
Abstract
Stroke is one of the main causes of neurological disability worldwide and the second cause of death in people over 65 years old, resulting in great economic and social burden. Ischemic stroke accounts for 85% of total cases, and the approved therapies are based on re-establishment of blood flow, and do not directly target brain parenchyma. Thus, novel therapies are urgently needed. In this review, limb remote ischemic conditioning (RIC) is revised and discussed as a potential therapy against ischemic stroke. The review targets both (i) fundamental research based on experimental models and (ii) clinical research based on clinical trials and human interventional studies with healthy volunteers. Moreover, it also presents two approaches concerning RIC mechanisms in stroke: (i) description of the underlying cerebral cellular and molecular mechanisms triggered by limb RIC that promote neuroprotection against stroke induced damage and (ii) the identification of signaling factors involved in inter-organ communication following RIC procedure. Limb to brain remote signaling can occur via circulating biochemical factors, immune cells, and/or stimulation of autonomic nervous system. In this review, these three hypotheses are explored in both humans and experimental models. Finally, the challenges involved in translating experimentally generated scientific knowledge to a clinical setting are also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke is one of the main causes of death and disability worldwide, with high social and economic burden [1], being 85% ischemic and 15% hemorrhagic. Ischemic stroke results from a vessel blockage resulting in lack of cerebral blood supply, limiting oxygen and nutrient availability in the brain. The only approved therapies are based on the re-establishment of blood flow in the ischemic area, either by lysis of thrombi with recombinant tissue-activated plasminogen (thrombolysis) or by mechanical removal (endovascular treatment) [2]. Both treatments present limitations: risk of hemorrhage transformation, narrow selection criteria including short time window or need for advanced imaging, and no direct action on brain parenchyma [3,4,5]. Thus, innovative conceptual and methodological approaches targeting brain parenchyma are urgently needed.
The ischemic brain tissue can be divided in two regions: (i) the ischemic core, where severe ischemia rapidly results in cell necrosis and tissue loss; and (ii) the ischemic penumbra, a surrounding rim of hypoperfused tissue that may remain viable for several hours, or even days. Thus, the main aim of any acute stroke treatment is to salvage the penumbra area by re-establishment of blood flow and/or by limiting neural cell death and neuronal dysfunction. Over the last three decades, great efforts have been made to develop new therapeutic drugs targeting penumbra, namely minocycline, natalizumab, fingolimod, or uric acid, among others. Despite promising pre-clinical results, clinical trials were disappointing [4, 6].
An alternative strategy may be to take advantage of endogenous mechanisms to protect the brain from ischemia. Indeed, the brain can activate several different responses to stress and trigger mechanisms of defense against ischemia. Hormesis, for example, or conditioning (also known as preconditioning), is based on the activation and strengthening of endogenous defense mechanisms. In fact, hormesis or conditioning is a procedure by which a noxious stimulus (such as ischemia), below the threshold of damage, is applied to a tissue or system. Without causing any lesion, conditioning promotes a cellular protective state (tolerance or cytoprotection) against more severe noxious stimuli given beyond the threshold of damage [7].
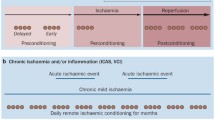
In 1986, it was demonstrated for the first time the cardioprotective effect of ischemic preconditioning [8]. Five minutes of occlusion followed by 5-min reperfusion of the left anterior descending artery decreased cardiac tissue lesion from 40 min of ischemia [8]. Since then, much experimental and clinical data has been generated concerning cardioprotection and more recently neuroprotection has also been explored mainly in animal models [7, 9]. This protective effect of ischemic preconditioning can also be found when applied to a distant organ or tissue and is called remote ischemic conditioning. It was first described that 5-min period of circumflex branch artery occlusion decreased infarct size in a remote myocardium region [10]. The concept of limb remote ischemic preconditioning was first explored in 1997 by Oxman and colleagues for promoting cardioprotection against reperfusion tachyarrhythmia [11]. For the last two decades, the remote ischemic conditioning procedure has received much attention due to its non-invasive nature, safety, and feasibility in the clinical setting (Fig. 1).

Limb remote ischemic conditioning (RIC) and neuroprotection.Scheme for RIC applied in the arm and the potential signaling that confers neuroprotection
In the context of stroke, conditioning can be divided in three categories depending on timing: preconditioning, per-conditioning, and post-conditioning [7, 12] (Fig. 2). Limb RIC can be applied at several distinct time points, which in turn induce early- (hours) and late- (days) phase conditioning responses. Pre-RIC might be applied as a protective conditioning in the event of a future stroke in at-risk individuals. Per-RIC refers to RIC applied during the early acute stroke phase before reperfusion. Rapid post-RIC might provide neuroprotection when applied immediately after ischemic stroke and initial reperfusion, and delayed post-RIC could be equally applied days after a stroke. Herein, the used nomenclature is remote ischemic conditioning (RIC) in order to encompass pre-, per-, and post-conditioning.

Timing for ischemic conditioning.There are three different times for the application of ischemic conditioning in the context of ischemic stroke: pre-conditioning (before ischemia), per-conditioning (after ischemia before reperfusion), and post-conditioning (after the onset of reperfusion)
Major Molecular and Cellular Mechanisms Following Cerebral Ischemia and Reperfusion
Remote ischemic conditioning can target different cellular and molecular processes occurring during ischemia and reperfusion. Cerebral ischemia causes excitotoxicity, oxidative and nitrosative stress, neuroinflammation, and bioenergy catastrophe, which are associated with neuronal dysfunction, neural cell death, blood–brain barrier permeabilization, and tissue loss (Fig. 3).

Chronological cellular and molecular consequences of ischemic stroke and the associated processes involved in neuroprotection.Following ischemic stroke, there is rapid generation of excitotoxicity, necrosis, oxidative, and nitrosative stress in the core of stroke. Later on (hours up to few days), there is apoptosis, neuroinflammation, bioenergy catastrophe, BBB permeabilization, and still oxidative and nitrosative stress, which are more associated with penumbra area of stroke
Excitotoxicity was the first identified molecular mechanism following brain ischemia and is due to excessive and rapid release of the excitatory amino acid glutamate, without the necessary reuptake by astrocytes. Glutamate leads to the activation of N-methyl-D-aspartate (NMDA), amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA), and kainate receptors, which in turn promotes great Ca2+ influx [13]. Intracellular Ca2+ accumulation activates lytic enzymes (proteases) and promotes mitochondrial dysfunction. Mitochondria are key organelles for bioenergy production, control of cell death, and generation of reactive oxygen species (ROS); thus, their dysfunction promotes unbalanced bioenergy status, apoptosis and necrosis, and oxidative stress [13]. Likewise, activation of enzymes such as nitric oxide synthase, cyclooxygenase, or NADPH oxidase is coupled with excitotoxicity [4, 14]. Paradoxically, during reperfusion, when blood flow is reestablished, there is a great increase on ROS and reactive nitrogen species (RNS) formation, probably due to the unbalanced re-establishment of oxidative metabolism and by overwhelming the endogenous antioxidant defenses [8]. Exacerbated inflammation occurs in the microvasculature and in the brain parenchyma, including the release of pro-inflammatory and neurotoxic factors along with the accumulation of leukocytes [4]. In response to stress, microglia (the brain resident immune cells) become reactive releasing neurotoxic factors such as TNF-α, IL-1, ROS, or RNS [15]. Nevertheless, microglia are also key players in the clearance of apoptotic cells by phagocytosis, which in turn reduces inflammation and neurotoxicity, promoting re-generation of brain parenchyma [16, 17]. Likewise, oxidative stress, neural cell death, and/or neuroinflammation lead to increased permeability of blood–brain barrier, which in turn exacerbate inflammation by infiltration of more circulating immune cells and pro-inflammatory factors. The main events occurring during ischemia reperfusion are described in Fig. 3.
With our review, we aimed to cover experimental models (the “Experimental Models” section) and clinical trials and human interventional studies (the “Human Studies and Clinical Trials of Remote Ischemic Conditioning (RIC)” section). Two main aspects of RIC are approached: (i) RIC-induced protective effect in the brain following stroke and its potential underlying molecular and cellular mechanisms (“Molecular and Cellular Mechanisms Underlying RIC-Induced Neuroprotection in the Ischemic Brain” section for models and “RIC Clinical Trials in Neurological Disease: Potential Cerebral Effects” section for humans) and (ii) how inter-organ communication occurs between limb and brain (“Inter-organ Communication: Signaling Between Remote Ischemic Conditioned Limb and the Brain” section and “Mechanisms of RIC in the Inter-organ Communication” section for experimental models and humans, respectively). The rationale for the review is represented in Fig. 4.

Rationale of the review.Organization of data generated by experimental models or human-based studies (including clinical trials). Data were divided in two: description of the underlying cerebral cellular and molecular mechanisms triggered by limb RIC that promote neuroprotection against stroke-induced damage and the identification of signaling factors involved in inter-organ communication following RIC procedure
Experimental Models
In this part of the review, we summarize the cellular and molecular neuroprotective signals and mechanisms, derived from experimental evidence, which support limb RIC as neuroprotective in order to characterize the specific physiology of limb remote preconditioning that will support its translation to future clinical trials. It is organized in two main parts: (i) which are the cerebral mechanisms activated by RIC and (ii) which are the RIC-activated blood circulating factors or autonomic nervous system components that may in turn drive protection in the ischemic brain.
Introduction to Experimental Models
Although the first application in rats of limb RIC as a cardioprotective strategy was shown in 1997, it was only in 2004 that RIC was explored for neuroprotective purposes [18]. Neuroprotection by remote ischemic preconditioning performed on a limb was first demonstrated in a rat model of experimental brain ischemia to induce neuroprotection against ischemia–reperfusion injury, by application of 3 cycles of 10-min RIC and 10-min reperfusion [18]. Neural protection via RIC has since been observed in both young male and female rodent models [19,20,21] and aged male rats [22].
Molecular and Cellular Mechanisms Underlying RIC-Induced Neuroprotection in the Ischemic Brain
Herein, the RIC-induced cerebral pathways, gene expression, or neural cellular responses that can potentially play a role in neuroprotection are described. The main information is summarized in Table 1 and Fig. 5.


A Pathways and gene expression that are altered by limb pre-RIC following ischemic stroke and that are related to neuroprotection in experimental models.There is a timeline with the altered pathways and gene expression that occurs when limb RIC is applied before the ischemic stroke (pre-RIC) in comparison with ischemic stroke without RIC. When pre-RIC is induced up to 1h before ischemic stroke, the altered events are represented in the upper part of the figure. In the lower part of the figure, there are the events occurring when pre-RIC is induced between 1h and 3 days before ischemic stroke. In the right hand side, the required conditions for pre-RIC to protect the brain against ischemic stroke are described. The altered pathways and different gene expressions are described accordingly with brain region that is represented by different colors. The reference number is described in Tables 1 and 2. The used symbols are for upregulated/increased and for downregulated/reduced. B Pathways and gene expression that are altered by limb per- and post-RIC following ischemic stroke and that are related to neuroprotection in experimental models.There is a timeline with the altered pathways and gene expression that occurs when limb RIC is applied after the ischemic insult before reperfusion (per-RIC) and after ischemia and reperfusion up to 1h (rapid post-RIC) or at later stages (post-RIC). In all three cases, alterations in pathways and gene expression are compared with ischemic stroke without RIC treatment. In the right hand side, the required conditions for per- and post-RIC to protect the brain against ischemic stroke are described. The altered pathways and different gene expressions are described accordingly with brain region that is represented by different colors. The reference number is described in Tables 1 and 2. The used symbols are for upregulated/increased and for downregulated/reduced
Involvement of Protein Synthesis and Transcriptional Regulation
The protein synthesis inhibitor cycloheximide was shown to block the neuroprotective effects of limb post-RIC in rat [23] indicating that de novo protein synthesis is required for neuroprotection via limb RIC.
Activation of the transcription factor hypoxia-inducible factor 1α (HIF-1α) is triggered by lack of oxygen and coordinates adaptation to hypoxia via transcriptional regulation of more than 200 genes. HIF-1α mRNA expression is upregulated in ischemic cerebral cortex 24 h after IS but declines to control level within 3 days, but protein remains in both neurons and astrocytes for up to 7 days post-ischemia in a rat model of experimental ischemic stroke (IS) [24]. Post-RIC, applied immediately after IS, inhibited HIF-1α mRNA expression in ischemic cerebral cortex by around 50% within 24 h, and protein up to the third day after IS [24]. In another rat model using pre-RIC, 1 h before IS, overall, the protein expression of HIF-1α was increased in whole brain extracts after IS, and increased a further twofold in the pre-RIC model [30]. In aged rats, HIF1α and HIF2α are also upregulated in ischemic penumbra 48 h after IS but not when pre-RIC is applied 24 h before IS [22]. HIF1α was also increased in whole blood after post-RIC [32]. Exogenous systemic activation of HIF-1α, by IP injection 60 min after reperfusion (RF), in a rat model of IS, mimics the neuroprotective effects as well as the pro- and anti-inflammatory cytokine levels in ischemic brain seen in post-RIC, while inhibition of HIF-1α in post-RIC abolished these effect [32].
Intravenous administration of a HIF-1α inhibitor has also been shown to reverse the neuroprotective effects of pre-RIC within 24–48 h, described as decreased neurological deficit scores, lower brain water content, and increase HSP70 protein expression, and reversed inflammatory cytokine profiles in brain and peripheral blood, as seen in rat after pre-RIC [22, 30].
The full significance to neuroprotection of HIF1α inhibition by post-RIC specifically in the ischemic cerebral core, but overall increase in pre-RIC whole brain extract, remains to be fully clarified; it may reflect differential action of different RIC procedures or regional brain differences [30].
