of MS might play a primary role to understand the basis of altered CNS homeostasis, observed during MS. In this review, we mainly discuss the role of CNS GJs, specifically the Cx43/Cx47 axis in the perspective of demyelination.
Similar content being viewed by others

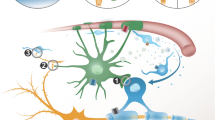
1 Background: Gap junctions in the central nervous system
Gap junctions (GJs), a group of cell-to-cell connecting channel proteins, play a pivotal role in maintaining homeostasis in different organs of vertebrates. In the central nervous system (CNS), GJs perform a crucial role in maintaining ionic buffering, small molecule exchange and nutrient homeostasis, which, in turn, help in maintenance of myelin and neuronal activity. GJs are present in at least four types of the CNS, which are astrocytes, ependymocytes/brain fibroblasts, oligodendrocytes, and neurons. GJ are made up of connexin (Cx) proteins. Different Cx proteins are named after their predicted molecular weight, which denotes specific type of Cx present in a specific cell. The neurons mainly express Cx36, which forms GJICs between the neuron only, in adult brain. Among the other cells, brain fibroblasts express mainly Cx43, whereas brain capillary endothelial cells (BCECs) mainly express Cx43 as well as Cx26 (Spray et al. 1991).
Oligodendrocytes form GJs mainly with astrocytes and dependent on them for homeostatic and nutrient support. In contrast, astrocytes form large numbers of gap junction intercellular communications (GJICs) with other astrocytes and also with oligodendrocytes. Together, the glial cells, mainly astrocytes connect with other brain cells to form a GJ connected network, named as ‘panglial syncytium’. As per the previous reports astrocytes mainly express three connexins (Cx43, Cx30, and Cx26) (Dermietzel et al. 1989; Kunzelmann et al. 1999; Nagy et al. 2001), and show CNS-specific regional variation in their expression (Nagy et al. 1999). The expression of Cx26 in astrocytes is debated in recent studies, as deletion of both Cx43 and Cx30 in astrocytes cause severe pathological conditions due to severe disruption of homeostasis (Lutz et al. 2009) and lacZ was not expressed in astrocytes under Cx26 promoter in mouse (Filippov et al. 2003). Astrocytes are primarily connected by Cx43/Cx43 homotypic channels. Cx43, which is the most abundant Cx in CNS, is strongly expressed throughout the white matter (Giaume and McCarthy 1996; Iacobas et al. 2003). These GJs connect the brain parenchyma to the capillaries through astrocytic end-feet, forming a complete network. Another astrocytic GJ protein, Cx30 is mainly observed in mainly grey matter regions. Immunostaining and functional studies show that another astrocytic GJ protein, Cx30, mainly forms homotypic Cx30/Cx30 channels in the gray matter. However, expression of Cx30 in certain white matter areas is observed, representing an inter-astrocytic Cx30/Cx30 channels and also participation with oligodendrocytes by heterotypic channels (Nagy et al. 1999; Rash et al. 2001; Rouach et al. 2002; Rozental et al. 2000).
Oligodendrocytes mainly express Cx29, Cx32 and Cx47. Cx32/Cx32 homotypic channels appear to form GJs along the myelinated fibers in the white matter, and also expressed in Schmidt–Lantermann incisures and paranodes near to the nodes of Ranvier. These channels mostly form intracellular GJ within the myelin sheath. In contrast to that, the Cx32 channels present in the oligodendrocytes inside gray matter form heterotypic GJ channel with astrocytes mediated by astrocyte/oligodendrocyte Cx30/Cx32 channels, where it is additionally expressed primarily in perikaryonic regions and proximal processes of oligodendrocytes (Orthmann-Murphy et al. 2008). In contrast, in normaly developed myelin, Cx29 is generally not abundant in oligodendrocyte cell bodies. It is observed in small plaques along oligodendrocytic processes, particularly at the myelin sheaths enwrapping smaller axons and in the juxtaparanodal region of neurons. Cx29 does not majorly colocalize with Cx32 (Altevogt et al. 2002). Cx29 hemichannels are also observed along the adaxonal membrane of the small myelinated fibers and in the internode, which is present in both gray and white matter. Cx29 is recently shown to be incapable of forming functional GJICs with other cells (Nagy et al. 2003).
Oligodendrocytes are solely dependent on astrocytes for GJ mediated homeostasis maintenance. In any case, intra-oligodendrocytic GJs appear to be concentrated at paranodes. Thus, astrocyte/oligodendrocyte GJ coupling is more crucial near oligodendrocytic somata and proximal processes. This case, astrocyte/oligodendrocyte GJs are mainly formed by Cx43/Cx47 mediated channel and Cx43/Cx47 outnumbers Cx30/Cx32 channels at oligodendrocyte somata (Orthmann-Murphy et al. 2008; Wasseff and Scherer 2011). The Cx47 (GJA12; another α-Cx) is primarily observed in the proximal processes of the oligodendrocytes and the oligodendrocyte somata. Cx47 is also present on the outer layer of the myelin sheath, present both in the white and gray matter of the CNS. Cx43/Cx47 channels couple oligodendrocytes with astrocytic processes. In mice, Cx47 is primarily found in the myelinating cells of CNS, and it is expressed solely by oligodendrocytes. Thus, Cx47 is very crucial in CNS homeostasis. Cx47 was previously believed to be by neurons, (Teubner et al. 2001). Later, it was shown to be mainly observed most frequently but not exclusively in cells of WM region like the deep cerebellar white matter, the corpus callosum, the spinal cord white matter, and the optic nerve (Menichella et al. 2003; Odermatt et al. 2003). Large numbers of Cx47-positive cells is also seen in the anterior commissure, the optic chiasma and the striatum. This GJ network mediates distribution of the excess K + ions and glutamate produced during neuronal activity, and also putatively provides a lactate shuttle and mediates Ca2 + wave propagation.
This study aims at understanding of alteration of nervous system-specific GJ proteins and its role in demyelinating diseases. In different neuroinflammatory and neurodegenerative conditions, the Cx proteins are altered. Current investigations precisely show that alteration of these Cx proteins could have significant impact on tissue homeostasis. The human CNS demyelinating disease, multiple sclerosis (MS) is one of such neuroinflammatory disease but the role of GJs in the chronic progression of MS was not lucid till the recent past. Animal model based studies were helpful to explore the mechanism of initial alteration and elucidate the significant role of Cxs in chronic progression of MS. This study mainly focuses on the current insights on the GJs and its involvement in CNS homeostasis and maintenance of oligodendrocyte/myelin health, with a specific emphasis to the existing investigations involving different models of MS.
Oligodendrocytes are mainly dependent on astrocytes for maintaining ionic and nutrient homeostasis, which is primarily controlled by GJIC. Conditional deletion of both oligodendrocytic GJ proteins, Cx32 and Cx47 demonstrated that astrocyte/oligodendrocyte GJ coupling is heterotypic, and this is mainly mediated by Cx43/Cx47 and Cx30/Cx32 channels, forming a ‘glial syncytium’ (Orthmann-Murphy et al. 2007).
An important initial study elucidating the role and localization of Cx47 showed that Cx47 expression is similarly regulated, compared to the myelin-associated genes and it partially colocalizes with Cx32 in oligodendrocytes. The temporal expression profile of Cx47 mRNA was similar to expression profile other myelin related genes like proteolipid protein (Plp), which encodes for a major constituent of compact myelin. In the md rat, which carries a mutation in the Plp gene, the levels of Cx47 and Cx32 were much lower compared to control. In the gray matter, the majority of Cx47 signal was reported to colocalize with Cx32. Few of the dispersed puncta were solely positive for Cx32. Thus, in overall, Cx32 and Cx47 expression highly colocalizes, which is consistent with the hypothesis that these GJ proteins might provide similar and redundant functions. In contrast to Cx32 knockout mice, where no demyelination was detected in CNS, the double knockout (for both Cx32 and Cx47) mice exhibited severe tonic seizures, associated with abnormal limb movement and loss of consciousness. Extensive CNS pathological alterations was observed, which was confined to myelinated axons. Numerous myelinated fiber tracts with markedly enlarged extracellular spaces were observed in between the axon and its myelin sheath. In this condition of CNS, macrophages were observed to contain myelin debris. The demyelinated and hypomyelinated axons were observed with enlarged spaces of periaxonal oligodendrocyte cytoplasm. Consistent pronounced loss of myelinated axons was evident in the optic nerve (Menichella et al. 2003).