Another transcription factor that regulates endogenous antioxidant capacity is nuclear factor erythroid 2-related factor 2 (Nrf2/NFE2L2), which was upregulated in ischemic cortex, by post-RIC in the CD1 mouse model 24 h after IS [26]. In the nucleus, Nrf2 binds to the antioxidant response element (ARE) on target genes initiating expression of cytoprotective genes including NAD(P)H quinone dehydrogenase 1 (NQO1), a reductant, and heme oxygenase-1 (HO-1/HMOX1) that degrades heme into iron ions, biliverdin, and antiapoptotic CO. Expression of both NQO1 and HO-1 is upregulated by post-RIC 24 h after IS [26]. However, inhibition of HO-1 in post-RIC abolished the neuroprotective effects of post-RIC [28], including the upregulation of BDNF (involved in hippocampal neurogenesis and brain plasticity); hippocampal structural abnormalities; and the decrease in the pro-inflammatory cytokine TNFα which are triggered by post-RIC [28]. The protein expression of cAMP response element-binding protein (CREB), which was reduced in whole brain at 72 h in a rat model of IS, was rescued by post-RIC [29]; this may account for the increased expression of BDNF observed after post-RIC, as BDNF harbors CREB response elements on its promoter. Increased glycogen synthase kinase 3 beta (GSK3β) protein expression is observed after IS in rat brain model after 72 h, being partly rescued by post-RIC [29].
The glial water channel aquaporin-4 (AQP4), a downstream target of hypoxia-inducible factor 1 alpha (HIF-1α) and inflammatory cytokines, is upregulated in ischemic hemisphere of rat model of IS, at 48 h, and downregulated by pre-RIC [25]. This could be attributed to HIF1α inhibition and explain reduced edema by pre-RIC.
Several pathways that are activated by cytokines or growth factors and regulate gene expression have been shown to be involved in limb RIC. A twofold increase in phosphorylated STAT3 was observed in rat ischemic penumbra 24 h after middle cerebral artery occlusion (MCAO), rising to fourfold after 3 days and still at 2.5 fold after 14 days [31, 34], while 24 h after post-RIC pSTAT3 is initially higher than in IS, but by 3 days, the levels are down to half that in IS, decreasing even more after 14 days [31, 34]. This suggests that endogenous neuroprotection may act via activation of the pSTAT3 transcription factor at early stages (24 h after IS) but deactivation 3–14 days after IS.
The neuroprotective effects of post-RIC on penumbra region may also be linked to suppression of another transcription factor p-ATF2 via the p38 MAPK-ATF2 pathway [27]. Mechanistically, p-ATF2 would activate transcription of genes by binding cAMP-responsive elements (CRE) and might also be a histone acetyltransferase, thus directly affecting chromatin.
Also implicated in limb RIC neuroprotection against IS in rats are NF-κB pathways [33] that regulate the transcription of over 150 genes controlling inflammation, immune cell development, cell cycle, proliferation, and cell death, including the expression of cytokines, chemokines, immunoreceptors, and regulators of proliferation and apoptosis. NF-kB can be activated by a variety of intracellular and extracellular signals, including TNFα. In the context of limb RIC neuroprotection against IS in rat, NF-kB pathways activated by limb RIC were shown to be dependent on activation of upstream Notch1 signaling and downstream Notch regulated transcription of Hes1 [33]. Curiously, in the latter model, in the absence of limb pre-RIC, inhibition of Notch1 by itself reduced infarct volume, improved neurological deficit score, and attenuated apoptosis in hippocampus, evidence that neuroprotection by Notch1 activation or inhibition is not linear.
Neuroglobin
Neuroglobin (Ngb) is a major player in mediating neuroprotective effects of limb RIC. Ngb, an oxygen-binding heme protein found in the brain that can also be detected in serum [68], is increased in rodent brain neurons following hypoxia suggesting that it may play a role in the brain’s response to hypoxic-ischemic injury. In rat, pre-RIC, increased Ngb expression, was associated with neuroprotection, and improved mitochondrial Na+-K+-ATPase activity and mitochondrial membrane potential after global ischemia [35]. In rat, per-RIC, increased Ngb in peri-infarct region, was only observed 1 h after IS [36], while daily repeated post-RIC for 14 days resulted in sustaining increased levels of Ngb. The redox state of Ngb during acute hypoxia regulates the stability of the transcription factors HIF-1α and Nrf2 as well as release of cytochrome c from mitochondria that triggers apoptosis [69].
Purinergic Signaling
Adenosine plays an important role in the endogenous neuroprotective mechanisms induced by RIC. While extracellular ATP is in the nanomolar range, intracellular ATP is in the millimolar range, so that damage to cell membranes during trauma (like an ischemic event) leads to massive increase in extracellular ATP, and rapid formation of adenosine which can activate four distinct subtypes of G-protein-coupled receptors, named A1, A2A, A2B, and A3 [70, 71]. Adenosine A1 and A2A receptors in particular have inhibitory functions in most tissues including the brain, slowing metabolic activity and reducing synaptic vesicle release [72] placing brain cells in a neuroprotective quiescent state.
Circulating adenosine is also an endogenous distress signal that modulates tissue damage and repair [72, 73]. In rat Hu et al. 2012 showed that limb pre-RIC, performed 1 h before experimental IS, resulted in reduced systemic and cerebral inflammation and oxidative stress just 1 h after the IS, and that this was dependent on activation of adenosine A1 receptors throughout the organism [37]. Another study has also shown that an adenosine A1 receptor antagonist dose-dependently blocked pre-RIC-induced brain ischemic tolerance and delayed neuronal cell death up to 7 days post-IS [39], supporting the premise that adenosine signaling through adenosine A1 receptors is required for pre-RIC neuroprotection. The same study showed that femoral injection of adenosine, 10 min prior to IS, mimics the neuroprotective effects of pre-RIC, dose-dependently attenuating neuronal cell death and increasing expression of both p-p38 MAPK and pERK in CA1 hippocampus 12 h after IS [39].
Brain Blood Flow and Blood Vessel Development
Limb RIC increases brain blood flow and promotes blood vessel development. Transient localized ischemic stroke, by its very nature, causes reduced regional cerebral blood flow, which in turn leads to oxidative stress, while the reperfusion that follows aggravates the ischemic damage through increased production of reactive oxygen species (ROS). In a rat model of MCAO, both per-RIC and post-RIC were shown to prevent collateral vessel collapse and increase collateral flow into the middle cerebral artery which may help rescue the penumbral region, the marginal zone around the ischemic core, which can be in part rescued by RIC [40, 41]. Post-RIC applied daily for 7 days increased focal cerebral blood flow in the ischemic ipsilateral hemisphere [43]; after 14 days, arteriogenesis was observed, specifically in the form of growth of functional leptomeningeal collateral arteries [43] suggesting that RIC reinforces the development of supplemental networks of vessels to compensate defective blood flow.
Ischemic stroke and RIC also affect expression of matrix metalloproteinases (MMPs) in the brain, which may be linked to changes in tissue and/or vessel remodeling. Ischemic stroke modeled in rat by itself increases expression of MMP-2 and MMP-9 [42], while per-RIC, immediately after 1-h MCAO, significantly reduced MMP-9 but not MMP-2 expression and activity in ipsilateral ischemic hemisphere after 24 h [42].
Post-RIC performed 2 days after IS in rat resulted in upregulation of endogenous tissue kallikrein (TK), detected in plasma from day 3 for 21 days, an enzyme which produces Lys-bradykinin, a potent vasodilator that contributes to hypotension in systemic circulation [46]; the latter study also showed that the selective bradykinin B2 receptor antagonist HOE-140 reverses the neuroprotective effects of post-RIC.
Chronic post-RIC, applied daily for 3 weeks in rat, increased capillary density and diameter, angiogenesis, arteriogenesis and collaterals, as well as expression of pericytes colocalized with cerebral blood vessels, concomitant with increased endothelial cell progenitors in blood [45]. In this context, the highly conserved Notch signaling pathway, regulating cell proliferation, differentiation, and apoptosis, appears to be central for RIC neuroprotection via activation of blood vessel plasticity. Chronic limb post-RIC in rat for 21 days was shown to increase angiogenesis in CA1 hippocampus [44], and in 14 days, it increased arteriogenesis concomitant with increased Notch1 and NICD expression in the ischemic brain arteries in rat after IS [43]. Pre-activation of Notch1 and downstream NF-κB pathways in neurons also appears to be required for successful neuroprotective effect of pre-RIC against cerebral I/R injury [33].
Vascular cognitive impairment (VCI) is caused by brain hypoperfusion; it presents similar mechanisms to ischemic stroke and constitutes a major aging-related public health concern, with no available treatment. Bilateral common carotid artery stenosis is used as a model of VCI, since it induces hypoperfusion by decreasing cerebral blood flow, which in turn causes accumulation of amyloid and promotes white matter loss [74]. When post-RIC was applied daily for 2 weeks, there was an improvement in cerebral blood flow and a reduction of neurodegenerative features such as amyloid beta accumulation, inflammation and cell death [74].
Antioxidant
Neuroprotection by limb RIC acts by reducing oxidative stress. The brain’s high oxidative metabolic rate and high lipid content make it a vulnerable target for ischemia-derived ROS that causes general oxidative damage and lipid peroxidation. In rat, limb pre-RIC applied 1 h before MCAO was shown to reduce mitochondrial and overall oxidative stress, lipid peroxidation, oxidative DNA damage, and oxidative damage to proteins in whole brain 24 h later [30, 37]. Combination of per-RIC and chronic post-RIC for 3 days also reduced ROS in peri-infarct region but did not have an additive effect when used in combination [36]. In a mouse CD1 model of IS, post-RIC reduced oxidative stress, as determined by increased activity of super oxide dismutase, and reduced lipid peroxidation, as determined by lower levels of MDA (malondialdehyde) in ischemic cortex 24 h after IS [26]. In a rat post-RIC model, 72 h later, lower lipid peroxidation (MDA), and nitrite, and increased antioxidant GSH were observed, witness to post-RIC effects on reducing oxidative stress even after 3 days [29]. In a cellular model, 10 min of post-RIC human plasma (high molecular weight dialysate hydrophobic fraction) has also been shown to reduce apoptosis and oxidative stress of human neural stem cells in culture when subjected 24 h of oxygen glucose deprivation [75].
Neuronal Cell Death and Apoptosis
Marked increase in neuronal apoptosis is observed in the hippocampal CA1 region in animal models of transient global ischemia that mimic ischemia/reperfusion (IS/RF) injury and delayed neuronal death induced by a cerebral ischemic insult such as an ischemic stroke. Several studies have shown that limb remote ischemic conditioning (limb RIC) reduces neuronal apoptosis in the CA1 hippocampus region (72 h after IS/RF), when applied before experimental stroke as limb pre-RIC [18], and in whole brain at 48 h [51] concomitant with improved glucose metabolism, or at 24 h [30] witnessed by increased Bcl2 and reduced caspases 3 and 9. Likewise, immediately after stroke (MCAO 90 min) as limb rapid post-RIC (3 cycles: 5 min RIC + 4 min RF) [57]; or in delayed post-RIC, 3 h and 6 h after focal IS/RF [57], as evidenced by DNA fragmentation and or apoptotic body formation. Very significant neuroprotection in CA1 hippocampal region in rat was also observed by a single 20-min limb RIC, applied as very delayed post-RIC 2 days after a global 10-min ischemic insult [21]. Increased Bcl-2/Bax ratio has been consistently observed in rat 24–48 h after post-RIC [34, 55] indication that reduced neuronal cell death in limb RIC is at least in part regulated by reducing apoptosis. Combination of per-RIC and chronic post-RIC for 14 days does not have an added effect on reduced neural apoptosis in peri-infarct region [36]. Reduced neuronal cell death was also observed after 21 days of chronic post-RIC [44]. In rat, increased production over 72 h of the pro-apoptotic protein Bim was observed in neurons, which was reversed by pre-RIC in salvage area [48].
Mitochondrial function and integrity in CA1 hippocampus region are compromised by ischemia/reperfusion. This has been observed in rat by deterioration of mitochondrial membrane potential, a marker of neuronal cell death, and reduced mitochondrial Na + -K + -ATPase activity, both of which are improved by pre-RIC [35].
In a rat model of delayed post-RIC, up to 6 h, blockade of potassium channels was shown to reduce the neuroprotective and antiapoptotic effects of post-RIC on the brain, while potassium channel activation mimicked the antiapoptotic and neuroprotective effects of post-RIC [57]
MAPK, AMPK, and PI3K-Akt Pathways
Several mitogen-activated protein kinase (MAPK) cascades have been shown to play a role in neural ischemic conditioning. pERK was decreased after IS in mouse [76] and aged rats [22] while pre-RIC greatly increased pERK levels in ischemic ipsilateral hemisphere [22, 76], as did post-RIC though to a lesser extent [76]. Neuroprotection was also shown to be dependent on MEK-1 dependent increase in pERK1/2, as observed in CA1 hippocampus region 6 h after pre-RIC and 8-min MCAO, peaking at 12 h and returning to normal after 5 days [52]. Ischemic tolerance induced by pre-RIC has also been shown to be dependent on activation of the p38 MAPK cascade at 6 h, peaking at 12 h and normalizing 1 day after pre-RIC in CA1 hippocampus [53] at least partly by downstream upregulation of HSP 70 expression which is detected 1 day later and peaks 2 days after pre-RIC [54]. Limb pre-RIC-induced upregulation of both p-p38 MAPK and pERK in CA1 hippocampus 12 h after IS appears to be adenosine dependent [39]. These activations may also be triggered by cytokines or growth factors inducing activation of p38 MAPK or ERK1/2 that translocate to the nucleus to trans-activate transcription factors regulating expression of genes involved in several pathways involving differentiation and cell survival.
AMPK is activated when AMP/ATP or ADP/ATP ratios in cells rise due to physiological stresses, including ischemia. In rat, AMPK protein in whole brain [30] and p-AMPK in ischemic ipsilateral hemisphere [42] were increased 24 h after MCAO alone, and increased further in pre-RIC performed 1 h before MCAO [30] or after per-RIC [42]. Inhibition of AMPK reversed the neuroprotective effects of pre-RIC, namely, the better neurological deficit scores, lower brain water content, and increase HSP70 protein expression observed after pre-RIC [30].
In a post-RIC mouse model of IS, AMPK pathway was also activated in the cerebral cortex, 12 h after RF, as witnessed by increased p-AMPK, p-ACC, and p-ULK1 and decreased p-mTOR [64]. In per-RIC performed immediately after 1-h MCAO in rat, p-AMPK was increased more than twofold after 24-h reperfusion [42].
Limb RIC also interferes with PI3K-Akt pathway. Results relating to activation of Akt in IS and RIC are not always consistent. Reduction of pAkt was observed in ischemic hemisphere in mouse brain 48 h after experimental embolic stroke (2-h eMCAO) [20], 45-min MCAO [76], or 90-min MCAO in aged rats [22], which was partially rescued by pre-RIC 24 h before MCAO, per-RIC (2 h after eMCAO), or tPA (4 h after eMCAO), per-RIC and tPA together having an added effect on pAkt rescue [20] but not by pre-RIC or post-RIC in a diabetic mouse model of IS [76] or in presence of HIF inhibitor [22]. In a rat model using 8-min global cerebral ischemia, no change was observed in pAkt in hippocampal CA1 region 48 h after reperfusion, while rapid post-RIC-induced activation of pAkt [55]. However, in a rat model of post-RIC performed immediately after 2-h MCAO, both pAkt1(Thr308) and pAkt1(Ser473) were highly upregulated 24 h after post-RIC [62]. In aged rats, pAkt was increased 48 h after IS and pre-RIC had no effect [22]. PI3K/Akt enhanced signaling in limb RIC may be responsible for decreased blood–brain barrier permeability via fibulin-5 [62], as pAkt-dependent fibulin-5 overexpression was observed 24 h after post-RIC with concomitant fibulin-5-dependent claudin-5 and occludin post-RIC upregulation [62, 65]. Post-RIC in rat also reduced the increase in matrix metalloproteinase 9 (MMP-9) that is observed after IS which is implicated in BBB breakdown [65].
Autophagy
In a mouse model of ischemic stroke, limb post-RIC activates autophagy in cerebral cortex of right hemisphere, as witnessed by increased LC1-II/LC3-I, Beclin-1, and Atg7, and decreased SQSTM1/P62 at 12 h, and reduces apoptosis [64]. Inhibition of autophagy using intracerebroventricular (ICV) injection of 3-methyladenine (3-MA) reverses the neuroprotective effects of limb post-RIC observed after 12 h [64] as witnessed by increased neurological deficit score, brain water content, infarct volume, and apoptosis. Post-RIC-induced autophagy, in cerebral cortex of right hemisphere, was also dependent on AMPK pathway activation, as demonstrated by reversion of the post-RIC observed increase in p-AMPKα, p-ACC, and p-ULK1, and decrease in p-mTOR, using AMPK inhibitor compound C [64].
Experimental results measuring autophagy can vary according to the brain region observed. In a rat model, 2-h MCAO did not change amount of autophagosomes in penumbral tissue 24 h after RF, but immediate rapid post-RIC (3–4 cycles: 10 min RIC + 10 min RF) increased amount of autophagosomes and autophagy-lysosomal pathway in penumbral tissue 24 h after reperfusion [58] in a p(S473)Akt/p(S9)GSK3β-dependent manner [56]. In an identical model of rapid post-RIC (3 cycles: 15 min RIC + 15 min RF), MCAO increased amount of autophagosomes in ischemic cortex which was decreased 24 h after post-RIC [63]. Therefore, it appears that in penumbral tissue, autophagy is activated only by post-RIC, whereas in ischemic cortex, autophagy is activated during I/R and reduced by post-RIC. In rat ischemic hemisphere, both per-RIC and post-RIC promote Akt-dependent S70 phosphorylation of Bcl-2 triggering dissociation of Bcl-2/Beclin1 complex and Beclin-1-dependent autophagosome formation [59].
In rat ischemic cortex of the right middle cerebral artery region, autophagy was also shown to be increased during acute ischemia/reperfusion injury via reduced mTOR/p70S6K signaling [63]. Conversely, autophagy was reduced in rat, in response to limb post-RIC during reperfusion, via increased mTOR/p70S6K signaling [63]. The question remains as to how signals from the limb post-RIC trigger this reduced autophagy. Limb post-RIC, in a rat model of chronic cerebral ischemia, was shown to attenuate neural damage in cortex and hippocampus via activation of transcription factor EB (TFEB), a driver of autophagy via the autophagolysosome pathway [50].
Neurogenesis
In addition to reducing infarction size and improving functional outcome, post-RIC in rat has been shown to induced neurogenesis for up to 28 days, in rat after 90-min IS, in the hippocampus region where adult neurogenesis occurs, the subgranular zone (SGZ), and the subventricular zone (SVZ) [61]. Rapid post-RIC in rat was also shown to promote synaptogenesis in ischemic penumbra and to increase expression, in the infarct cortex, of Post-Synaptic Density Protein 95 (PSD95) that is critical for synaptogenesis and synaptic plasticity, Growth Associated Protein 43 (GAP43), a major component of growth cones of elongating axons, and synapsin involved in regulation of axonogenesis and synaptogenesis [49].
Nitric Oxide Synthase (NOS)/Nitric Oxide signaling
In rat, experimental ischemic stroke alone does not change endothelial nitric oxide synthase (eNOS) expression in ischemic ipsilateral hemisphere compared to contralateral hemisphere, but per-RIC performed immediately after 1-h MCAO, resulted in eNOS increase after 24-h reperfusion in ischemic hemisphere compared to MCAO alone [42]. In another rat model using 8-min global cerebral ischemia, limb pre-RIC increased NO and NOS activity following a double peak pattern in both serum (0 h and 48 h) and CA1 hippocampal region (6 h and 48 h) [77]. Limb rapid post-RIC increased eNOS expression and activation in hippocampal CA1 region 48 h after ischemic stroke (both p-eNOS(S1177) and eNOS), along with increased pAkt, in a PI3K-dependent manner, the neuroprotective effects (reduced apoptosis and increased neuronal density in CA1 and reduced behavioral deficits) of rapid post-RIC being abolished by both non-selective NOS or PI3K inhibitors [55], indicating that eNOS activation in this CA1 region occurs, at least in part, via PI3K dependent phosphorylation of pAkt. In chronic post-RIC, applied daily, plasma nitrite levels were increased at 3 weeks, but no longer at 1 month or 4 months [45]. In a model of cerebral hypoperfusion, p-eNOS rose in CA1 hippocampus at day 1 and maintained for 2 weeks declining at 3 weeks, but chronic post-RIC applied daily for 28 days sustained p-eNOS beyond 4 weeks [44].
In another rat model of focal ischemia, iNOS expression doubled, from 1 to 24 h after ischemia, in pooled brain tissue (pooled ischemic core, penumbra, and cortex), and pre-RIC completely inhibited iNOS expression below normal levels [78], indication that RIC blocks the pro-inflammatory expression of iNOS in IS. Twenty-four hours after stroke, a reduced nitrite/nitrate ratio was observed in rat whole brain with limb pre-RIC applied 1 h before 2-h MCAO, indicating reduced inflammation in the whole brain [37]. These results indicate that limb RIC globally reduces the pro-inflammatory iNOS/NO production, but locally (e.g., in hippocampal C1 region), PI3K-Akt/p-eNOS/NO is involved in signaling that triggers conditioning pathways of neuroprotection that reduce apoptosis and increase neuronal density in CA1.
Neuroinflammation
Limb RIC reduces brain neuroinflammation. Oxidative stress precipitated by ischemic stroke in the brain results in chronic expression of pro-inflammatory genes, which can further aggravate neuronal injury. In rat, limb pre-RIC applied immediately before ischemia was shown to inhibit brain edema and blood–brain barrier permeability measured 2 days after stroke [78]. Rat models of ischemic stroke have shown that pro-inflammatory markers and cytokines such as MPO (myeloperoxidase), TNFα, IL-1β, and IL-6, known to be releases by activated microglia, are all highly increased in whole brain tissue 24 h after IS [30, 34]; IL-1β and IFNγ are increased in ischemic penumbra 48 h after IS but not with pre-RIC 24 h before IS, while no changes were observed in IL-6, TNFα, IL-4, and IL-10 [22]; but 72 h after IS, TNFα [29] was increased in whole brain homogenate. In another study, 48 h after IS in rat cortical penumbra surrounding the ischemic core, IL-1β and IL-6 are not changed, but IL-1β is decreased by post-RIC; while IS-induced decrease of anti-inflammatory cytokines IL-4 and IL-10 in cortical penumbra is reversed by post-RIC [32]. An increase in IL-6 and TNFα in ischemic ipsilateral hemisphere was observed 48 h after reperfusion in a diabetic mouse model of IS, which for IL-6 was partially compensated for by pre-RIC but not post-RIC [76], while no changes were observed in IL-1β, IFN-γ, or IL-4 before or after IS or RIC.
Pre-RIC, applied 1 h before IS, reverses this increase for MPO and IL-6, and significantly reduced this increase for IL-1β and TNFα [30]. Reduction in TNFα was also observed in ischemic penumbra after 1 h using rapid post-RIC [34] and in whole brain 72 h after post-RIC [29]. In ischemic brain 48 h after post-RIC, anti-inflammatory cytokines IL-4 and IL-10 are increased, and pro-inflammatory cytokines IL-1β and IFNγ are decreased while no change is seen in IL-6 [32]. In whole brain, IL-6 and IL-1β mRNA are markedly increased by ischemic stroke after 48 h, while post-RIC attenuates this increase [79]. IL-4 mRNA appears to be decreased in whole brain after ischemia and markedly increased 48 h after post-RIC [79]. In ischemic ipsilateral hemisphere of a diabetic IS model, 48 h after reperfusion, helper T cells (CD4 +), cytotoxic T cells (CD8a +), and NK cells increase is compensated by pre-RIC, while post-RIC also markedly decreased cytotoxic T cells and NK cells, having no effect of helper T cells [76].
After ischemic stroke in rat, increased production over 72 h of lipocalin 2 (LCN2) from reactive astrocytes in salvage area is reversed by pre-RIC [48]. Lipocalin has been heralded as a therapeutic target to reduce neuroinflammation and neuronal cell death in brain injury [80].
In mouse model of ischemic stroke, post-RIC was shown to induce changes in astrocyte protoplasmic (glutamine synthetase, GS expressing) or fibrous (GFAP) types [31]. GS expression is upregulated by IS after 3 days but no longer after 14 days, while post-RIC upregulates GS expression only after 14 days. GFAP, specifically GFAPα, in ipsilateral side of brain, is increased almost twofold 3 days later after IS, with increase maintained at 50% 14 days later [31]. This is reversed by post-RIC but only at 3 days. This occurs with no concomitant change of the GFAPδ isoform, resulting in increased GFAPα/GFAPδ ratio in post-RIC brain that reflects altered astrocyte intermediate filament networks [31].
Inflammatory processes in non-infectious reactions can be triggered through Toll-like receptors (TLRs) through interaction with endogenous molecules released from damaged tissues or dead cells. These TLR inflammatory pathways can drive early- and late-phase cytokine or chemokine production via NF-kB. In rat experimental IS, both TLR4 and NF-kB mRNA levels are highly overexpressed in ischemic cortex, 1 day after IS, with levels returning to normal after 7 days, but protein levels were still maintained at 7 days in neurons but not astrocytes in peripheral ischemic tissue [60], evidence of a pro-inflammatory response to IS driven by neurons in the brain; when post-RIC was applied, both TLR4 and NF-kB expression were inhibited 1 or 7 days later, evidence of a long-lasting anti-inflammatory effect of post-RIC [60].
In rat, pre-RIC inhibited expression of Tim-3 and Galectin-9 measured 24-h post-stroke in the brain (pooled ischemic core, penumbra, cortex) which increase after IS and may regulate cell death in lymphocytes [78].
Final Remarks on Experimental Models and RIC Time-frames
The vast majority of experimental animal models demonstrating RIC-induced neural protection are of young male adult rodents, with only a few studies using female rodents [19,20,21] and aged male rats [22]. In relation to co-morbidities, although diabetes is a key co-morbidity present in stroke patients, in particular in aged ones, only one study [76] performed MCAO in a diabetic mouse model. In the latter, Liu and colleagues showed that neither pre-RIC nor post-RIC rescued IS-induced pAkt reduction in ischemic hemisphere, as seen in other studies in young adult models, which stresses the need to clarify how dysmetabolism may interfere with RIC-induced protection and signaling. More experimental model studies are needed to clarify the effects sex, age, and co-morbidities on the beneficial effects of RIC observed so far in young adult models.
The precise RIC protocol that might afford neuroprotection against stroke in humans is likely to require optimization. The dose (acute or chronic) and the time point of limb RIC application will only be effective in precise and limited frames. In experimental models of limb post-RIC, reduced infarction was observed after 2 days when RIC was applied up to 3 h after stroke, but not if RIC was applied after 6 h or 2 months later when only behavioral outcome was ameliorated [23]. In the gerbil brain, for example, induction of ischemic tolerance was shown to require at least 2 min of ischemic preconditioning, at least 1 day before a damaging cerebral ischemic event [81], with a 2-day interval providing even more neuroprotection. This early study also showed that two episodes of preconditioning on consecutive days provided complete tolerance to 5-min cerebral ischemia after 2 days. A later systematic analysis of post-RIC time courses revealed that the total cumulative time of repeated limb occlusion/reperfusions may be fundamental for effective remote post-conditioning, with the maximum protective effect attained in a rat model with 2 cycles of 15 min each, and 2 or 3 cycles of 10 min each and a total time of 40 to 60 min [67].
In a mouse model of 45-min MCAO, which compared delayed pre-RIC (24 h before MCAO), early pre-RIC (just before MCAO), per-RIC (during MCAO), and post-RIC (just after MCAO), only the per-RIC procedure showed a clear neuroprotection possibly resulting from enhanced collateral circulation [66].
In a rat model, with ischemic preconditioning via middle cerebral artery occlusion, neuroprotection against 100 min of ischemic insult did not last beyond 7 days, while repeated post-RIC (chronic RIC), for 14 consecutive days, was associated with stronger neuroprotection against cerebral ischemia/reperfusion injury [36]. However, there seems to be a limit in the effectiveness of chronic RIC. Application of 1 month or 4 months daily limb RIC, in a model of vascular cognitive impairment and dementia, was equally effective [45]. The effective therapeutic window, including the cycles and duration of reperfusion and occlusion, which have been recently reviewed [82], remains to be determined precisely and is certain to impact on the degree and time window of neuroprotection.
Inter-organ Communication: Signaling Between Remote Ischemic Conditioned Limb and the Brain
Experimental models have also been explored to disclose how the communication between the ischemic conditioned limb and the brain occurs. The main hypotheses are the activation of the autonomous nervous system and/or the presence of signaling circulating factors in blood, namely biochemical molecules, immune cells, and/or exosomes carrying microRNAs. Data are described in the present subsection and summarized in Table 2.