Combined loss of Cx32 and Cx47 induced demyelination and oligodendrocyte cell death led to the hypothesis that Cx47 is crucial for myelination. Later, Cx43/Cx47 and Cx30/Cx32 channels were reported to have distinct single-channel properties, macroscopic appearance and different dye permeabilities. Cx30/Cx32 and Cx43/Cx47 channels are similarly permeable to AF 350 (a GJ permeable small molecule; charge 1) but differently permeable to Lucifer yellow (LY) (another GJ permeable small molecule; charge 2). So, other multivalent anions like ATP (Goldberg et al. 2002) or IP3 (Niessen et al. 2000) may also differentially pass through these channels. In addition to the charge, other factors which affect the permeability, are the molecular architecture of the channel pore and the size and shape of the permeating molecule (Harris 2007).
Replacement of the Cx47 gene with an enhanced green fluorescent protein (EGFP) reporter demonstrated that homozygous mutant mice had no gross morphological or behavioral abnormalities. But at the same time, ultrastructural investigations performed by electron microscopy revealed that a conspicuous vacuolation of nerve fibers was observed in the white matter regions, particularly at the myelination start site of the optic nerve, where the axons are first contacted by myelinating oligodendrocytes. In contrast, peripheral myelination was not affected in Cx47-deficient mice. These pathological features were worsened by double deletion of Cx32 and Cx47, which exhibited much more abundant vacuolation in nerve fibers than mice deficient for only Cx47. Hence, redundancy in functional perspective or compensatory regulation of oligodendrocytic Cx expression may explain the relatively mild phenotype observed in Cx32-deficient mice (Anzini et al. 1997; Nelles et al. 1996; Scherer et al. 1998) and Cx47-null mice (Odermatt et al. 2003). However, there was no significant alteration in Cx29 or Cx32 transcripts observed in Cx47 EGFP (−/−) mice, compared to wild-type mice. Cx47-deficient mice display myelination abnormalities, which includes sporadic vacuolation of nerve fibers, in and around the compact myelin or periaxonal space.
Cx30/Cx47 double knockout results in severe phenotypical alterations, which are characterized by the loss of astrocyte/oligodendrocyte functional GJ coupling and altered myelin pathology both in young and adult mice (Tress et al. 2012). Similarly, apoptosis of astrocytes, vacuolization and malformation of white matter region and death as early as by 16 weeks of age is reported for the animals, which are deficient in both Cx43 and Cx32. The underlying mechanism of these pathologies remained elusive by physiological characterization (Magnotti et al. 2011). The double knockout of Cx43 and Cx32 also resulted in profuse microglial activation, astrogliosis, and was associated with loss of myelin specifically in the forebrain region. The hindbrain region was moderately affected in adult mice. A strong reduction in the number of myelinating oligodendrocytes is associated with astrogliosis and prominent neuroinflammation. The activated microglia are reported to be involved in oligodendrocytic death either by internalization of damaged oligodendrocytes or by inducing oligodendrocyte necrosis occurring at the time of removal of cellular debris. Progressive demyelination is seen in the whole cortical region, and less dense myelinated tracts are observed in the thalamus of the Cx43/Cx32 double knockout mouse. Most importantly, immunostaining exhibited loss of oligodendrocytic Cx47 in the Cx43-deficient brain sections, which suggests that the stability of oligodendrocytic Cx47 GJ channels depend on astrocytic Cx43 expression, and unravels a novel importance of Cx43 mediated GJCs (May et al. 2013).
A double deficiency of Cx43 and Cx32 in mice induces loss of Cx47 mediated channels, whereas Cx47 mRNA levels remain unaltered. The absence of Cx43 leads to deficiency of Cx47 phosphorylation. A mutated Cx43, which is only delivered to plasma membrane but does not form functional channel, shows that presence of Cx43 at the cell surface is necessary and sufficient to normal expression, phosphorylation and stability of Cx47-mediated GJ plaques at cell surface (not depending on Cx43 GJC function on cell surface). Thus, after docking of an astrocytic Cx43 connexon to cell surface, it is predicted to interact with an oligodendrocytic Cx47 connexon, which might lead to a conformational change in the C-terminal region of Cx47. This phenomenon, in turn, might allow access for kinase(s), which exerts phosphorylation in the C-terminal domain of Cx47 and provides stabilization of Cx47 GJs at the cell surface. This way, presence of Cx43 at the cell surface might help the Cx43/Cx47 heterotypic channels to remain in the plasma membrane. The homotypic Cx47/Cx47 channels may not be formed in vivo. Thus, Cx43/Cx47 channels are exclusively important for astrocyte/oligodendrocyte cross-talk. Even if minute amounts of Cx47/Cx47 channels persist, that cannot serve a major function maintaining myelin integrity (May et al. 2013). Hence, Cx43 is important to control the stability and phosphorylation of Cx47 in GJ plaque, in vivo.
Thus, these GJ channels play a pivotal role in controlling panglial ionic homeostasis, especially, K+ buffering. The K+ ions, which are released from myelinated axons, are likely to accumulate in the periaxonal space. After that, it is probably dispersed by entering axons and oligodendrocytes via Na + K+-ATPases or possibly released via paranodal axoglial junctions. Once K+ enters the inner regions of an oligodendrocyte, it may disperse through the reflexive Cx32/Cx32 GJCs and then enter into the astrocytes via oligodendrocyte/astrocyte (Cx32/Cx30 and Cx47/Cx43) GJCs. Then the K+ is diffused away in astrocytic network by Cx43/Cx43 or Cx30/Cx30 channels. Cx32/Cx30 GJCs are primarily found on the outer layer of myelin sheaths and in the somatic region of oligodendrocytes, which are present mainly in the gray matter, compared to the white matter. In white matter regions, Cx47/Cx43 channels are primarily observed. Compared with Cx32/Cx30 GJCs, Cx47/Cx43 channels are more symmetrical in relation to the permeability properties. Cx47/Cx43 GJCs are mainly localized in the oligodendrocyte somata of the white matter regions, where they outnumber Cx32/Cx30 mediated heterotypic panglial GJCs. Thus, they might be involved in a fast dispersal of K+ ions from oligodendrocytes to astrocytic network in white matter and play a pivotal role in ionic buffering in these regions. The depletion of either Cx43 or Cx47 thus affects maintenance of white matter function. Along with that, nutrient homeostasis like lactate shunting might be affected. In addition, small molecule (like leukemia inhibitory factor, LIF) mediated signaling, which is essential to form myelin proteins (for example, myelin basic protein, MOG), also might be perturbed (Ishibashi et al. 2006). The functional importance of Cx43/Cx47 channels is primary underlying objective of these studies.
2 Expression and alteration of gap junctions in different diseases models
Not only mutations of GJs are associated with human neurodegenerative diseases but also in different other disease conditions and viral infections, GJs are reported to be altered. Although, alteration of Cx47 (being comparatively new Cx protein to be discovered) is relatively less explored, the alteration of Cx43 (most well-studied Cx) has been reported in different pathological conditions. Here we discuss a number of pathological conditions, where CNS Cx expression is altered.
In Lewis rat brain, Borna disease virus (BDV) infection induces dentate gyrus degeneration, where astroglial Cxs, Cx43 and Cx30 are downregulated during the course of persistent viral infection. BDV infection is also associated with astrogliosis (Koster-Patzlaff et al. 2007). In another study, human influenza virus was administered in E9 pregnant Balb/c mice and the virally exposed littermates showed significant decrease in brain Cx43 level at postnatal day 56. Although abnormal glial-neuronal communication is suggested to be associated with increased cell proliferation and decreased cell-to-cell communication, the mechanism of Cx43 alteration and role of Cx43 mediated cell-to-cell coupling in the growing brains of virus-challenged animals was not thoroughly studied (Fatemi et al. 2008). A cell culture based study demonstrated bovine papillomavirus type 4 (BPV-4) E8 protein is associated with reduction of GJIC, which was assumed to be mediated by binding to ductin (Faccini et al. 1996). Cx43 hemichannels are observed to be opened due to HIV infection in astrocytes, which results in dysregulated secretion of a soluble protein which inhibits Wnt signaling (dickkopf-1) (Orellana et al. 2014). In general, infection and inflammatory agents reduce Cx43 expression and function of GJs. However, despite of being inflammatory in nature, HIV is different because it sustains Cx43 expression and GJCs in astrocytes for the maintenance of persistent infection (Eugenin and Berman 2007). Functional GJC formation promotes the spread of toxic signals from a few HIV-infected astrocytes to uninfected glial cells. This alteration allows the spreading of toxic mediators, which dysregulate the glutamate and CCL2 secretion (Eugenin et al. 2011). Interestingly, the few HIV infected astrocytes are protected from being apoptotic by a viral infection-dependent mechanism, thereby acting as a viral reservoir within the CNS. Rous sarcoma virus (RSV)-induced transformation of the mammalian fibroblasts is associated with an early and profound disruption of GJCs and pp60v-src was predicted to directly regulate Cx43 channel closure upon infection (Crow et al. 1990). Herpes simplex virus (HSV-2) induces reduction of Cx expression and GJC formation by direct tyrosine phosphorylation of Cx43 (Castellano and Eugenin 2014; Fischer et al. 2001). Swine Flu virus infection also causes depletion of endothelial Cx43 expression in an extracellular signal dependent manner (mediated by c-Jun N-terminal kinase and other kinases) (Hsiao et al. 2010).
Cxs are reported to be important for different cell biological functions like antigen cross presentation upon viral infection. GJs enable coupled cells to exchange antigens, derived from viral peptides and trigger cytotoxic T cell mediated immune response, even when some cells were never directly exposed to the pathogen (Neijssen et al. 2005). Cx43 is also recruited to the immunologic synapse during T cell priming, suggesting that GJ and HCs also participate in the function of antigen presentation (Mendoza-Naranjo et al. 2011). Both Cx26 and Cx43 are expressed at the contact points between the radial glial fibers and migrating neurons, which in turn, provide dynamic adhesive contact points that interact with the internal cytoskeleton and help in glial-guided neuronal migration (Elias et al. 2007). Hence, proper migration of the cells is predicted to be important for both development and regeneration processes. Virus-induced downregulation of GJ proteins is predicted to have important consequences in vivo.
3 Human CNS demyelinating disease multiple sclerosis and its existing models
As described in the previous sections, Cxs are crucial for human myelination. MS is a human CNS demyelinating disease, which is characterized by foci of inflammation in the CNS leading to loss of myelin sheath, axonal loss, and reactive astrogliosis. The immunomodulatory therapies, restricting entry of peripheral immune cells, may be successful in partial disease protection in the relapsing-remitting phase, reducing the occurrence of focal lesions. In contrast, secondary progression and neurodegeneration cannot be restricted with these medications and are current potential challenges to therapies available, which mainly target the peripheral immune process. Although oligodendrocyte precursor cells (OPCs) are observed to be recruited to the MS lesions following demyelination and oligodendrocyte loss. Oligodendrocytic GJs are vital for generation and maintenance of CNS myelin, but their involvement in MS progression is relatively unexplored.
MS is characterized by two pathological hallmarks, which are demyelination and axonal loss. It is believed to be spontaneous, acquired, inflammatory disease by nature but the etiology of the disease is unknown. Studies show genetic traits controlling immune factors are of paramount importance to determine susceptibility to MS. Environmental factors are constantly being investigated that predispose the host to MS. Low vitamin D and CNS viral infections are hypothesized to be crucial for initiation and progression of the disease (Ascherio and Munger 2007a, b).
About 85% of patients of which women are majorly susceptible show initial occurrence of MS at the ages as early as 20 to 40 (with a sharp peak of the symptoms at about age 30) and intermittent episodes of neurological dysfunction are observed, which are termed as ‘attacks’ or ‘relapses’. The major symptoms include impairment of the motor nerve functions, blurred vision, accompanied with sensory disturbance (either tingling or loss of sensation). These symptoms typically remit at this period of relapsing-remitting MS, often to extent degree that neurological function returns to its normal functional level, but intermittent attacks are observed. Clinical investigations confirm MS progression by MRI scans which show abnormal signal and plaques of demyelination in the brain (the regions like cerebral cortex which is affected during early stages of MS, later in periventricular area and posterior fossa) and spinal cord white matter. Often, transient disruption of BBB is also observed. This phase of the disease is termed as relapsing-remitting MS (RR-MS), which is observed between 5 and 30 years. RR-MS is most commonly followed by secondary progressive MS (SP-MS), during which neurological function slowly worsens, with increased number of attacks. SP-MS is associated with severe motor function impairment with loss of ability to independently walk. However, MS pathology is unpredictable, with some patients showing little effect termed as ‘benign’ MS or primary progressive MS or severe pathological condition leading to death, termed as ‘Marburg’s variant of MS’ (Ransohoff 2012).
The pathological hallmarks of MS are characterized by infiltration of blood-borne immune cells to the CNS parenchyma (primarily lymphocytes and monocytes) and disruption of the BBB. CNS axons are demyelinated, which prominently involves the action of activated macrophages (derived from either resident microglia or peripheral monocytes). Targeting of the myelin occurs by multiple processes like myelin-specific antibodies, production of inflammatory cytokines by T cells, and activated microglia/macrophage mediated myelin stripping. Innate immune factors such as interleukin (IL)-1β, IL-6, as well as the adaptive-immune cytokines such as interferon (IFN)-γ, IL-17 and IL-23 performs important function. Presence of excess reactive oxygen (ROS) and nitrogen species (NOS), along with the prostaglandins and vasoactive factors functions synergistically along with the IL-mediated inflammation. Chemokines (mainly CCL2, CCL3, CCL4, CCL5, CXCL10, CXCL12 and CXCL13) are also involved in myelin degeneration (Frohman et al. 2006; Prineas and Graham 1981; Zhang et al. 2000). Large-scale oligodendrocyte loss, hypoxic tissue damage and an altered pattern of inflammation is associated with pathology of MS. In addition, activation of astrocytes (termed as ‘astrogliosis’), axonal loss and demyelination are the main pathological signs of this demyelinating disease.
As the etiology of MS still remains unclear, there are several animal models to study and investigate the pathology and cause-effect relationship of MS. An autoimmune model, experimental autoimmune encephalomyelitis (EAE), is the most widely applied model in the field of MS research. Myelin basic protein (MBP) was the first identified antigenic constituent of myelin, followed by many proteins like PLP and MOG, which was used to create EAE. In this model, the model animals moved from nonhuman primates to the larger rodents (rats, guinea pigs) to mice, taking advantage of ease of handling and genetic tools (transgenics and knockouts). Most studies are done with C57Bl/6 mice, where immunization is performed by subcutaneous injection of MOG peptide, emulsified in the Freund’s adjuvant and supplemented with Mycobacterium tuberculosis extract. Mice are injected with pertussis toxin as a booster on the day of immunization and 2 days thereafter. A RR-MS variant of the demyelinating disease is reproducible in EAE in some mouse strains, most prominently in SJL/J. Histopathology of EAE mouse spinal cord shows the white matter is more affected than gray matter, which is also seen in MS. Cells, rather than serum is important for disease progression, and T cell clones are found to mediate paralytic inflammation and also react to short peptides of myelin proteins. Transfer of these T-cell clones, by adoptive transfer, can solely transfer the disease to non-immunized mice. Predominantly CD4 + T cells are important and spinal cord is affected out of proportion to brain regions in EAE (Bernard et al. 1976; Waksman and Adams 1962; Yasuda et al. 1975). Major MS drugs available currently including natalizumab, glatiramer acetate (a mixture of oligomeric peptides) and fingolimod were mainly studied in EAE, but these drugs are targeted towards blocking adaptive-immune response and show less efficacy in SP-MS. The EAE model is silent on few questions in MS research such as limited insight into MS disease progression and remyelination phenomena in MS (Pelletier and Hafler 2012; Steinman and Zamvil 2006; Yednock et al. 1992). In contrast to MS, where pathology is mainly restricted to brain, EAE pathology is mainly spinal cord restricted. Moreover, EAE, being a completely autoimmune model, neither elucidate the role of innate immune cells/ B cells nor elaborate on the importance of oligodendrocytic death and neuroprotective approaches. The alteration of CNS GJs has been investigated in this model. The studies showed that there was an acute depletion of Cx43 in initial phase of disease, which was resolved during the chronic demyelinating phase. The oligodendrocytic Cx47 was reported to be reduced in and around the demyelinated plaques. However, the mechanism of GJ alteration was not completely investigated.