Autonomous Nervous System
The neuroprotective effects of limb RIC require afferent nerves and the blood factors. Experimental models show that the neuroprotective mechanisms afforded by limb RIC require complementary and interdependent neural elements and factors circulating in the bloodstream [23, 78]. Afferent sensory nerve blocker capsaicin or ganglion blocker hexamethonium completely abrogated the neuroprotective effects of limb pre- and post-RIC in rat [23, 78], suggesting that limb pre- and post-RIC procedures activate an essential neural afferent pathway. However, RIC neural transmission pathways may not be significant in per-RIC, when the RIC procedure is performed during ischemia, before reperfusion [84], giving added importance to other factors.
Circulating Blood Factors
Blood factors, engendered by RIC, have also been shown to provide a degree of neuroprotection. Plasma proteomics in a rat model revealed that RIC results in significant changes in plasma protein profiles [95, 96]. Platelet-derived microparticles (MP) extracted from healthy rat RIC plasma, and injected before reperfusion into rats subjected to 90-min MCAO, had reduced infarction area 24 h later, albeit to a lesser degree than per-RIC itself [83]. Likewise, in a rat model using 8-min global cerebral ischemia, limb pre-RIC increased NO levels and NOS activity following an double peak pattern, both in serum (0 h and 48 h) and CA1 hippocampal region (6 h and 48 h) [77]. In chronic post-RIC, applied daily, plasma nitrite levels were increased at 3 weeks, but no longer at 1 month or 4 months [45].
Cytokines circulating in blood are also altered by ischemic stroke and limb RIC. In mouse model, limb pre-RIC applied 1 h before MCAO [37] reduced TNFα in plasma measured 24 h later, indicating that limb pre-RIC initially reduces systemic inflammation. In rat, TNFα mRNA was increased in whole blood 48 h after IS [79] with similar trends observed in whole brain. However, in serum, 3 days after IS, TNFα levels are normal, while pre-RIC TNFα levels in serum after 3 days were markedly increased [85]. In the same study, serum interleukin 6 (IL-6) was increased after IS in rat [85] and diabetic mouse [76], while pre-RIC further exacerbated this increase at 3 days after IS in rat [85], and pre-RIC and post-RIC mitigated the increase in diabetic mouse at 48 h [76] with no change in IL-1β, IFN-γ, TNF-α, or IL-4. However, post-RIC in another study of rat IS markedly reduced IL-6 mRNA in both whole blood and whole brain 48 h later [79]. Two days after brain ischemia in rat, protein levels of anti-inflammatory cytokines IL-4 and IL-10 are reduced in plasma and brain in one study [32] but IL-10 mRNA is increased in blood after IS. When RIC is applied in the absence of cerebral ischemia in rat, plasma shows a 50% increase in protein levels of anti-inflammatory cytokines IL-4 and IL-10 with no change in pro-inflammatory cytokines IL-1β, IL-6, and IFN-γ at 48 h [32]. However, when post-RIC is performed just after ischemic stroke, 48 h later, plasma protein levels of anti-inflammatory cytokines IL-4 and IL-10 are 2- to threefold increased [32], but mRNA of IL-10 is decreased in whole blood [79]. Pro-inflammatory cytokines IL-1β, IL-6, and IFN-γ were decreased in plasma after post-RIC [32]; while systemic inhibition of HIF1α reverses both the neuroprotective effects of post-RIC and protein cytokine profiles in plasma [32]. In aged rats, IL-1β, IL-6, and IFN-γ were increased 48 h after IS but not when pre-RIC was applied 24 h before, while no changes were observed in TNFα, IL-4, and IL-10 [22].
Exosomes and MicroRNAs
Exosomes and MicroRNAs may be involved in neuroprotective effects of limb RIC. Endothelial-derived exosomes have been shown to provide a degree of protection against ischemia/reperfusion injury in neuronal cells, including reducing apoptosis and increasing proliferation [89]. Pre-RIC plasma exosomes from mice are rich in HIF-1α, CD63, TSG101, and CD81, and attenuate infarct size and neurological function when infused into IS model [92]. Exosomes extracted from human RIC plasma reduced DNA methylation and provided a degree of tolerance to oxygen/glucose deprivation in vitro in SH-SY5Y neuroblastoma cells [94]. Fifty differentially expressed microRNAs have been identified in human plasma collected immediately after RIC, including miR-126, which was upregulated and is postulated to target DNA (cytosine-5)-methyltransferase 3B (DNMT3B) which may in part explain the RIC exosome induced reduction in DNA methylation [94].
MicroRNAs (miRNAs) are also candidate molecules that could be involved in induction of limb RIC neuroprotection. miRNAs can be found in blood linked to the carrier protein Argonaute-2 or inside exosomes that are produced by a variety of cells both of which can traverse the blood–brain barrier [97, 98]. In mouse plasma, 13 miRNAs were downregulated and 18 upregulated 24 h after limb RIC performed on abdominal aorta [99]. However, different RIC protocols can result in different miRNA expression profiles [100]. In rat, presumed muscle-derived miR-29a was upregulated in plasma after limb RIC was performed once a day for 4 weeks [91]. In both mouse and human, miR-144 was upregulated in plasma after limb RIC 4 cycles of 5-min ischemia/reperfusion [90]; and in an MCAO stroke mouse model, miR-144 introduced into circulation targeted PTEN resulting in increased Akt pathway activity and inhibiting of apoptosis in brain [93].
Circulating Blood Cells
Limb RIC causes changes in blood cell populations that may have direct neuroprotective effects. In a mouse model of IS, pre-RIC resulted in increased levels of 2,3-biphosphoglycerate (2,3-BPG) in circulating erythrocytes after 24 h [87]. In fact, 2,3-BPG facilitates the dissociation of oxygen-hemoglobin bound, shifting the oxygen dissociation curve toward increased oxygen delivery to ischemic brain tissue [87].
RIC also alters circulating immune cell composition which appears to be dependent on an intact spleen [86, 88]. In addition, whereas normally splenic lymphocytes positively correlate with circulating B and T lymphocytes, following RIC, a negative correlation with circulating T lymphocytes was observed. In rat experimental ischemic stroke, limb pre-RIC increased splenic volume, total lymphocytes and the percentages of cytotoxic and natural killer T cells, and B lymphocytes in the spleen after 3 days [86], whereas an increase in CD8 + cytotoxic T cells is seen after 1 h and is maintained after 3 days. In the blood, leukocyte populations are also altered by RIC. The percentage of CD4 + helper T cells is reduced by IS in rat and pre-RIC does not change this. In the same pre-RIC model, however, the downregulation in CD8 + cytotoxic T cells, NKT cells, B cells, and noninflammatory resident monocytes, triggered by IS, appears to be partially rescued by pre-RIC, 3 days after IS [85]. In a mouse model of IS, after 2 days, CD4 + helper T cells are decreased in the spleen and lymph nodes, and post-RIC completely reverses this, with increasing trend also observed in blood [79]. Cytotoxic CD8 + T cells and NKT cells appear reduced in lymph nodes, spleen, and blood after IS, with this trend being reversed by post-RIC [79]. B cells and NK cells on the contrary appear to be increased by IS in the lymph nodes, spleen, and blood after 24 h, with this trend being reversed by post-RIC [79]. In a diabetic IS stroke mouse model, the decrease in blood helper T cells (CD4 +) and cytotoxic T cells (CD8 +) was reversed by pre-RIC but unaltered by post-RIC after 48 h, while no changes were observed in B cells or NK cells [76]. In mouse, IS-induced reduction in splenic monocytes/macrophages is reversed by rapid post-RIC within 24 h [47].
Noninflammatory monocytes were also increased after IS in mouse blood but not in the spleen or lymph nodes, while post-RIC reversed this; no changes in inflammatory monocytes were observed in the blood, spleen, or lymph nodes after IS and/or post-RIC [79, 88]. Although post-RIC did not change relative values of pro-inflammatory or anti-inflammatory monocyte subsets in spleen, pro-inflammatory monocytes are increased in blood. In vitro cultured splenocytes treated with post-RIC serum converted from anti-inflammatory monocytes into pro-inflammatory monocytes, with a greater shift observed with serum from severe stroke with post-RIC [88]. This supports the notion that limb post-RIC produces circulating factors that induce the spleen to produce pro-inflammatory monocytes into circulation that can infiltrate the brain and have a protective role. In rat brain, contralateral to stroke lesion or in ipsilateral hemisphere, post-RIC did not affect resident microglia measured 3 days after stroke; however, in ipsilateral ischemic hemisphere, there was increased infiltration of pro-inflammatory monocytes, and reduced mRNA expression of MCP-1 and IL-1β [88].
In mouse, blood monocyte depletion reversed the neuroprotective effects of rapid post-RIC on infarct volume and brain monocytes/macrophages [47]. In the same model, rapid post-RIC attenuates the large increase of granulocyte colony‐stimulating factor (G-CSF) in ischemic cortex, and the reduction in plasma G-CSF and splenic tissue monocytes/macrophages induced by ischemic stroke [47], and this regulation of inflammatory stimuli increases leptomeningeal collateral circulation [47].
Human Studies and Clinical Trials of Remote Ischemic Conditioning (RIC)
Introduction
Similarly to experimental research settings, the first clinical studies about RIC were made in myocardial infarction patients. RIC procedure was applied in ST-segment elevation myocardial infarction, and has reported reductions in infarcted area, some improvement of left-ventricle ejection fractions, reduction of creatinine-kinase myocardial plasma release or decreased troponin I levels [101,102,103,104,105]. Likewise, in patients with stable ischemic heart failure chronically treated with RIC (2 procedures per day during 6 weeks) improved left-ventricle ejection fraction volume and decreased B type natriuretic peptide levels were registered [106]. In rheumatic heart disease patients undergoing valve replacement, remote ischemic conditioning decreased the release of serum cardiac troponin I and also improved liver and lung biomarkers, indicating a potential systemic protective effect [107]. Recently, it was also found that strength training in healthy volunteers along with 8 sessions of RIC procedure improved muscle strength in wrist extensor muscle [108].
Herein, the main focus is to review the potential protective effects of RIC targeting the brain in clinical settings. Because there is not as much data for ischemic stroke as in experimental models and also because the cellular mechanisms RIC-mediated neuroprotection are similar for several different disorders, our review also highlights other neurological diseases besides ischemic stroke.
RIC Clinical Trials in Neurological Disease: Potential Cerebral Effects
In neurological disease, RIC-based strategies have been tested for safety and efficacy in patients with acute ischemic stroke, hemorrhagic stroke, middle cerebral or carotid artery stenosis, and spinal cord lesion. RIC has also been evaluated for cerebral complications associated with coronary artery bypass grafting. Clinical trial RIC data relating to ischemic stroke and other cerebral diseases is summarized in Tables 3 and 4, respectively.
Ischemic Stroke
In acute ischemic stroke, a proof-of-concept clinical trial with 171 stroke patients showed that RIC is safe and can be applied in the pre-hospital setting [113, 114]. In this study, the primary outcome was penumbral salvage assessed on multimodal magnetic resonance imaging and secondary outcome was infarct growth at 24-h and 1-month follow-up. Despite most results were neutral, RIC should have potential benefit in reducing the risk of infarction growth after 1 month [113, 114].
RECAST (Remote Ischemic Conditioning After Stroke Trial) was a pilot randomized placebo controlled phase II trial in acute ischemic stroke. RECAST was first developed with 26 patients and had a primary outcome of feasibility and tolerability and a secondary outcome including blood biomarkers and 90-day NIHSS [110]. Despite RIC increased plasma concentration of heat shock protein-27 (HSP-27), no plasma level changes in S100ß, troponin-T, HSP-60, HSP-70, HSP-90, or matrix metalloproteinase-9 (MMP-9) were observed [110]. Finally, RIC improved neurological outcome assessed by a decrease on 90-day NIHSS [110]. RECAST was further developed as a phase IIb clinical trial with 60 acute ischemic stroke patients randomized 1:1 to receive RIC [111]. Again, feasibility and safety were warranted, but no changes in NIHSS or in plasma levels of neuron-specific enolase or MMP-9, except of S100ß plasma concentration that decreased in RIC group [111]. RECAST-3 is the phase III prospective randomized multicenter clinical trial that is in progress and will include 1300 participants [112]. In this phase III, four doses of RIC (4 cycles of 5 min of ischemia) will be applied: the first one within 6 h or less of stroke onset, second dose at 2 h after the first dose, and doses 3 and 4 will be applied at the second day after stroke onset.
Another pilot study was performed to assess feasibility and safety of RIC as adjuvant therapy along with thrombectomy for acute ischemic stroke [119]. Twenty patients were treated with RIC procedure at 3 time points: immediately before canalization, immediately after canalization, and 7 days after thrombectomy. No RIC-induced effect was found on intracranial pressure, cranial perfusion, mean arterial pressure, or middle artery systolic flow velocity [119].
Symptomatic intracranial arterial stenosis is a major cause of stroke. Daily and bilateral application of RIC (5 cycles of 5 min of ischemia and 5 min of reperfusion) for about 300 days reduced (from 26.7 to 7.9%) the stroke recurrence in patients with intracranial arterial stenosis [121]. Likewise, in octo- and nonagenarians, the same study was performed for 180 days with a reduction of stroke recurrence in RIC group patients [122]. Nevertheless, a single procedure of RIC (5 cycles of 5 min of ischemia and 5 min of reperfusion) did not alter heart rate, oxygenation index, or mean flow velocity in patients with middle cerebral artery stenosis, a reduction in blood pressure in healthy volunteers was the single RIC effect found [123]. Thus, these data suggest that chronic application of RIC might present more efficient results in neuroprotection than a single acute treatment.
The potential protection of RIC against ischemic injury in patients with severe carotid artery stenosis undergoing carotid artery stenting was tested [131]. Two RIC procedures applied daily during 2 weeks before carotid artery stenting significantly decreased the incidence of new ischemic lesions assessed by new diffusion-weighted imaging lesions 48 h after stenting. Nevertheless, RIC did not change plasma concentrations of neuron-specific enolase or S100ß nor clinical outcomes after 6 months [131].
RESCUE BRAIN was a French multicenter randomized clinical trial of remote ischemic per-conditioning in the treatment of acute ischemic stroke, which included 188 patients receiving either remote ischemic per-conditioning or sham procedure during or after reperfusion treatments, up to 6 h after the stroke onset. Infarct volume at 24 h was the primary outcome, but no significant changes were observed between intervention and control groups [115]. Likewise, at 3 months, no significant difference was found in mortality or symptomatic intracerebral hemorrhage [115].
RESIST is a clinical trial of remote ischemic per- and post-conditioning for treatment of acute stroke ongoing in Denmark whose protocol was described in [109]. Authors intend to include 1000 patients with diagnosis of acute ischemic stroke and intracerebral hemorrhage along with 1500 patients with a pre-hospital presumed stroke, with the 3-month follow-up modified ranking scale score as the primary outcome [109]. Since this clinical trial is larger than the previous ones, it holds expectations for potential beneficial outcomes of RIC.
REPOST is another randomized clinical trial for remote ischemic post-conditioning (RIPostC) in ischemic stroke that is ongoing in the Netherlands [117]. In this clinical study, RIPostC procedure is applied to stroke patients twice a day during hospitalization; primary outcome is infarct size assessed by MRI diffusion-weighted image at the end of hospitalization [117]. In fact, one may speculate that remote conditioning triggers mild responses by the organism, which would need chronic application for generating beneficial effects. Accordingly, Liu and colleagues have described a protocol, which is ongoing and only includes patients with acute minor ischemic stroke or transient ischemic attack [118]. In this clinical trial, RIC is used as adjuvant treatment for secondary stroke prevention. Again, it may be considered that RIC might present a mild and long-term effect, which can be more efficient for less severe ischemic events.
Other clinical trials are in progress, namely REMOTE-CAT (NCT03375762), REVISE 2 (NCT030445055), and RICE PAC (NCT03152799).
Another pilot study with 49 acute stroke patients assessed the effect of RIC in combination with treatment with intravenous thrombolysis [120]. RIC procedure was applied within 6 to 24 h of thrombolysis treatment. The two groups were followed up for 90 days and no differences between RIC and sham group were found regarding blood pressure, hemorrhage transformation, or adverse effects. Nevertheless, the levels of high sensitivity C reactive protein were lower for RIC group [120].
Finally, in stroke survivors, the application of RIC (5 cycles of 5 min of ischemia) at their paretic leg every other day for 2 weeks improved the self-selected walking speed and increased the time to fatigue, when compared the sham RIC applied patients [132]. Although not being a direct cerebral effect, RIC may improve stroke patient quality of life.
Subarachnoid Hemorrhage and Intracerebral Hemorrhage
RIC was first assessed in a small pilot study with 4 patients with aneurysmal subarachnoid hemorrhage (SAH). Three to four sessions of 4 cycles were applied during 2 to 12 non-consecutive days following SAH [125]. In the 4 tested patients, RIC appeared to increase the mean of intracranial pressure and decrease the mean velocities in the middle cerebral artery. Brain microdialysis revealed that RIC might reduce lactate/pyruvate ratio and glycerol levels [125]. One year later, same authors performed a similar study, considered phase I clinical trial with 20 aneurysmal SAH patients [126]. In this study, the primary outcome was the development of deep venous thrombosis, bruising, or injury to the conditioned limb. None of these outcomes was registered, meaning the clinical trial is safe and feasible [126]. The main conclusion is the need for subsequent larger clinical trials. A similar study was performed by Koch and colleagues with 34 aneurysmal SAH patients for safety and feasibility purposes [127]. The analyzed parameters were transcranial Doppler signs of vasospasm, delayed cerebral ischemia, and 3-month modified Rankin scale with no statistical difference between control and treated groups. Herein, the authors also tested different times of ischemia during RIC procedure, namely 5, 7.5, and 10 min of ischemia with no difference on outcomes or potential injury [127].
Finally, another pilot study will be performed for RIC in the treatment of intracerebral hemorrhage (RICH-1) [128]. Patients will be daily treated within 24–48 h of the onset for one week. Primary outcome is safety and secondary outcome will be the volume of hematoma and perihematomal edema, functional outcomes, and the incidence of adverse events. Moreover, cranial tomography will be performed at days 1, 3, 7, and 14 after stroke onset [128].
Other Potential Cerebral Beneficial Effects of RIC
Remote ischemic preconditioning (RIPC) was tested in patients with cervical spondylotic myelopathy prior to undergoing elective decompression surgery, including 20 patients in each group: intervention and control [124]. While RIPC has reduced serum levels of neuron-specific enolase and S100ß, no effect was found in the median nerve somatosensory-evoked potential [124]. This suggests some effects of RIPC on neurological complications following spinal cord surgery, but many more studies are needed.
Long-term administration of RIC procedure was tested in patients with mild cognitive impairment related to small vessel disease. In fact, 5 cycles of RIC were applied twice a day during 1 year in 14 patients, while 16 patients corresponded to control group without any treatment [129]. After 1 year, RIC reduced the volume of white matter hyperintensities and improved the visuospatial and executive abilities, while no difference was found in the number of lacunes between the two groups. The major effects were found in plasma characteristics, namely RIC promoted marked reduction in plasma triglycerides, total cholesterol, low-density lipoprotein, and homocysteine [129]. Despite the reduced number of patients, this is a promising study since RIC might act more efficiently when applied in a long-term (chronic) manner due to its mild immediate effect. Likewise, in patients with subcortical ischemic vascular dementia, chronic RIC has been tested as daily applications for 6 months [130]. This randomized study included 37 patients: 18 underwent 5 cycles of brief bilateral upper limb compression at 200 mmHg; 9 controls underwent the same procedure with compression at 60 mmHg; and 10 controls had no procedure. RIC-treated patients showed cognitive improvement assessed by neuropsychological tests [130].
Following coronary artery bypass grafting (CABG), silent episodes of brain ischemia can occur; thus, RIC procedure holds potential as a pre-operatory preventive treatment. RIC or sham procedure were tested in 70 patients undergoing CABG, with structural brain magnetic resonance imaging (MRI) for connectivity analysis and neurological evaluation [133]. No difference between groups was found in new ischemic brain lesions or functional connectivity profile; however, RIC reduced the pooled volume of ischemic brain lesions [133].
Finally, young healthy adults underwent a cognitive and motor training along with 7-day daily RIC procedure or sham procedure for the evaluation of cognitive and motor learning. In fact, RIC promoted cognitive and motor learning enhancement, which can potentially by used in rehabilitation of cerebral injured patients [134].
Mechanisms of RIC in the Inter-organ Communication
Most of these clinical trials are mainly focused on disclosing the potential beneficial cerebral and cardiovascular effect of RIC in humans. Nevertheless, some of them were also devoted to describe the underlying mechanism of remote protection and inter-organ communication, as described in the following section. Furthermore, we also review several publications concerning human interventional studies in healthy volunteers for the assessment of RIC-associated mechanisms, which are mainly based on modulation of autonomic nervous system or humoral factors. Likewise, other RIC-induced responses can be associated with microcirculation, endothelial function, or inflammation control. These responses can be considered beneficial consequences of RIC or as signaling events for inter-organ communication and neuroprotection.
Autonomic Nervous System
Data generated in experimental models pointed to the involvement of autonomic nervous system in RIC signaling and inter-organ communication. In fact, blockade of opioid receptors or muscarinic receptors, bilateral vagotomy, or spinal cord resection abolished RIC protection in animal models [135,136,137]. In healthy volunteers, ischemia–reperfusion (IR) procedure was applied in forearm for promoting lesion which was assessed by the activation of muscle sympathetic nerve activity, finger reactive hyperemia, and oxidative stress (reduced glutathione (GSH) in erythrocytes). Pre-treatment with 2 cycles of 5 min of RIC in one leg delayed the IR-induced increase on muscle sympathetic nerve activity measured in contralateral leg by microneurography [138]. Likewise, RIC limited the increase on erythrocyte-derived GSH levels during ischemia and promoted vasodilation [138]. Other studies used heart rate variability (HRV) to follow autonomic nervous system response to RIC in healthy volunteers. In a pilot study using young and senior (> 60 years old) sub-groups, HRV analysis demonstrated that the non-linear parameter SD2 (associated with long-term HRV) increased significantly in response to RIC procedure, suggesting the activation of both sympathetic and parasympathetic nervous system, in particular via the slow sympathetic response to the baroreceptors stimulation and via fast vagal parasympathetic response [139]. In addition, another study with 50 subjects analyzed HRV is several time points: 1 h before RIC procedure and 1, 3, 3, 6, 9, 12, and 24 h after. Similarly, the time domain SDNN and the non-linear SD2 parameters increased 1 h after RIC, while 12 h later only SD2 increase [140]. Khaliulin and colleagues found a reduction on porphyrin fluorescence in the blood, as well as an increase in power of very low frequency band in ECG recording; both facts indicate an increased stress resistance response [141].
Humoral Factors
In human healthy subjects, RIC procedure increased the levels of nitrite in plasma and platelet rich plasma [142]. Of note, nitrite is an oxidation product of NO, being an indirect measure of NO. Although NO is associated with vasomodulation, Dezfulian and colleagues have demonstrated that RIC-derived human plasma protects myocytes against cell death induced by hypoxia/reoxygenation in an in vitro cell model [142]. Moreover, RIC reduced platelet mitochondrial respiration and increased mitochondrial anion superoxide production, which can be associated to NO binding to mitochondrial respiratory chain reaction complexes [142]. Exosomes-mediated intercellular communication may be another factor involved in RIC signaling to distant organs due to the ability of transferring microRNAs, lipids, and proteins. In fact, exosomal miRNA 126 was found in plasma of healthy volunteers subjected to RIC procedure [94]. Similar to miRNA 126-induced neuroprotection against stroke in experimental models [143], exosomes isolated from RIC-derived human plasma prevented neuronal cell death using the in vitro model of SH-SY5Y cells [94].
A microarray for the analysis of blood gene expression was done 15 min and 24 h after RIC procedure in 4 healthy volunteers subject to RIC, with the identification of 169 genes differently expressed [144]. Konstantinov and colleagues highlighted the potential anti-inflammatory pattern of gene expression derived from RIC, pointing to the upregulation of anti-inflammatory heat shock protein (HSP) 70 and calpastatin genes, while downregulation of immunity response genes, namely TLR4 and TNF signaling pathway TNFR6 [144]. Proteomic studies based on 2 dimensional difference in gel electrophoresis (2DIGE) and mass spectrometry (MS) were performed to identify potential modifications on the composition of circulating plasma proteins following RIC [145]. Six proteins were found to change in response to RIC when analysis was done by 2DIGE, while 48 proteins were found in MS analyses, being only 3 of them common in both analysis. This sort of large proteomic approaches must follow further validations with different analytical techniques, as well as functional validation using experimental models. In another study, three cycles (5 min ischemia and 5 min of reperfusion) of RIC were applied in 60 healthy young volunteers followed by proteomic analysis at 1, 3, 8, 24, and 48 h [146]. Fourteen proteins were found differently present in the serum, most of them (11 proteins) at 1 h after RIC. These proteins are involved in mechanisms related to coagulation, apoptosis, lipid metabolism, and immune system [146]. Furthermore, mass spectrometry proteomic analysis was done in plasma derived from children with cyanotic heart disease undergoing cardiopulmonary bypass surgery treated or not with remote ischemic preconditioning (RIPreC). Protein pattern in the plasma was different only 6 h after RIPreC with the increased concentration of six proteins: inter-alpha globulin inhibitor, fibrinogen preproprotein, complement-C3 precursor, complement C4B, apolipoprotein B100, and urinary proteinase inhibitor [147]. In fact, the large heterogeneity of reported data and lack of reproducibility might be due to inter-donor variability, age, and different data analysis processes [148].
RIC procedure applied in traumatic brain injury or cervical spondylotic myelopathy patients reduced the levels of neuron-specific enolase (NSE) and S100ß, which are serum biomarkers for acute brain injury [124, 149]. Despite being a good indication of RIC-induced neuroprotection, NSE and S100ß may be not involved in neuroprotection signaling triggered by RIC.
Finally, recent metabolomic profiling of human cerebrospinal fluid after daily RIC, applied during 3 consecutive days, showed increased creatine, ethanolamine, and isobutyrate, and decreased phenylalanine, hypoxanthine, and tyrosine [150]; beyond being evidence of altered pathways of energy and amino-acids metabolism in the central nervous system, the significance of these results to RIC-induced neuroprotection remains to be determined.
Endothelial Function and Blood Circulation
Endothelial function and blood circulation (vasodilation), which are key events in response and protection against ischemia and reperfusion, can be assessed by blood flow-mediated dilation of the brachial artery or by skin microcirculation using cutaneous vascular conductance. Several studies in clinical settings were conducted to evaluate blood circulation and endothelial function in response to RIC. In the arm, 20 min of ischemia followed by reperfusion decreased endothelial function, which was assessed by blood flow-mediated dilation. The ischemia–reperfusion-induced decrease of flow-mediated dilation was partially reverted by RIC procedure in the contralateral arm or leg, applied before (pre) or after (post) ischemia [151, 152]. Moreover, RIC-triggered improvement of flow-mediated dilation was reverted by pharmacological treatment with glibenclamide, a sulfonylurea that inhibits ATP-sensitive potassium channels, indicating that endothelial protection depends on KATP channel activation in humans [151]. Likewise, administration of the autonomic ganglion blocker trimetaphan also reduced the RIC-induced vasoprotection [152]. In addition, RIC procedure in the arm of healthy subjects improved microcirculation assessed in the thigh, in particular at the third cycle tissue oxygen saturation and capillary blood flow increased [153]. Likewise, 7 days daily based RIC improved flow-mediated dilation and skin microcirculation (cutaneous vascular conductance assessment) in the RIC arm and the contralateral arm, suggesting a local and distant effect of RIC [154]. Finally, 1 week of twice a day, RIC application improved coronary microcirculation in healthy and heart failure patients [155]. Coronary microcirculation was measured by transthoracic Doppler echocardiography assessed by coronary flow reserve [155].
RIC Controversies and Remaining Questions
Translation of Knowledge from Experimental Models and Basic Research to Clinical Settings
RIC-based clinical trials have just begun and much data is still needed in order to reveal the real potential protective role of RIC in clinical settings. Nevertheless, one can partially predict less successful results in clinical trials than experimental models, with a complex translation of knowledge due to the features of the animals used. In fact, in the majority of models, the animals are young, in some cases adolescent, without any other co-morbidities that usually are associated with stroke patients, namely hypertension, diabetes, atherosclerosis, etc. Although the maintenance and use of aged animals are highly expensive, this should be taken into account when performing pre-clinical basic research about age-related disease.