There are also the other models also to understand the multifaceted etiology of MS. Toxic models of MS involve administration of toxins to induce demyelination, which overcomes concerns about timing and localization of loss of myelin and enables the study of remyelination. Because enhancing remyelination is envisaged to be crucial for neuroprotection in MS and aims at halting the chronic progression of MS, toxin-induced models are important tool for current translational research. There are two models, which are used extensively. First, the copper chelator named cuprizone (2% in chow) is fed to susceptible strains of 4–6-week-old mice. Cuprizone induces dysfunction of mitochondrial complex IV, causing selective toxicity for oligodendrocytes. Oligodendrocytes present in the corpus callosum and hippocampus undergo apoptosis after 3 weeks of cuprizone treatment. After the toxin is discontinued, remyelination starts. This model provides insights into the damage and repair of myelin and determinants of oligodendrocyte cell death (Arnett et al. 2001; Matsushima and Morell 2001; Skripuletz et al. 2011). Another model uses microinjection of ethidium bromide or recently used lysophosphatidylcholine into white matter tracts which causes prompt demyelination, followed by remyelination. The model helps in examination of cell biological and molecular determinants of remyelination (Blakemore et al. 1977; Blakemore and Franklin 2008). In contrast, this model does not elucidate the role of immune cells, which is seen in MS and poses potential challenges for interpreting the responses of OPCs/stem cells, and other cellular mediators of remyelination, performing important function due to the dynamic nature of demyelination and remyelination. Alteration of GJ remodeling has not been addressed in this model, till date.
4 Viral models of multiple sclerosis
Current insights about potential infectious etiologies of MS suggests that MS is most likely to be caused by a virus because a good number of MS patients bear high concentrations of IgG in CSF and brain, which is manifested as oligoclonal bands. Many chronic inflammatory CNS disorders have an infectious etiology. In humans, several types of demyelinating encephalomyelitis are associated with viral infection, and in animal models infection with viruses induces demyelination during chronic infection. For example, paramyxovirus nucleocapsids and high concentrations of antibody to measles virus were found in brains of patients having subacute sclerosing panencephalitis (SSPE: a chronic neuroinflammatory disease of both grey and white matter) patients. Progressive multifocal leucoencephalopathy (PML), which is also a human demyelinating disease characterized by rapidly progressive dementia and motor deficit, was found to be caused by a Human papovavirus (JC virus) infection in the oligodendrocytes in a patient with PML. However, till date, no reproducible viral infection has been isolated from the CNS of MS patients. A possible role of human herpesvirus type 6 (HHV-6) and establishment of a latent CNS infection in man has been associated with MS, as HHV-6 protein and DNA have been identified in the neuroglial cells present in ‘active’ MS lesions. Oligoclonal bands, associated with MS, have shown features of antigen-driven response: like clonal amplification and extensive somatic hypermutations. Thus it is strongly predicted that a virus might be reactivated after years of latency and induce oligodendrocyte damage or could initiate immunopathology, which might lead to demyelination. Hence, viral models of MS are of prime importance to study immunopathogenesis of MS as well as direct oligodendrocyte damage. Along with these, viral models of MS elucidate direct virus induced alteration of neuronal and glial cells, and viral-induced ‘neuroinflammation’. In general, viral infection induced demyelination simultaneously uses two mechanisms: direct infection to neural and/or glial cells and immune-mediated (both innate and adaptive) tissue injury (Gilden 2005).
In animals, there are multiple viruses which induce demyelination. For example, a few strains of mouse hepatitis virus (MHV) or Theiler’s murine encephalomyelitis virus (TMEV) infection in mouse, canine distemper virus infection in dogs, and Visna virus and caprine arthritis-encephalitis virus infection in sheep and goats all induce demyelination. Each of these viruses is able to establish persistent infection in their host, so that there is sustained virus replication over a long period of time, but without killing the host. Even when the reproductive virus is in CNS, inflammation/demyelination is observed. There are several viral models of MS in which picornavirus TMEV and certain strains of the coronavirus MHV infection in mice have given useful mechanistic information on MS. In these models, successful infection is a prerequisite for demyelination, and the cause/effect relationship between viral infection and demyelination, makes these models suitable for exploring the etiology and pathogenesis of MS. Virus induced demyelination is observed in the chronic disease phase, which is associated with viral persistence. A biphasic disease in the CNS, consisting of early acute meningoencephalitis and late chronic demyelination is caused by the infection of some strain of TMEV or MHV in the susceptible strains of mice. Importantly, similar to MS patients, these models do not exclude the factor of genetic predisposition. For example, TMEV infection induced demyelination develops only in SJL/J mice, but not in C57Bl/6 mice and MHV-A59 induced demyelination is studied in C57Bl/6 mice only. TMEV-induced demyelination mainly relies on CD8+ autoreactive cytotoxic T cells or regulatory T cells. In addition, the antibody against TMEV cross-reacts with oligodendroglial galactocerebroside (GalC), and passive transfer of anti-TMEV antibody is able produce demyelination similar to adoptive transfer of EAE. An acute focal demyelination can be observed upon intracranial inoculation with a TMEV-infected macrophage cell line, and depletion of macrophage cells cures TMEV-induced demyelination. There are also close similarities between TMEV-induced demyelinating disease in animals and MS in humans, like neuropathological similarities, including axonal damage and remyelination, involvement of immune system and paucity of T-cell apoptosis in demyelinating disease (Lipton and Canto 1976; Lipton and Dal Canto 1976; Wroblewska et al. 1977). In contrast, this model does little to describe the role of the innate immune system and initial immuno-pathogenesis. The pathogenesis of TMEV-induced demyelination differs from that observed in MS because persistent viral infection in the CNS of the MS patients has not been demonstrated.
Another model of virus induced demyelination is mediated by neurotropic strains of MHV, which belongs to the coronavirus family. The MHV-induced disease is dependent on several factors including the age and strain of the mouse, the infectious strain of MHV is being used, and the route of virus inoculation. Closely related strains of MHV differ in viral tropism and pathogenic properties. All strains are hepatotropic (e.g., MHV-2), some are primarily neurotropic (e.g., JHM, MHV-4: which induce severe encephalitis); while others (e.g., MHV-A59 and MHV3) are equally hepatotropic and neurotropic (Houtman and Fleming 1996b; Lavi et al. 1984; Stohlman and Weiner 1981). Highly neurovirulent MHV-JHM or JHMV strain also suggests that MHV-induced demyelination is primarily immune mediated. The demyelination can be completely eliminated by elimination of functional T and B cells in RAG knockout mice, which can be further reversed upon transfer of splenocytes from immunocompetent mice. CD4+ or CD8+ T cells suffice for MHV-JHM induced demyelination (Houtman and Fleming 1996a; Knobler et al. 1981; Sussman et al. 1989).