In addition, basic research-associated technology (proteomic, metabolomics, transcriptomics, etc.) should be more explored to identify potential biochemical factors and circulating cells (leukocytes and erythrocytes) involved in the communication between the remote ischemic conditioned limb and the brain in human subjects. Taking advantage of the mild invasive procedure, such as blood collection, it is possible to investigate the RIC-associated molecular and cellular underlying mechanisms within human samples. Thus, a deep knowledge about the signaling mechanisms of RIC in humans could be reached.
Real Therapeutic Properties of RIC in Ischemic Stroke Clinical Settings
For acute ischemic stroke, several clinical trials have revealed the feasibility and safety of RIC. Although the first results regarding the beneficial outcomes of RIC are not yet clear, there are some clues indicating the potential beneficial effects of RIC. Thus, ongoing clinical trials are still promising. For example, RESIST, Danish clinical trial of remote ischemic per- and post-conditioning, will include 1000 stroke patients [109].
Patient selection may be a key factor to access the therapeutical potential of RIC, since age and co-morbidities may influence RIC efficacy. Since RIC triggers endogenous defense mechanism, one could argue that its effect are mild, and therefore only beneficial in non-severe strokes. On the other hand, RIC impact on cerebral blood flow and on brain parenchyma can be dependent on the presence of salvageable penumbra. Thus, it would be important to correlate RIC improvement in patient’s outcome with the existence of salvageable penumbra. Also, RIC can have reduced impact in patients with early recanalization, but useful in those with longer treatments delays or failed recanalization therapies.
Finally, how co-morbidities can influence the beneficial effect of RIC is still unclear. Most of the completed RIC clinical studies are pilot studies that only provide preliminary data. Thus, larger clinical trials, allowing subgroup analysis, are needed for understanding the potential influence of co-morbidities such as diabetes, atherosclerosis, hypertension, age, and others on the benefits RIC treatment.
RIC Application for Chronic Cerebral Diseases
RIC may also be more efficient in long-term applications in chronic diseases. In fact, Meng and colleagues have demonstrated that daily application of RIC during 300 or 180 days (the last one for octo- and nonagenarians) decreased the recurrence of stroke in patients with intracranial arterial stenosis [121, 122]. RIC was also tested for 1 year in patients with mild cognitive impairment caused by small vessel disease, improving some visuospatial and executive abilities and decreasing the volume of white matter hyperintensities, despite being a pilot study with only 29 patients [129]. A second study with 37 patients with vascular dementia, that applied RIC daily during 6 months, also reported an improvement in cognitive functions [130]. Likewise, chronic application of post-RIC in an experimental model of vascular cognitive impairment also improved cerebral blood flow and protected the brain parenchyma [74]. Thus, one can speculate that chronic RIC treatment holds great beneficial potential if applied chronically for patients with high risk of cerebral vascular diseases, including vascular dementia and white matter disease, among others. As a secondary stroke prevention treatment, RIC beneficial effects may also depend on ischemic stroke etiology, being potentially more effective in patients with large vessel disease when compared with cardioembolic stroke.
Concluding Remarks and Perspectives
Pre-clinical studies showed the potential neuroprotective effect of RIC and disclose its associated pathways and signaling. This therapy is based on the promotion of physiological protective mechanisms, and can inspire new targeted therapies built upon these same principals. In the translation to clinical practice, RIC results are modest so far. Type of RIC, clinical application, and patient selection criteria are of utmost importance in order to establish the RIC role in patient care. If efficient, RIC will most certainly be applied as an add-on therapy, together in other strategies for acute stroke treatment or stroke prevention.
Data and Materials Availability
Not applicable.
Code Availability
Not applicable.
References
Feigin VL, Roth GA, Naghavi M et al (2016) Global burden of stroke and risk factors in 188 countries, during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol 15:913–924. https://doi.org/10.1016/S1474-4422(16)30073-4
Powers WJ, Rabinstein AA, Ackerson T et al (2018) 2018 Guidelines for the Early Management of Patients With Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 49. https://doi.org/10.1161/STR.0000000000000158
Yepes M, Roussel BD, Ali C, Vivien D (2009) Tissue-type plasminogen activator in the ischemic brain: more than a thrombolytic. Trends Neurosci 32:48–55. https://doi.org/10.1016/j.tins.2008.09.006
Chamorro Á, Dirnagl U, Urra X, Planas AM (2016) Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol 15:869–881. https://doi.org/10.1016/S1474-4422(16)00114-9
Lansberg MG, Straka M, Kemp S et al (2012) MRI profile and response to endovascular reperfusion after stroke (DEFUSE 2): a prospective cohort study. Lancet Neurol 11:860–867. https://doi.org/10.1016/S1474-4422(12)70203-X
Iadecola C, Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14:1363–1368. https://doi.org/10.1038/nn.2953
Dirnagl U, Becker K, Meisel A (2009) Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 8:398–412
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136. https://doi.org/10.1161/01.CIR.74.5.1124
Hess DC, Blauenfeldt RA, Andersen G et al (2015) Remote ischaemic conditioning-a new paradigm of self-protection in the brain. Nat Rev Neurol 11:698–710
Przyklenk K, Bauer B, Ovize M et al (1993) Regional ischemic “preconditioning” protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 87:893–899. https://doi.org/10.1161/01.CIR.87.3.893
Oxman T, Arad M, Klein R et al (1997) Limb ischemia preconditions the heart against reperfusion tachyarrhythmia. Am J Physiol Circ Physiol 273:H1707–H1712. https://doi.org/10.1152/ajpheart.1997.273.4.H1707
Stetler RA, Leak RK, Gan Y et al (2014) Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol 114:58–83. https://doi.org/10.1016/j.pneurobio.2013.11.005
Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: Mechanisms in search of treatments. Neuron 67:181–198. https://doi.org/10.1016/j.neuron.2010.07.002
Fukuyama N, Takizawa S, Ishida H et al (1998) Peroxynitrite Formation in Focal Cerebral Ischemia—Reperfusion in Rats Occurs Predominantly in the Peri-Infarct Region. J Cereb Blood Flow Metab 18:123–129. https://doi.org/10.1097/00004647-199802000-00001
Jayaraj RL, Azimullah S, Beiram R et al (2019) Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation 16:142. https://doi.org/10.1186/s12974-019-1516-2
Kawabori M, Kacimi R, Kauppinen T et al (2015) Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) Deficiency Attenuates Phagocytic Activities of Microglia and Exacerbates Ischemic Damage in Experimental Stroke. J Neurosci 35:3384–3396. https://doi.org/10.1523/JNEUROSCI.2620-14.2015
Deczkowska A, Keren-Shaul H, Weiner A et al (2018) Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173:1073–1081. https://doi.org/10.1016/j.cell.2018.05.003
Zhao HG, Bin Li W, Li QJ et al (2004) Limb ischemic preconditioning attenuates apoptosis of pyramidal neurons in the CA1 hippocampus induced by cerebral ischemia-reperfusion in rats. Acta Physiol Sin 56:407–412
Hoda MN, Bhatia K, Hafez SS et al (2014) Remote Ischemic Perconditioning is Effective After Embolic Stroke in Ovariectomized Female Mice. Transl Stroke Res. https://doi.org/10.1007/s12975-013-0318-6
Hoda MN, Siddiqui S, Herberg S et al (2012) Remote ischemic perconditioning is effective alone and in combination with intravenous tissue-type plasminogen activator in murine model of embolic stroke. Stroke 43:2794–2799. https://doi.org/10.1161/STROKEAHA.112.660373
Burda R, Danielisova V, Gottlieb M et al (2014) Delayed remote ischemic postconditioning protects against transient cerebral ischemia/reperfusion as well as kainate-induced injury in rats. Acta Histochem. https://doi.org/10.1016/j.acthis.2014.04.011
Du X, Yang J, Liu C et al (2020) Hypoxia-Inducible Factor 1α and 2α Have Beneficial Effects in Remote Ischemic Preconditioning Against Stroke by Modulating Inflammatory Responses in Aged Rats. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2020.00054
Ren C, Yan Z, Wei D et al (2009) Limb remote ischemic postconditioning protects against focal ischemia in rats. Brain Res. https://doi.org/10.1016/j.brainres.2009.07.029
Zong Y, Jiang L, Zhang M et al (2015) Limb remote ischemic postconditioning protects cerebral ischemia from injury associated with expression of HIF-1aα in rats. BMC Neurosci. https://doi.org/10.1186/s12868-015-0235-6
Chen Q, Wu F, Peng X et al (2016) Limb remote ischemic preconditioning protects against cerebral ischemia through down-regulation of aquaporin-4. Int J Clin Exp Med 9:13878–13889
Li P, Su L, Li X et al (2016) Remote limb ischemic postconditioning protects mouse brain against cerebral ischemia/reperfusion injury via upregulating expression of Nrf2, HO-1 and NQO-1 in mice. Int J Neurosci. https://doi.org/10.3109/00207454.2015.1042973
Li H, Zhou S, Wu L et al (2015) The role of p38MAPK signal pathway in the neuroprotective mechanism of limb postconditioning against rat cerebral ischemia/reperfusion injury. J Neurol Sci. https://doi.org/10.1016/j.jns.2015.08.004
Ramagiri S, Taliyan R (2017) Protective effect of remote limb post conditioning via upregulation of heme oxygenase-1/BDNF pathway in rat model of cerebral ischemic reperfusion injury. Brain Res. https://doi.org/10.1016/j.brainres.2017.05.016
Ramagiri S, Taliyan R (2017) Remote limb ischemic post conditioning during early reperfusion alleviates cerebral ischemic reperfusion injury via GSK-3β/CREB/ BDNF pathway. Eur J Pharmacol. https://doi.org/10.1016/j.ejphar.2017.03.028
Xia M, Ding Q, Zhang Z, Feng Q (2017) Remote Limb Ischemic Preconditioning Protects Rats Against Cerebral Ischemia via HIF-1α/AMPK/HSP70 Pathway. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-016-0444-2
Cheng X, Zhao H, Yan F et al (2018) Limb remote ischemic post-conditioning mitigates brain recovery in a mouse model of ischemic stroke by regulating reactive astrocytic plasticity. Brain Res. https://doi.org/10.1016/j.brainres.2018.02.019
Yang J, Liu C, Du X et al (2018) Hypoxia inducible factor 1α plays a key role in remote ischemic preconditioning against stroke by modulating inflammatory responses in rats. J Am Heart Assoc. https://doi.org/10.1161/JAHA.117.007589
Liang W, Lin C, Yuan L et al (2019) Preactivation of Notch1 in remote ischemic preconditioning reduces cerebral ischemia-reperfusion injury through crosstalk with the NF-κB pathway. J Neuroinflammation. https://doi.org/10.1186/s12974-019-1570-9
Cheng Z, Li L, Mo X et al (2014) Non-invasive remote limb ischemic postconditioning protects rats against focal cerebral ischemia by upregulating STAT3 and reducing apoptosis. Int J Mol Med. https://doi.org/10.3892/ijmm.2014.1873
Li SQ, Bin LW, Zhang M et al (2013) The role of neuroglobin in the neuroprotection of limb ischemic preconditioning in rats. Mol Neurobiol 47:197–208
Ren C, Wang P, Wang B et al (2015) Limb remote ischemic per-conditioning in combination with post-conditioning reduces brain damage and promotes neuroglobin expression in the rat brain after ischemic stroke. Restor Neurol Neurosci 33:369–379. https://doi.org/10.3233/RNN-140413
Hu S, Dong H, Zhang H et al (2012) Noninvasive limb remote ischemic preconditioning contributes neuroprotective effects via activation of adenosine A1 receptor and redox status after transient focal cerebral ischemia in rats. Brain Res. https://doi.org/10.1016/j.brainres.2012.04.017
Brager AJ, Yang T, Ehlen JC et al (2016) Sleep is critical for remote preconditioning-induced neuroprotection. Sleep. https://doi.org/10.5665/sleep.6238
Yuan Q, Jia HX, Li SQ et al (2019) The role of adenosine in up-regulation of p38 MAPK and ERK during limb ischemic preconditioning-induced brain ischemic tolerance. Brain Res. https://doi.org/10.1016/j.brainres.2018.11.015
Ma J, Ma Y, Dong B et al (2017) Prevention of the collapse of pial collaterals by remote ischemic perconditioning during acute ischemic stroke. J Cereb Blood Flow Metab. https://doi.org/10.1177/0271678X16680636
Ma J, Ma Y, Shuaib A, Winship IR (2020) Improved collateral flow and reduced damage after remote ischemic perconditioning during distal middle cerebral artery occlusion in aged rats. Sci Rep. https://doi.org/10.1038/s41598-020-69122-8
Parray A, Ma Y, Alam M et al (2020) An increase in AMPK/e-NOS signaling and attenuation of MMP-9 may contribute to remote ischemic perconditioning associated neuroprotection in rat model of focal ischemia. Brain Res. https://doi.org/10.1016/j.brainres.2020.146860
Ren C, Li S, Wang B et al (2018) Limb remote ischemic conditioning increases Notch signaling activity and promotes arteriogenesis in the ischemic rat brain. Behav Brain Res. https://doi.org/10.1016/j.bbr.2016.10.036
Ren C, Li N, Li S et al (2018) Limb ischemic conditioning improved cognitive deficits via eNOS-dependent augmentation of angiogenesis after chronic cerebral hypoperfusion in rats. Aging Dis. https://doi.org/10.14336/AD.2017.1106
Khan MB, Hafez S, Hoda MN et al (2018) Chronic Remote Ischemic Conditioning Is Cerebroprotective and Induces Vascular Remodeling in a VCID Model. Transl Stroke Res. https://doi.org/10.1007/s12975-017-0555-1
Liang D, He XB, Wang Z et al (2018) Remote limb ischemic postconditioning promotes motor function recovery in a rat model of ischemic stroke via the up-regulation of endogenous tissue kallikrein. CNS Neurosci Ther. https://doi.org/10.1111/cns.12813
Zhang Y, Ma L, Ren C et al (2019) Immediate remote ischemic postconditioning reduces cerebral damage in ischemic stroke mice by enhancing leptomeningeal collateral circulation. J Cell Physiol. https://doi.org/10.1002/jcp.27858
Liu M, Chen J, Zhang S, Ren C (2018) Downregulation of lipocalin-2 and Bim expression after remote limb preconditioning in the ischemic rat brain. Brain Res. https://doi.org/10.1016/j.brainres.2017.11.003
Wang Y, Zhang Z, Zhang L et al (2018) RLIPostC protects against cerebral ischemia through improved synaptogenesis in rats. Brain Inj. https://doi.org/10.1080/02699052.2018.1483029
Li Z, Cui X, Lv H et al (2020) Remote ischemic postconditioning attenuates damage in rats with chronic cerebral ischemia by upregulating the autophagolysosome pathway via the activation of TFEB. Exp Mol Pathol. https://doi.org/10.1016/j.yexmp.2020.104475
Lv J, Guan W, You Q et al (2020) RIPC provides neuroprotection against ischemic stroke by suppressing apoptosis via the mitochondrial pathway. Sci Rep. https://doi.org/10.1038/s41598-020-62336-w
Jin RL, Bin LW, Li QJ et al (2006) The role of extracellular signal-regulated kinases in the neuroprotection of limb ischemic preconditioning. Neurosci Res. https://doi.org/10.1016/j.neures.2006.01.006
Sun XC, Bin LW, Li QJ et al (2006) Limb ischemic preconditioning induces brain ischemic tolerance via p38 MAPK. Brain Res. https://doi.org/10.1016/j.brainres.2006.02.041
Sun XC, Xian XH, Bin LW et al (2010) Activation of p38 MAPK participates in brain ischemic tolerance induced by limb ischemic preconditioning by up-regulating HSP 70. Exp Neurol. https://doi.org/10.1016/j.expneurol.2010.04.009
Peng B, Guo QL, He ZJ et al (2012) Remote ischemic postconditioning protects the brain from global cerebral ischemia/reperfusion injury by up-regulating endothelial nitric oxide synthase through the PI3K/Akt pathway. Brain Res. https://doi.org/10.1016/j.brainres.2012.01.033
Qi ZF, Luo YM, Liu XR et al (2012) AKT/GSK3β-Dependent Autophagy Contributes to the Neuroprotection of Limb Remote Ischemic Postconditioning in the Transient Cerebral Ischemic Rat Model. CNS Neurosci Ther. https://doi.org/10.1111/cns.12016
Sun J, Tong L, Luan Q et al (2012) Protective effect of delayed remote limb ischemic postconditioning: Role of mitochondrial K ATP channels in a rat model of focal cerebral ischemic reperfusion injury. J Cereb Blood Flow Metab. https://doi.org/10.1038/jcbfm.2011.199
Su J, Zhang T, Wang K et al (2014) Autophagy activation contributes to the neuroprotection of remote ischemic perconditioning against focal cerebral ischemia in rats. Neurochem Res. https://doi.org/10.1007/s11064-014-1396-x
Qi Z, Dong W, Shi W et al (2015) Bcl-2 Phosphorylation Triggers Autophagy Switch and Reduces Mitochondrial Damage in Limb Remote Ischemic Conditioned Rats After Ischemic Stroke. Transl Stroke Res. https://doi.org/10.1007/s12975-015-0393-y
Qi W, Zhou F, Li S et al (2016) Remote ischemic postconditioning protects ischemic brain from injury in rats with focal cerebral ischemia/reperfusion associated with suppression of TLR4 and NF-κB expression. NeuroReport. https://doi.org/10.1097/WNR.0000000000000553
Huang D, Liu H, Qu Y, Wang P (2017) Non-invasive remote ischemic postconditioning stimulates neurogenesis during the recovery phase after cerebral ischemia. Metab Brain Dis. https://doi.org/10.1007/s11011-017-0068-3
Zhang W, Wang Y, Bi G (2017) Limb remote ischaemic postconditioning-induced elevation of fibulin-5 confers neuroprotection to rats with cerebral ischaemia/reperfusion injury: Activation of the AKT pathway. Clin Exp Pharmacol Physiol. https://doi.org/10.1111/1440-1681.12742
Chen Gz, Shan Xy, Li Xs, Tao Hm (2018) Remote ischemic postconditioning protects the brain from focal ischemia/reperfusion injury by inhibiting autophagy through the mTOR/p70S6K pathway. Neurol Res. https://doi.org/10.1080/01616412.2018.1424696
Guo H, Zhao L, Wang B et al (2018) Remote limb ischemic postconditioning protects against cerebral ischemia-reperfusion injury by activating AMPK-dependent autophagy. Brain Res Bull. https://doi.org/10.1016/j.brainresbull.2018.02.013
Li J, Hu XS, Zhou FF et al (2018) Limb remote ischemic postconditioning protects integrity of the blood-brain barrier after stroke. Neural Regen Res. https://doi.org/10.4103/1673-5374.237122
Kitagawa K, Saitoh M, Ishizuka K, Shimizu S (2018) Remote Limb Ischemic Conditioning during Cerebral Ischemia Reduces Infarct Size through Enhanced Collateral Circulation in Murine Focal Cerebral Ischemia. J Stroke Cerebrovasc Dis. https://doi.org/10.1016/j.jstrokecerebrovasdis.2017.09.068
Xu C, Yi C, Guo H et al (2012) Limb remote ischemic postconditioning is effective but also time-course-limited in protecting the brain from I/R injury. Turkish J Med Sci. https://doi.org/10.3906/sag-1106-2
Xue L, Chen H, Lu K et al (2017) Clinical significance of changes in serum neuroglobin and HIF-1α concentrations during the early-phase of acute ischemic stroke. J Neurol Sci. https://doi.org/10.1016/j.jns.2017.01.039
Hota KB, Hota SK, Srivastava RB, Singh SB (2012) Neuroglobin regulates hypoxic response of neuronal cells through Hif-1α- and Nrf2-mediated mechanism. J Cereb Blood Flow Metab. https://doi.org/10.1038/jcbfm.2012.21
Borea PA, Varani K, Vincenzi F et al (2015) The a3 adenosine receptor: History and perspectives. Pharmacol Rev. https://doi.org/10.1124/pr.113.008540
Chen JF, Eltzschig HK, Fredholm BB (2013) Adenosine receptors as drug targets-what are the challenges? Nat Rev Drug Discov. https://doi.org/10.1038/nrd3955
Fredholm BB (2007) Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 14(7):1315–1323
Trautmann A (2009) Extracellular ATP in the immune system: More than just a “danger signal.” Sci Signal. https://doi.org/10.1126/scisignal.256pe6
Khan MB, Hoda MN, Vaibhav K et al (2015) Remote Ischemic Postconditioning: Harnessing Endogenous Protection in a Murine Model of Vascular Cognitive Impairment. Transl Stroke Res 6:69–77. https://doi.org/10.1007/s12975-014-0374-6
Motomura A, Shimizu M, Kato A et al (2017) Remote ischemic preconditioning protects human neural stem cells from oxidative stress. Apoptosis. https://doi.org/10.1007/s10495-017-1425-8
Liu C, Yang J, Zhang C et al (2020) Remote ischemic conditioning reduced cerebral ischemic injury by modulating inflammatory responses and ERK activity in type 2 diabetic mice. Neurochem Int. https://doi.org/10.1016/j.neuint.2020.104690
Zhao HG, Sun XC, Xian XH et al (2007) The role of nitric oxide in the neuroprotection of limb ischemic preconditioning in rats. Neurochem Res. https://doi.org/10.1007/s11064-007-9381-2
Wei D, Ren C, Chen X, Zhao H (2012) The Chronic Protective Effects of Limb Remote Preconditioning and the Underlying Mechanisms Involved in Inflammatory Factors in Rat Stroke. PLoS ONE 7:e30892. https://doi.org/10.1371/journal.pone.0030892
Liu C, Yang J, Zhang C et al (2019) The changes of systemic immune responses during the neuroprotection induced by remote ischemic postconditioning against focal cerebral ischemia in mice. Neurol Res. https://doi.org/10.1080/01616412.2018.1523037
Suk K (2016) Lipocalin-2 as a therapeutic target for brain injury: An astrocentric perspective. Prog Neurobiol. https://doi.org/10.1016/j.pneurobio.2016.08.001
Kitagawa K, Matsumoto M, Tagaya M et al (1990) “Ischemic tolerance” phenomenon found in the brain. Brain Res. https://doi.org/10.1016/0006-8993(90)90189-I
Yang J, Shakil F, Cho S (2019) Peripheral Mechanisms of Remote Ischemic Conditioning. Cond Med 2:61–68
Shan LY, Li JZ, Zu LY et al (2013) Platelet-derived microparticles are implicated in remote ischemia conditioning in a rat model of cerebral infarction. CNS Neurosci Ther. https://doi.org/10.1111/cns.12199
Ren C, Liu K, Li N et al (2015) Neural transmission pathways are involved in the neuroprotection induced by post- but not perischemic limb remote conditioning. Brain Circ. https://doi.org/10.4103/2394-8108.172897
Liu ZJ, Chen C, Li XR et al (2016) Remote Ischemic Preconditioning-Mediated Neuroprotection against Stroke is Associated with Significant Alterations in Peripheral Immune Responses. CNS Neurosci Ther. https://doi.org/10.1111/cns.12448
Chen C, Jiang W, Liu Z et al (2018) Splenic responses play an important role in remote ischemic preconditioning-mediated neuroprotection against stroke. J Neuroinflammation. https://doi.org/10.1186/s12974-018-1190-9
Wang L, Ren C, Li Y et al (2020) Remote ischemic conditioning enhances oxygen supply to ischemic brain tissue in a mouse model of stroke: Role of elevated 2,3-biphosphoglycerate in erythrocytes. J Cereb Blood Flow Metab. https://doi.org/10.1177/0271678X20952264
Yang J, Balkaya M, Beltran C et al (2019) Remote Postischemic Conditioning Promotes Stroke Recovery by Shifting Circulating Monocytes to CCR2+ Proinflammatory Subset. J Neurosci. https://doi.org/10.1523/JNEUROSCI.2699-18.2019
Xiao B, Chai Y, Lv S et al (2017) Endothelial cell-derived exosomes protect SH-SY5Y nerve cells against ischemia/reperfusion injury. Int J Mol Med. https://doi.org/10.3892/ijmm.2017.3106
Li J, Rohailla S, Gelber N et al (2014) MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res Cardiol 109:423. https://doi.org/10.1007/s00395-014-0423-z
Yamaguchi T, Izumi Y, Nakamura Y et al (2015) Repeated remote ischemic conditioning attenuates left ventricular remodeling via exosome-mediated intercellular communication on chronic heart failure after myocardial infarction. Int J Cardiol. https://doi.org/10.1016/j.ijcard.2014.10.144
Li Y, Ren C, Li H et al (2019) Role of exosomes induced by remote ischemic preconditioning in neuroprotection against cerebral ischemia. NeuroReport. https://doi.org/10.1097/WNR.0000000000001280
Zhong SJ, Cui MM, Gao YT et al (2020) MicroRNA-144 promotes remote limb ischemic preconditioning-mediated neuroprotection against ischemic stroke via PTEN/Akt pathway. Acta Neurol Belg. https://doi.org/10.1007/s13760-020-01500-5
Cui J, Liu N, Chang Z et al (2020) Exosomal MicroRNA-126 from RIPC Serum Is Involved in Hypoxia Tolerance in SH-SY5Y Cells by Downregulating DNMT3B. Mol Ther - Nucleic Acids 20:649–660. https://doi.org/10.1016/j.omtn.2020.04.008
Hibert P, Prunier-Mirebeau D, Beseme O et al (2014) Modifications in rat plasma proteome after remote ischemic preconditioning (RIPC) stimulus: Identification by a SELDI-TOF-MS approach. PLoS ONE. https://doi.org/10.1371/journal.pone.0085669
Hibert P, Prunier-Mirebeau D, Beseme O et al (2013) Apolipoprotein A-I Is a Potential Mediator of Remote Ischemic Preconditioning. PLoS ONE. https://doi.org/10.1371/journal.pone.0077211
Zhuang X, Xiang X, Grizzle W et al (2011) Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. https://doi.org/10.1038/mt.2011.164
Ferreira R, Santos T, Amar A et al (2014) Argonaute-2 promotes miR-18a entry in human brain endothelial cells. J Am Heart Assoc. https://doi.org/10.1161/JAHA.114.000968
Ueno K, Samura M, Nakamura T et al (2016) Increased plasma VEGF levels following ischemic preconditioning are associated with downregulation of miRNA-762 and miR-3072-5p. Sci Rep. https://doi.org/10.1038/srep36758
Duan X, Ji B, Wang X et al (2012) Expression of MicroRNA-1 and MicroRNA-21 in different protocols of ischemic conditioning in an isolated rat heart model. Cardiol. https://doi.org/10.1159/000338149
Crimi G, Pica S, Raineri C et al (2013) Remote Ischemic Post-Conditioning of the Lower Limb During Primary Percutaneous Coronary Intervention Safely Reduces Enzymatic Infarct Size in Anterior Myocardial Infarction. JACC Cardiovasc Interv 6:1055–1063. https://doi.org/10.1016/j.jcin.2013.05.011
Munk K, Andersen NH, Schmidt MR et al (2010) Remote Ischemic Conditioning in Patients With Myocardial Infarction Treated With Primary Angioplasty: Impact on Left Ventricular Function Assessed by Comprehensive Echocardiography and Gated Single-Photon Emission CT. Circ Cardiovasc Imaging 3:656–662. https://doi.org/10.1161/CIRCIMAGING.110.957340
Prunier F, Angoulvant D, Saint Etienne C et al (2014) The RIPOST-MI study, assessing remote ischemic perconditioning alone or in combination with local ischemic postconditioning in ST-segment elevation myocardial infarction. Basic Res Cardiol 109:400. https://doi.org/10.1007/s00395-013-0400-y
Rentoukas I, Giannopoulos G, Kaoukis A et al (2010) Cardioprotective Role of Remote Ischemic Periconditioning in Primary Percutaneous Coronary Intervention. JACC Cardiovasc Interv 3:49–55. https://doi.org/10.1016/j.jcin.2009.10.015