The neurotropic strain, MHV-A59 induces a biphasic disease, where hepatitis and meningoencephalitis are observed in the acute phase of infection (day 5–6 p.i.) and chronic demyelination and axonal loss are observed in the chronic phase of disease (day 30 p.i.). Demyelination is histopathologically observable or it is accompanied by chronic hind limb paralysis. Both MHV-JHM and MHV-A59 cause inflammatory demyelination in the CNS (which is mainly scored on basis of spinal cord histopathology, as spinal cord has a defined clear grey/white matter structures) whereas MHV3 only causes vasculitis. The spike gene is of primary determinant of demyelination, as it is a major determinant of tropism and virulence CNS cells (Bender and Weiss 2010). MHV-A59-induced demyelination develops in the absence of B and T cells (Matthews et al. 2002a, b). Furthermore, depletion of pan-T cells after the acute stage of infection does not reduce demyelination. MHV-A59 induced demyelination is majorly caused by activation of microglia and this model elucidates the crucial role of the innate immune system in this neuroinflammatory disease. Different related strains of MHV induce demyelination via different mechanisms. For example, induced demyelination is believed to be the result of lytic infection of oligodendrocytes. It is noteworthy that some strains of MHV infection can induce demyelination in the absence of intact immune responses. Current research uncovers that direct virus induced CNS cell damage or virus-persistence induced altered cellular physiology is a key player of virus-induced demyelination. In contrast, MHV-2, a weakly neurotropic virus (closely related to MHV-A59), differs in the capability of persisting in CNS and cannot cause demyelination (Das Sarma et al. 2000). For MHV-A59, viral genome persists in the white matter of infected mice during the chronic demyelinating phase and it is suggested that glial cells, specifically astrocytes, may be the site of viral persistence during the disease (Das Sarma et al. 2008; Lavi et al. 1987). In contrast, how astrocytes take part in virus-induced demyelination and which molecules are affected due to persistent viral infection remains largely unknown. The neuropathological hallmarks and pathophysiology exerted by demyelinating stain of MHV in acute and chronic stage of inflammation is depicted in figure 1.

MHV infection as a model of gliopathy and demyelination. The demyelinating stains of MHVs cause meningitis (A) and encephalitis (B, C). The chronic infection in CNS is majorly restricted to brain and causes demyelination (D) and axonal loss (E). During acute phase, brain astrocytes are infected as demonstrated by colocalization of GFAP and viral-N staining (F).
A key feature of demyelinating strains of MHVs is reported to be their specific utilization of microtubule (MT)-network. Herpes simplex virus 1 (HSV-1) is able to utilize the MT network for cellular trafficking of virions and viral glycoproteins to deliver the virus to its release sites (Mingo et al. 2012). Vaccinia virus is reported to take the help of cytoskeletal elements like both MT networks and actin filament for viral egression (Hollinshead et al. 2001). Adenovirus entry to the host cells is also mediated by MT-network and associated molecular motors, which are used for retrograde transport (Yea et al. 2007). The adeno-associated virus (AAV) also displays unidirectional retrograde movement on MTs, from the cell periphery to the nuclei (Xiao and Samulski 2012). Virus mediated utilization of cytoskeletal network also can disrupt normal cellular processes and trafficking. For example, NSP-4, a rotavirus membrane glycoprotein, binds to the MTs and arrests normal cellular ER-to-Golgi trafficking (Xu et al. 2000). Ebola Virus Matrix Protein VP40 interacts directly with MTs. Many viruses also use the associated molecular motors for trafficking to the cell surface (Ruthel et al. 2005). It has been shown that the Hantaan virus (a negative stranded RNA virus) nucleocapsid protein takes the help of MTs for intracellular trafficking and the retrograde movement occurs via molecular motors such as dynein (Ramanathan et al. 2007). In addition, during adenovirus infection, cytoplasmic dynein is reported to mediate interaction between viral capsid and MTs (Kelkar et al. 2004). A neurovirulant strain of MHV, MHV-JHM, specifically uses the MT network for transneuronal spread and viral trafficking (Pasick et al. 1994). Though RSA59, a demyelinating recombinant strain of MHV-A59, is shown to specifically use MT networks for intercellular spread, direct cell-biological alteration associated with viral trafficking was not shown (Biswas and Das Sarma 2014).
As the glial cells, like astrocytes and oligodendrocytes are believed to be the primary sites of viral persistence, the alteration of glial cell function have a high impact in this viral model of demyelination. As described, the demyelinating strain of MHV, MHV-A59, has two major pathological peaks: peak of inflammation (acute phase: day 5 p.i.) and peak of demyelination (day 30 p.i.). (Das Sarma et al. 2000; Lavi et al. 1984, 1986). In this viral model of MS, MHV-A59 infects neurons and other glial cells. Previous studies as well as recent findings showed that astrocytes were infected, in vivo, in MHV-A59 infected C57Bl/6 CNS. Although astrocytes were primarily uninfected in spinal cords upon infection with both demyelinating and non-demyelinating strains of MHV, demyelinating recombinant strain of MHV (Kenyon et al. 2015), RSA59 was able to infect astrocytes in brain (Das Sarma et al. 2008). Similarly, in primary astrocyte culture, MHV-A59 is reported to induce persistent viral infection. Viral infection in primary astrocytes continues to be present for a long period of time, without showing obvious cytopathic effects (CPE) and cell death, even at a high dose of viral inoculum (Lavi et al. 1987). These preliminary studies did not elucidate the role of persistent viral infection in astrocytes and whether they were directly involved in producing demyelinating disease. Pathological and functional changes of astrocytes and astrogliosis are associated with MS. As discussed in the previous section, astrocytes are important in maintenance of cell-to-cell communication and CNS homeostasis, which is mediated by GJCs. Altered GJ communication in panglial system, which is mainly mediated by astrocytes, are believed to crucially involved in expansion of demyelinated plaques (Markoullis et al. 2012). Based on these basic findings, infection with demyelinating strain of MHV is hypothesized to remodel GJ expression in astrocytes, which, in turn, is predicted to be involved in initiation and progression of demyelinating disease.
5 Alteration of gap junctions in viral model of multiple sclerosis
Previous studies demonstrated that alteration of GJ proteins affect myelin formation, structure and function. Specifically, alteration of Cx43 and Cx47 is highly associated with CNS dysmyelination, and perturbation of oligodendrocyte function. In human demyelinating diseases also the GJ expression and function is retarded. Alteration of Cx protein expression is evident in MS patient tissues as well as in EAE. The most abundant GJ protein in the CNS, Cx43 is initially downregulated and partially expressed in normal level due to astrogliosis in chronic demyelinating phase. Whereas, oligodendrocytic coupling partner of Cx43, Cx47 expression is mainly reduced during chronic phase of demyelination. The loss of Cx43/Cx47 mediated GJCs is hypothesized to be a basis of perturbed astrocyte/oligodendrocyte homeostasis and playing a pivotal role in chronic expansion of demyelinated plaques. Importantly, the GJs are also observed to be localized in the intracellular compartment, which demonstrates the GJ protein trafficking, channel formation and function is restricted. All these studies are limited to elaborate the mechanism of initial loss of Cx43 during acute inflammation, the restriction of GJ proteins in the intracellular compartments and its role in chronic neuroinflammation.
In a viral model of MS, MHV-A59 infects astrocytes in vivo. Astrocytes are predicted to be major sites of viral persistence but the role of astrocytes in demyelination is not well understood. This model of virus induced demyelination is utilized to understand the basic role of astrocytes in the perspective of altered localization, expression and function of GJ proteins in the panglial network. Establishment of primary astrocyte culture provided an excellent platform to understand the cell biological basis of altered Cx43 expression and localization during acute neuroinflammation. MHV-A59 infection in astrocytes induced a reduced Cx43 protein and RNA expression. The depletion of Cx43 mRNA might be due to short half-life of Cx43 and presence of AU-rich region in the untranslated region (UTR) of Cx43 (Basu et al. 2015). In addition, the synthesized Cx43 was restricted in ER/ERGIC (figure 2). Therefore, the GJ plaque formation and functional homotypic Cx43/Cx43 mediated channel formation between astrocytes were diminished significantly (Basu et al. 2015). Cx43 is also altered in the meningeal fibroblast, an important part of BBB and this alteration has important consequences during the MHV induced neuroiflammation (Bose et al. 2018).