White SK, Frohlich GM, Sado DM et al (2015) Remote Ischemic Conditioning Reduces Myocardial Infarct Size and Edema in Patients With ST-Segment Elevation Myocardial Infarction. JACC Cardiovasc Interv 8:178–188. https://doi.org/10.1016/j.jcin.2014.05.015
Chen L, Zhou Q, Jin H et al (2018) Effects of Remote Ischaemic Conditioning on Heart Rate Variability and Cardiac Function in Patients With Mild Ischaemic Heart Failure. Hear Lung Circ 27:477–483. https://doi.org/10.1016/j.hlc.2017.03.164
Hu Q, Luo W, Huang L et al (2016) Multiorgan protection of remote ischemic perconditioning in valve replacement surgery. J Surg Res 200:13–20. https://doi.org/10.1016/j.jss.2015.06.053
Surkar SM, Bland MD, Mattlage AE et al (2020) Effects of remote limb ischemic conditioning on muscle strength in healthy young adults: A randomized controlled trial. PLoS ONE 15:e0227263. https://doi.org/10.1371/journal.pone.0227263
Blauenfeldt RA, Hjort N, Gude MF et al (2020) A multicentre, randomised, sham-controlled trial on REmote iSchemic conditioning In patients with acute STroke (RESIST) – Rationale and study design. Eur Stroke J 5:94–101. https://doi.org/10.1177/2396987319884408
England TJ, Hedstrom A, O’Sullivan S et al (2017) RECAST (Remote Ischemic Conditioning After Stroke Trial). Stroke 48:1412–1415. https://doi.org/10.1161/STROKEAHA.116.016429
England TJ, Hedstrom A, O’Sullivan SE et al (2019) Remote Ischemic Conditioning After Stroke Trial 2: A Phase IIb Randomized Controlled Trial in Hyperacute Stroke. J Am Heart Assoc 8.https://doi.org/10.1161/JAHA.119.013572
England T (2021) Remote Ischaemic Conditioning After Stroke 3 (RECAST-3): A multicentre randomised controlled trial. https://fundingawards.nihr.ac.uk/award/NIHR128240
Hougaard KD, Hjort N, Zeidler D et al (2013) Remote ischemic perconditioning in thrombolysed stroke patients: Randomized study of activating endogenous neuroprotection - design and MRI measurements. Int J Stroke 8:141–146. https://doi.org/10.1111/j.1747-4949.2012.00786.x
Hougaard KD, Hjort N, Zeidler D et al (2014) Remote ischemic perconditioning as an adjunct therapy to thrombolysis in patients with acute ischemic stroke: A randomized trial. Stroke 45:159–167. https://doi.org/10.1161/STROKEAHA.113.001346
Pico F, Lapergue B, Ferrigno M et al (2020) Effect of In-Hospital Remote Ischemic Perconditioning on Brain Infarction Growth and Clinical Outcomes in Patients With Acute Ischemic Stroke. JAMA Neurol 77:725. https://doi.org/10.1001/jamaneurol.2020.0326
Pico F, Rosso C, Meseguer E et al (2016) A multicenter, randomized trial on neuroprotection with remote ischemic per-conditioning during acute ischemic stroke: the REmote iSchemic Conditioning in acUtE BRAin INfarction study protocol. Int J Stroke 11:938–943. https://doi.org/10.1177/1747493016660098
Landman T, Schoon Y, Warlé M et al (2019) The effect of repeated remote ischemic postconditioning on infarct size in patients with an ischemic stroke (REPOST): study protocol for a randomized clinical trial. Trials 20:167. https://doi.org/10.1186/s13063-019-3264-0
Liu S-M, Zhao W-L, Song H-Q et al (2018) Rationale and Study Design for a Single-Arm Phase IIa Study Investigating Feasibility of Preventing Ischemic Cerebrovascular Events in High-Risk Patients with Acute Non-disabling Ischemic Cerebrovascular Events Using Remote Ischemic Conditioning. Chin Med J (Engl) 131:347–351. https://doi.org/10.4103/0366-6999.223849
Zhao W, Che R, Li S et al (2018) Remote ischemic conditioning for acute stroke patients treated with thrombectomy. Ann Clin Transl Neurol 5:850–856. https://doi.org/10.1002/acn3.588
He Y, Guo Z, Qin C et al (2020) Remote ischemic conditioning combined with intravenous thrombolysis for acute ischemic stroke. Ann Clin Transl Neurol 7:972–979. https://doi.org/10.1002/acn3.51063
Meng R, Asmaro K, Meng L et al (2012) Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology 79:1853–1861. https://doi.org/10.1212/WNL.0b013e318271f76a
Meng R, Ding Y, Asmaro K et al (2015) Ischemic Conditioning Is Safe and Effective for Octo- and Nonagenarians in Stroke Prevention and Treatment. Neurotherapeutics 12:667–677. https://doi.org/10.1007/s13311-015-0358-6
Li S, Ma C, Shao G et al (2015) Safety and Feasibility of Remote Limb Ischemic Preconditioning in Patients with Unilateral Middle Cerebral Artery Stenosis and Healthy Volunteers. Cell Transplant 24:1901–1911. https://doi.org/10.3727/096368914X683520
Hu S, Dong H, Li Y et al (2010) Effects of Remote Ischemic Preconditioning on Biochemical Markers and Neurologic Outcomes in Patients Undergoing Elective Cervical Decompression Surgery. J Neurosurg Anesthesiol 22:46–52. https://doi.org/10.1097/ANA.0b013e3181c572bd
Gonzalez NR, Hamilton R, Bilgin-Freiert A et al (2013) Cerebral Hemodynamic and Metabolic Effects of Remote Ischemic Preconditioning in Patients with Subarachnoid Hemorrhage. Cerebral Vasospasm: Neurovascular Events After Subarachnoid Hemorrhage. Springer Vienna, Vienna, pp 193–198
Gonzalez NR, Connolly M, Dusick JR et al (2014) Phase I Clinical Trial for the Feasibility and Safety of Remote Ischemic Conditioning for Aneurysmal Subarachnoid Hemorrhage. Neurosurgery 75:590–598. https://doi.org/10.1227/NEU.0000000000000514
Koch S, Katsnelson M, Dong C, Perez-Pinzon M (2011) Remote Ischemic Limb Preconditioning After Subarachnoid Hemorrhage. Stroke 42:1387–1391. https://doi.org/10.1161/STROKEAHA.110.605840
Zhao W, Jiang F, Li S et al (2020) Remote Ischemic Conditioning for Intracerebral Hemorrhage (RICH-1): Rationale and Study Protocol for a Pilot Open-Label Randomized Controlled Trial. Front Neurol 11.https://doi.org/10.3389/fneur.2020.00313
Wang Y, Meng R, Song H et al (2017) Remote Ischemic Conditioning May Improve Outcomes of Patients With Cerebral Small-Vessel Disease. Stroke 48:3064–3072. https://doi.org/10.1161/STROKEAHA.117.017691
Liao Z, Bu Y, Li M et al (2019) Remote ischemic conditioning improves cognition in patients with subcortical ischemic vascular dementia. BMC Neurol 19:206. https://doi.org/10.1186/s12883-019-1435-y
Zhao W, Meng R, Ma C et al (2017) Safety and Efficacy of Remote Ischemic Preconditioning in Patients With Severe Carotid Artery Stenosis Before Carotid Artery Stenting. Circulation 135:1325–1335. https://doi.org/10.1161/CIRCULATIONAHA.116.024807
Durand MJ, Boerger TF, Nguyen JN et al (2019) Two weeks of ischemic conditioning improves walking speed and reduces neuromuscular fatigability in chronic stroke survivors. J Appl Physiol 126:755–763. https://doi.org/10.1152/japplphysiol.00772.2018
Gasparovic H, Kopjar T, Rados M et al (2019) Impact of remote ischemic preconditioning preceding coronary artery bypass grafting on inducing neuroprotection. J Thorac Cardiovasc Surg 157:1466-1476.e3. https://doi.org/10.1016/j.jtcvs.2018.08.116
Cherry-Allen KM, Gidday JM, Lee J-M et al (2017) Remote Limb Ischemic Conditioning at Two Cuff Inflation Pressures Yields Learning Enhancements in Healthy Adults. J Mot Behav 49:337–348. https://doi.org/10.1080/00222895.2016.1204268
Donato M, Buchholz B, Rodríguez M et al (2013) Role of the parasympathetic nervous system in cardioprotection by remote hindlimb ischaemic preconditioning. Exp Physiol 98:425–434. https://doi.org/10.1113/expphysiol.2012.066217
Mei B, Li W, Cheng X et al (2017) Activating mu-opioid receptors in the spinal cord mediates the cardioprotective effect of remote preconditioning of trauma. Cardiol J 24:314–323. https://doi.org/10.5603/CJ.a2016.0062
Basalay MV, Mastitskaya S, Mrochek A et al (2016) Glucagon-like peptide-1 (GLP-1) mediates cardioprotection by remote ischaemic conditioning. Cardiovasc Res 112:669–676. https://doi.org/10.1093/cvr/cvw216
Lambert EA, Thomas CJ, Hemmes R et al (2016) Sympathetic nervous response to ischemia-reperfusion injury in humans is altered with remote ischemic preconditioning. Am J Physiol Circ Physiol 311:H364–H370. https://doi.org/10.1152/ajpheart.00369.2016
Noronha Osório D, Viana-Soares R, Marto JP et al (2019) Autonomic nervous system response to remote ischemic conditioning: Heart rate variability assessment. BMC Cardiovasc Disord 19.https://doi.org/10.1186/s12872-019-1181-5
Qu Y, Liu J, Guo Z-N et al (2020) The Impact of Remote Ischaemic Conditioning on Beat-to-Beat Heart Rate Variability Circadian Rhythm in Healthy Adults. Hear Lung Circ. https://doi.org/10.1016/j.hlc.2020.08.017
Khaliulin I, Fleishman AN, Shumeiko NI et al (2019) Neuro-autonomic changes induced by remote ischemic preconditioning (RIPC) in healthy young adults: Implications for stress. Neurobiol Stress 11:100189. https://doi.org/10.1016/j.ynstr.2019.100189
Dezfulian C, Taft M, Corey C et al (2017) Biochemical signaling by remote ischemic conditioning of the arm versus thigh: Is one raise of the cuff enough? Redox Biol 12:491–498. https://doi.org/10.1016/j.redox.2017.03.010
Yang J, Zhang X, Chen X et al (2017) Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol Ther - Nucleic Acids 7:278–287. https://doi.org/10.1016/j.omtn.2017.04.010
Konstantinov IE, Arab S, Kharbanda RK et al (2004) The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol Genomics 19:143–150. https://doi.org/10.1152/physiolgenomics.00046.2004
Hepponstall M, Ignjatovic V, Binos S et al (2012) Remote Ischemic Preconditioning (RIPC) Modifies Plasma Proteome in Humans. PLoS ONE 7:e48284. https://doi.org/10.1371/journal.pone.0048284
Pang T, Zhao Y, Zhang N-R et al (2013) Transient Limb Ischemia Alters Serum Protein Expression in Healthy Volunteers: Complement C3 and Vitronectin May Be Involved in Organ Protection Induced by Remote Ischemic Preconditioning. Oxid Med Cell Longev 2013:1–9. https://doi.org/10.1155/2013/859056
Hepponstall M, Ignjatovic V, Binos S et al (2015) Remote Ischemic Preconditioning (RIPC) Modifies the Plasma Proteome in Children Undergoing Repair of Tetralogy of Fallot: A Randomized Controlled Trial. PLoS ONE 10:e0122778. https://doi.org/10.1371/journal.pone.0122778
Helgeland E, Breivik LE, Vaudel M et al (2014) Exploring the Human Plasma Proteome for Humoral Mediators of Remote Ischemic Preconditioning - A Word of Caution. PLoS ONE 9:e109279. https://doi.org/10.1371/journal.pone.0109279
Joseph B, Pandit V, Zangbar B et al (2015) Secondary brain injury in trauma patients. J Trauma Acute Care Surg 78:698–705. https://doi.org/10.1097/TA.0000000000000584
Wang H, He Z, Zhang Y, Zhang J (2018) 1H NMR metabolic signature of cerebrospinal fluid following repetitive lower-limb remote ischemia preconditioning. Neurochem Int. https://doi.org/10.1016/j.neuint.2018.02.009
Loukogeorgakis SP, Williams R, Panagiotidou AT et al (2007) Transient Limb Ischemia Induces Remote Preconditioning and Remote Postconditioning in Humans by a K ATP Channel-Dependent Mechanism. Circulation 116:1386–1395. https://doi.org/10.1161/CIRCULATIONAHA.106.653782
Loukogeorgakis SP, Panagiotidou AT, Broadhead MW et al (2005) Remote Ischemic Preconditioning Provides Early and Late Protection Against Endothelial Ischemia-Reperfusion Injury in Humans. J Am Coll Cardiol 46:450–456. https://doi.org/10.1016/j.jacc.2005.04.044
Kraemer R, Lorenzen J, Kabbani M et al (2011) Acute effects of remote ischemic preconditioning on cutaneous microcirculation - A controlled prospective cohort study. BMC Surg. https://doi.org/10.1186/1471-2482-11-32
Jones H, Hopkins N, Bailey TG et al (2014) Seven-Day Remote Ischemic Preconditioning Improves Local and Systemic Endothelial Function and Microcirculation in Healthy Humans. Am J Hypertens 27:918–925. https://doi.org/10.1093/ajh/hpu004
Fukuda S, Kono Y, Hanatani A et al (2014) Remote ischemic conditioning improves coronary microcirculation in healthy subjects and patients with heart failure. Drug Des Devel Ther 1175.https://doi.org/10.2147/DDDT.S68715
Acknowledgements
The authors would like to acknowledge Dr Cláudia Queiroga for her work, ideas, and fruitful discussions in the beginning of this project, and the funding agency that supported the work “Fundação para a Ciência e Tecnologia” (FCT) with 4 projects: Applied Molecular Biosciences Unit-UCIBIO (UID/Multi/04378/2019); iNOVA4Health, Programme in Translational Medicine (UID/Multi/04462/2013); LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy; and PTDC/MEC-NEU/28750/2017.
Funding
This work was supported by funding from the Portuguese Fundação para a Ciência e Tecnologia (FCT) in the scope of the project UIDP/04378/2020 and UIDB/04378/2020 of the Research Unit on Applied Molecular Biosciences, UCIBIO; the project LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy, i4HB; the project iNOVA4Health, Programme in Translational Medicine (UID/Multi/04462/2013); and the grant PTDC/MEC-NEU/28750/2017.
Author information
Authors and Affiliations
Contributions
All authors participate in the bibliographic research, writing, and revising of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mollet, I., Marto, J.P., Mendonça, M. et al. Remote but not Distant: a Review on Experimental Models and Clinical Trials in Remote Ischemic Conditioning as Potential Therapy in Ischemic Stroke. Mol Neurobiol 59, 294–325 (2022). https://doi.org/10.1007/s12035-021-02585-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02585-6