Neurotropic demyelinating strain of mouse hepatitis virus (MHV-A59) infection leads to downregulation and intracellular retention of Connexin43 in neonatal mouse brain derived primary astrocytes. The illustration is an amalgamation of immunofluorescence images of primary astrocytes, stained with either MHV-A59 nucleocapsid (N; green) and Connexin43 (Cx43; red in large central panel and top left inset) or GFAP (green) and Cx43 (red in bottom right inset). Nuclei were counterstained with DAPI (blue). The large central panel is MHV-A59 infected primary astrocytes where Cx43 was retained in the intracellular compartment, specifically in the virus infected cells. In the same culture, the cells which were not infected by MHV-A59, Cx43 was present as prominent puncta at the surface of two adjacent cells. The image is modified using Adobe Photoshop for a better understanding of Cx43 localization in uninfected and infected cells. The top left inset is a magnified infected single astrocyte where intracellular compartment retained Cx43 mostly colocalized with anti-N staining in a perinuclear compartment. The bottom right inset is a magnified uninfected single mock infected cell where discrete Cx43 puncta were present at the cell surface of GFAP positive astrocyte.
The understanding of primary molecular mechanism was the most important aspect of this study. Previous reports, demonstrating that demyelinating strains of MHV specifically used MT network for viral trafficking was hypothesized to be involved in altered localization of Cx43, as Cx43 is highly dependent on MT network to get delivered to the cell surface. It was also seen that Cx43/MT interaction was prominently perturbed in protein level and in the same time, MHV-A59 directly interacted with MT-network. Imaging based analyses evidently demonstrated viral particles replaced Cx43, at the cell surface and Cx43/MT colocalization was diminished in presence of demyelinating MHV-A59. Whether there is a direct competition for the molecular motors or associated glued and capping proteins are involved in this interaction demands further investigation.
The understanding of initial Cx43 expression, localization and function, which was a long-standing question in the field of MS (in the perspective of neuroinflammation and demyelination), raised an obvious question whether Cx43 is altered in vivo and it is associated with loss of oligodendrocytic GJ expression and loss of myelin. MHV-A59, which causes a clear biphasic disease in C57Bl/6 mice, served as an excellent model to assess the GJ expression both during acute inflammation, directly initiated by viral infection and also during chronic demyelination in absence of infectious viral particles in the system. Similar to that of observed in primary astrocyte culture in vitro, a reduced expression was observed specifically in and around the MHV-A59 infected area of mouse brain. The expression of Cx43 in total protein and RNA level was also reduced in acute phase. Cx47, being important in maintaining CNS myelination, was evaluated for expression during acute infection. MHV-A59 infection led to a small but significant downregulation of Cx47. The stability and expression of Cx47 is highly dependent on Cx43 surface expression in vivo. In the chronic demyelinating phase, the Cx43 expression was replenished back to its normal expression level. In contrast, Cx47 was sustained to be downregulated in the MHV-A59 infected brain. The persistent alteration of Cx47 was associated with loss of myelin marker PLP in the major white matter tracts of brain (Basu et al. 2017). The summary of the work and outcomes are shown in the figure 3.

Cell-to-cell communication and gliopathy in MHV induced demyelination. Demyelinating stain of MHV can infect astrocytes both in vivo and in vitro. This infection causes depletion of Cx43 expression and also restricts Cx43 protein trafficking to cell surface by a MT-dependent mechanism during acute phase of inflammation. The loss of Cx43 induces persistent loss of Cx47, which is associated with loss of myelin proteins.
6 Conclusion
The review reports show that virus infection can induce downregulation and alters MT-dependent trafficking of Cx43. Virus induced alteration in gap junctional intercellular channel formation is initiated with remodeling in astrocytes and meningeal fibroblasts and exerts panglial communication with oligodendrocytes mediated by Cx43/Cx47 channels. These findings finally give rise several questions like:
-
Whether the virus induced ER-stress and GJ associated chaperones could be involved in altered expression of GJ proteins, or which MT associated motor and glued proteins could be associated with MHV-A59 induced altered Cx43/MT interaction.
-
Which pathogenic or host factors selectively downregulates Cx43 mRNA and protein expression.
-
What is the putative molecular mechanism behind Cx43-induced persistent loss and destabilization of Cx47 in MHV-A59 induced model of MS.
-
Whether Astrocyte specific targeting and overexpression of GJ specific chaperones might be a fruitful approach to increase the delivery of Cx43 to the cell surface. It is plausible that improvement of Cx43 delivery and GJC formation in cell surface might be helpful in restoring the perturbed homeostasis during virus induced neuroinflammation and might induce cessation of chronic expansion of demyelinated plaques. However, spatio-temporal, tissue specific targeting of specific Cx protein in vivo, can pose potential challenges for therapeutic implication of targeting the Cx proteins.
Abbreviations
- AAV:
-
adeno-associated virus
- BBB:
-
blood brain barrier
- BCEC:
-
brain capillary endothelial cell
- BDV:
-
Borna disease virus
- BPV-4:
-
bovine papillomavirus type 4
- CNS:
-
central nervous system
- CMTX:
-
Charcot-Marie-Tooth disease
- CPE:
-
cytopathic effects
- Cx:
-
connexin
- EAE:
-
experimental autoimmune encephalomyelitis
- EGFP:
-
enhanced green fluorescent protein
- GalC:
-
Galactocerebroside
- GFAP:
-
glial fibrillary acidic protein
- GJ:
-
gap junctions
- GJIC:
-
gap junction intercellular communication
- HSV:
-
herpes simplex virus
- HHV:
-
human herpes virus
- HSVtk:
-
herpes simplex virus thymidine kinase
- Iba1:
-
ionized calcium-binding adapter molecule 1
- IL:
-
interleukin
- IFN:
-
interferon
- LIF:
-
leukemia inhibitory factor
- LY:
-
Lucifer yellow
- MBP:
-
Myelin basic protein
- MPTP:
-
Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- OPC:
-
oligodendrocyte precursor cells
- MHV:
-
mouse hepatitis virus
- MOG:
-
myelin basic protein
- MT:
-
microtubule
- MS:
-
multiple sclerosis
- NAWM:
-
normal appearing white matter
- ODDD:
-
oculodentodigital dysplasia
- Plp:
-
proteolipid protein gene
- PLP:
-
proteolipid protein
- PMLD:
-
Pelizaeus-Merzbacher-like disease
- RSV:
-
Rous sarcoma virus
- PML:
-
Progressive multifocal leucoencephalopathy
- RR-MS:
-
relapsing-remitting MS
- SP-MS:
-
secondary progressive MS
- SSPE:
-
subacute sclerosing panencephalitis
- TMEV:
-
Theiler’s murine encephalomyelitis virus
References
Altevogt BM, Kleopa KA, Postma FR, Scherer SS and Paul DL 2002 Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 22 6458–6470
Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U and Martini R 1997 Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin32. J. Neurosci. 17 4545–4551
Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK and Ting JP 2001 TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 4 1116–1122
Ascherio A and Munger KL 2007a Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann. Neurol. 61 288–299
Ascherio A and Munger KL. 2007b. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann. Neurol. 61 504–513
Basu R, Banerjee K, Bose A and Das Sarma J 2015 Mouse hepatitis virus infection remodels connexin43-mediated gap junction intercellular communication in vitro and in vivo. J. Virol. 90 2586–2599
Basu R, Bose A, Thomas D and Das Sarma J 2017 Microtubule assisted altered trafficking of astrocytic gap junction protein connexin43 is associated with depletion of connexin47 during mouse hepatitis virus infection. J. Biol. Chem. 292 14747–14763
Bender SJ and Weiss SR 2010 Pathogenesis of murine coronavirus in the central nervous system. J. Neuroimmune Pharmacol. 5 336–354
Bernard CC, Leydon J and Mackay IR 1976 T cell necessity in the pathogenesis of experimental autoimmune encephalomyelitis in mice. Eur. J. Immunol. 6 655–660
Biswas K and Das Sarma J 2014 Effect of microtubule disruption on neuronal spread and replication of demyelinating and nondemyelinating strains of mouse hepatitis virus in vitro. J. Virol. 88 3043–3047
Blakemore WF, Eames RA, Smith KJ and McDonald WI 1977 Remyelination in the spinal cord of the cat following intraspinal injections of lysolecithin. J. Neurol. Sci. 33 31–43
Blakemore WF and Franklin RJ 2008 Remyelination in experimental models of toxin-induced demyelination. Curr. Topics Microbiol. Immunol. 318 193–212
Bose A, Basu R, Maulik M and Das Sarma J 2018. Loss of Cx43-mediated functional gap junction communication in meningeal fibroblasts following mouse hepatitis virus infection. Mol. Neurobiol. 55 6558–6571
Castellano P and Eugenin EA 2014 Regulation of gap junction channels by infectious agents and inflammation in the CNS. Front. Cell. Neurosci. 8 122
Crow DS, Beyer EC, Paul DL, Kobe SS and Lau AF 1990 Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol. Cell. Biol. 10 1754–1763
Das Sarma J, Fu L, Tsai JC, Weiss SR and Lavi E 2000 Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J. Virol. 74 9206–9213
Das Sarma J, Iacono K, Gard L, Marek R, Kenyon LC, Koval M and Weiss SR 2008 Demyelinating and nondemyelinating strains of mouse hepatitis virus differ in their neural cell tropism. J. Virol. 82 5519–5526
Dermietzel R, Traub O, Hwang TK, Beyer E, Bennett MV, Spray DC and Willecke K 1989 Differential expression of three gap junction proteins in developing and mature brain tissues. Proc. Natl. Acad. Sci. USA 86 10148–10152
Elias LA, Wang DD and Kriegstein AR 2007 Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 448 901–907
Eugenin EA and Berman JW 2007 Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J. Neurosci. 27 12844–12850
Eugenin EA, Clements JE, Zink MC and Berman JW 2011 Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J. Neurosci. 31 9456–9465
Faccini AM, Cairney M, Ashrafi GH, Finbow ME, Campo MS and Pitts JD 1996 The bovine papillomavirus type 4 E8 protein binds to ductin and causes loss of gap junctional intercellular communication in primary fibroblasts. J. Virol. 70 9041–9045
Fatemi SH, Folsom TD, Reutiman TJ and Sidwell RW 2008 Viral regulation of aquaporin 4, connexin 43, microcephalin and nucleolin. Schizophr. Res. 98 163–177
Filippov MA, Hormuzdi SG, Fuchs EC and Monyer H 2003 A reporter allele for investigating connexin26 gene expression in the mouse brain. Eur. J. Neurosci. 18 3183–3192
Fischer NO, Mbuy GN and Woodruff RI 2001 HSV-2 disrupts gap junctional intercellular communication between mammalian cells in vitro. J. Virol. Methods 91 157–166
Frohman EM, Racke MK and Raine CS 2006 Multiple sclerosis–the plaque and its pathogenesis. N. Engl. J. Med. 354 942–955
Giaume C and McCarthy KD 1996 Control of gap-junctional communication in astrocytic networks. Trends Neurosci. 19 319–325
Gilden DH 2005 Infectious causes of multiple sclerosis. Lancet 4 195–202
Goldberg GS, Moreno AP and Lampe PD 2002 Gap junctions between cells expressing connexin43 or 32 show inverse permselectivity to adenosine and ATP. J. Biol. Chem. 277 36725–36730
Harris AL 2007 Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 94 120–143
Hollinshead M, Rodger G, H. Van Eijl, Law M, Hollinshead R, Vaux DJ and Smith GL 2001 Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 154 389–402
Houtman JJ and Fleming JO 1996a Dissociation of demyelination and viral clearance in congenitally immunodeficient mice infected with murine coronavirus JHM. J. Neurovirol. 2 101–110
Houtman JJ and Fleming JO 1996b Pathogenesis of mouse hepatitis virus-induced demyelination. J. Neurovirol. 2 361–376
Hsiao HJ, Liu PA, Yeh HI and Wang CY 2010 Classical swine fever virus down-regulates endothelial connexin43 gap junctions. Arch. Virol. 155 1107–1116
Iacobas DA, Urban-Maldonado M, Iacobas S, Scemes E and Spray DC 2003 Array analysis of gene expression in connexin-43 null astrocytes. Physiol. Genomics 15 177–190
Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL and Fields RD 2006 Astrocytes promote myelination in response to electrical impulses. Neuron 49 823–832
Kelkar SA, Pfister KK, Crystal RG and Leopold PL 2004 Cytoplasmic dynein mediates adenovirus binding to microtubules. J. Virol. 78 10122–10132
Kenyon LC, Biswas K, Shindler KS, Nabar M, Stout M, Hingley ST, Grinspan JB and Das Sarma J 2015 Gliopathy of demyelinating and non-demyelinating strains of mouse hepatitis virus. Front. Cell Neurosci. 9 488
Knobler RL, Haspel MV and Oldstone MB 1981 Mouse hepatitis virus type 4 (JHM strains) induced fatal central nervous system disease. I. genetic control and murine neuron as the susceptible site of disease. J. Exp. Med. 153 832–843
Koster-Patzlaff C, Hosseini SM and Reuss B 2007 Persistent Borna Disease Virus infection changes expression and function of astroglial gap junctions in vivo and in vitro. Brain Res. 1184 316–332
Kunzelmann P, Schroder W, Traub O, Steinhauser C, Dermietzel R and Willecke K 1999 Late onset and increasing expression of the gap junction protein connexin30 in adult murine brain and long-term cultured astrocytes. Glia 25 111–119
Lavi E, Gilden DH, Highkin MK and Weiss SR 1986 The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab. Anim. Sci. 36 130–135
Lavi E, Gilden DH, Wroblewska Z, Rorke LB and Weiss SR 1984 Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34 597–603
Lavi E, Suzumura A, Hirayama M, Highkin MK, Dambach DM, Silberberg DH and Weiss SR 1987 Coronavirus mouse hepatitis virus (MHV)-A59 causes a persistent, productive infection in primary glial cell cultures. Microbial Pathogenesis 3 79–86
Lipton HL and Canto MC 1976 Theiler’s virus-induced central nervous system disease in mice. UCLA Forum Med. Sci. 19 263–277
Lipton HL and Dal MC Canto 1976 Theiler’s virus-induced demyelination: prevention by immunosuppression. Science 192 62–64
Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS and Brosnan CF 2009 Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci. 29 7743–7752
Magnotti LM, Goodenough DA and Paul DL 2011 Deletion of oligodendrocyte Cx32 and astrocyte Cx43 causes white matter vacuolation, astrocyte loss and early mortality. Glia 59 1064–1074
Markoullis K, Sargiannidou I, Schiza N, Hadjisavvas A, Roncaroli F, Reynolds R and Kleopa KA 2012 Gap junction pathology in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol. 123 873–886
Matsushima GK and Morell P 2001 The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 11 107–116
Matthews AE, Lavi E, Weiss SR and Paterson Y. 2002a. Neither B cells nor T cells are required for CNS demyelination in mice persistently infected with MHV-A59. J. Neurovirol. 8 257–264
Matthews AE, Weiss SR and Paterson Y 2002b Murine hepatitis virus–a model for virus-induced CNS demyelination. J. Neurovirol. 8 76–85
May D, Tress O, Seifert G and Willecke K 2013 Connexin47 protein phosphorylation and stability in oligodendrocytes depend on expression of Connexin43 protein in astrocytes. J. Neurosci. 33 7985–7996
Mendoza-Naranjo A, Bouma G, Pereda C, Ramirez M, Webb KF, Tittarelli A, Lopez MN, Kalergis AM, Thrasher AJ, Becker DL and Salazar-Onfray F 2011 Functional gap junctions accumulate at the immunological synapse and contribute to T cell activation. J. Immunol. 187 3121–3132
Menichella DM, Goodenough DA, Sirkowski E, Scherer SS and Paul DL 2003 Connexins are critical for normal myelination in the CNS. J. Neurosci. 23 5963–5973
Mingo RM, Han J, Newcomb WW and Brown JC 2012 Replication of herpes simplex virus: egress of progeny virus at specialized cell membrane sites. J. Virol. 86 7084–7097
Nagy JI, Ionescu AV, Lynn BD and Rash JE 2003 Connexin29 and connexin32 at oligodendrocyte and astrocyte gap junctions and in myelin of the mouse central nervous system. J. Comp. Neurol. 464 356–370
Nagy JI, Li X, Rempel J, Stelmack G, Patel D, Staines WA, Yasumura T and Rash JE 2001 Connexin26 in adult rodent central nervous system: demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J. Comp. Neurol. 441 302–323
Nagy JI, Patel D, Ochalski PA and Stelmack GL 1999 Connexin30 in rodent, cat and human brain: selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience 88 447–468
Neijssen J, Herberts C, Drijfhout JW, Reits E, Janssen L and Neefjes J 2005 Cross-presentation by intercellular peptide transfer through gap junctions. Nature 434 83–88
Nelles E, Butzler C, Jung D, Temme A, Gabriel HD, Dahl U, Traub O, Stumpel F, Jungermann K, Zielasek J, Toyka KV, Dermietzel R and Willecke K 1996 Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc. Nat. Acad. Sci. USA 93 9565–9570
Niessen H, Harz H, Bedner P, Kramer K and Willecke K 2000 Selective permeability of different connexin channels to the second messenger inositol 1,4,5-trisphosphate. J. Cell Sci. 113 1365–1372
Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Bussow H, Schilling K, Steinhauser C and Willecke K 2003 Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 23 4549–4559
Orellana JA, Saez JC, Bennett MV, Berman JW, Morgello S and Eugenin EA 2014 HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J. Neurochem. 128 752–763
Orthmann-Murphy JL, Abrams CK and Scherer SS 2008 Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 35 101–116
Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS and Abrams CK 2007 Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 27 13949–13957
Pasick JM, Kalicharran K and Dales S 1994 Distribution and trafficking of JHM coronavirus structural proteins and virions in primary neurons and the OBL-21 neuronal cell line. J. Virol. 68 2915–2928
Pelletier D and Hafler DA 2012 Fingolimod for multiple sclerosis. N. Engl. J. Med. 366 339–347
Prineas JW and Graham JS 1981 Multiple sclerosis: capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann. Neurol. 10 149–158
Ramanathan HN, Chung DH, Plane SJ, Sztul E, Chu YK, Guttieri MC, M. McDowell, Ali G and Jonsson CB 2007 Dynein-dependent transport of the hantaan virus nucleocapsid protein to the endoplasmic reticulum-Golgi intermediate compartment. J. Virol. 81 8634–8647
Ransohoff RM 2012 Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat. Neurosci. 15 1074–1077
Rash JE, Yasumura T, Dudek FE and Nagy JI 2001 Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J. Neurosci. 21 1983–2000
Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, Blomstrand F and Giaume C 2002 Gap junctions and connexin expression in the normal and pathological central nervous system. Biol. Cell. 94 457–475
Rozental R, Giaume C and Spray DC 2000 Gap junctions in the nervous system. Brain Res. Brain Res. Rev. 32 11–15
Ruthel G, Demmin GL, Kallstrom G, Javid MP, Badie SS, Will AB, Nelle T, Schokman R, Nguyen TL, Carra JH, Bavari S and Aman MJ 2005 Association of ebola virus matrix protein VP40 with microtubules. J. Virol. 79 4709–4719
Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K and Bone LJ 1998 Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24 8–20
Skripuletz T, Gudi V, Hackstette D and Stangel M 2011 De- and remyelination in the CNS white and grey matter induced by cuprizone: the old, the new and the unexpected. Histol. Histopathol. 26 1585–1597
Spray DC, Moreno AP, Kessler JA and Dermietzel R 1991 Characterization of gap junctions between cultured leptomeningeal cells. Brain Res. 568 1–14
Steinman L and Zamvil SS 2006 How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 60 12–21
Stohlman SA and Weiner LP 1981 Chronic central nervous system demyelination in mice after JHM virus infection. Neurology 31 38–44
Sussman MA, Shubin RA, Kyuwa S and Stohlman SA 1989 T-cell-mediated clearance of mouse hepatitis virus strain JHM from the central nervous system. J. Virol. 63 3051–3056
Teubner B, Odermatt B, Guldenagel M, Sohl G, Degen J, Bukauskas F, Kronengold J, Verselis VK, Jung YT, Kozak CA, Schilling K and Willecke K 2001 Functional expression of the new gap junction gene connexin47 transcribed in mouse brain and spinal cord neurons. J. Neurosci. 21 1117–1126
Tress O, Maglione M, May D, Pivneva T, Richter N, Seyfarth J, Binder S, Zlomuzica A, Seifert G, Theis M, Dere E, Kettenmann H and Willecke K 2012 Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J. Neurosci. 32 7499–7518
Waksman BH and Adams RD 1962 A histologic study of the early lesion in experimental allergic encephalomyelitis in the guinea pig and rabbit. Am. J. Pathol. 41 135–162
Wasseff SK and Scherer SS 2011 Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol. Dis. 42 506–513
Wroblewska Z, Gilden DH, Wellish M, Rorke LB, Warren KG and Wolinsky JS 1977 Virus-specific intracytoplasmic inclusions in mouse brain produced by a newly isolated strain of Theiler virus. I. Virologic and morphologic studies. Lab. Invest. 37 595–602
Xiao PJ and Samulski RJ 2012 Cytoplasmic trafficking, endosomal escape and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. J. Virol. 86 10462–10473
Xu A, Bellamy AR and Taylor JA 2000 Immobilization of the early secretory pathway by a virus glycoprotein that binds to microtubules. EMBO J. 19 6465–6474
Yasuda T, Tsumita T, Nagai Y, Mitsuzawa E and Ohtani S 1975 Experimental allergic encephalomyelitis (EAE) in mice. I. Induction of EAE with mouse spinal cord homogenate and myelin basic protein. Jpn. J. Exp. Med. 45 423–427
Yea C, Dembowy J, Pacione L and Brown M 2007 Microtubule-mediated and microtubule-independent transport of adenovirus type 5 in HEK293 cells. J. Virol. 81 6899–6908
Yednock TA, Cannon C, Fritz LC, F. Sanchez-Madrid, Steinman L and Karin N 1992 Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 356 63–66
Zhang GX, Baker CM, Kolson DL and Rostami AM 2000 Chemokines and chemokine receptors in the pathogenesis of multiple sclerosis. Mult. Scler. 6 3–13
Acknowledgements
Supported in part by Research Grant (BT/PR14260/MED/30/437/2010 and BT/PR4530/MED/30/715/2012) from Department of Biotechnology, India, Research Grant (RG3774A2/1) from National Multiple Sclerosis Society (NMSS), USA and Indian Institute of Science Education and Research-Kolkata (IISER-K), India start up Fund to JDS. Council for Scientific and Industrial research (CSIR), India and DuPre Grant from Multiple Sclerosis International Federation (MSIF) provided the research support to RB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Neeraj Jain.
Corresponding editor: Neeraj Jain
Rights and permissions
About this article

Cite this article
Basu, R., Das Sarma, J. Connexin 43/47 channels are important for astrocyte/oligodendrocyte cross-talk in myelination and demyelination. J Biosci 43, 1055–1068 (2018). https://doi.org/10.1007/s12038-018-9811-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12038-018-9811-0