
Abstract
Recent studies have identified roles for complement in synaptic pruning, both physiological during development and pathological in Alzheimer’s disease (AD). These reports suggest that C1q initiates complement activation on synapses and C3 fragments then tag them for removal by microglia. There is an urgent need to characterise these processes in rodent AD models; this requires the development of reagents and methods for detection and quantification of rodent C1q in fluids and pathological tissues. These will enable better evaluation of the role of C1q in disease and its value as disease biomarker. We describe the generation in C1q-deficient mice of novel monoclonal antibodies against mouse and rat C1q that enabled development of a sensitive, specific, and quantitative ELISA for mouse and rat C1q capable of measuring C1q in biological fluids and tissue extracts. Serum C1q levels were measured in wild-type (WT), C1q knockout (KO), C3 KO, C7 KO, Crry KO, and 3xTg and APPNL-G-F AD model mice through ageing. C1q levels significantly decreased in WT, APPNL-G-F, and C7 KO mice with ageing. C1q levels were reduced in APPNL-G-F compared to WT at all ages and in 3xTg at 12 months; C3 KO and C7 KO, but not Crry KO mice, also demonstrated significantly lower C1q levels compared to matched WT. In brain homogenates, C1q levels increased with age in both WT and APPNL-G-F mice. This robust and adaptable assay for quantification of mouse and rat C1q provides a vital tool for investigating the expression of C1q in rodent models of AD and other complement-driven pathologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a severe neurodegenerative disorder characterised by amyloid-ß (Aβ) plaques, neurofibrillary tangles of hyperphosphorylated tau, neuroinflammation, synaptic loss, and cognitive decline. Multiple lines of evidence, genetic and experimental, have implicated the complement system in the aetiology of AD. Genome-wide association studies (GWAS) have repeatedly associated single nucleotide polymorphisms (SNPs) in genes encoding complement regulators clusterin (CLU) and complement receptor 1 (CR1) with risk of late-onset AD [1,2,3,4]. Biomarker studies have identified altered levels of complement proteins and activation products in plasma and/or cerebrospinal fluid (CSF) that distinguish AD patients from controls and predict progression from mild cognitive impairment (MCI) to AD [5,6,7,8,9]. Microarray studies identified upregulation of numerous complement genes in AD patients versus age-matched controls [10]. Immunohistochemical analysis of post-mortem AD brains identified deposition of complement proteins and activation products, including C1q, C3, and C4, in and around plaques and tangles [11,12,13]. Furthermore, deletion of C3 or suppression of C3 activation in multiple AD mouse models increased amyloid burden, indicating that complement is involved in amyloid clearance [14,15,16].
The complement system is the innate immune system’s primary defence against invading pathogens. In excess of 30 soluble and membrane-bound proteins, three distinct pathways coordinate to promote inflammation and destruction of pathogens. The classical pathway of complement activation is initiated when the circulating C1 complex binds to target surfaces via its C1q subunit, a heterohexameric defence collagen that recognises immunoglobulin Fc domains [17]. The other C1 components C1r and C1s associate with the collagen stalks of C1q, the latter catalysing proteolysis of C4 and C2 to form the classical pathway C3 convertase [18]; this then cleaves the central complement component C3 into C3a and C3b with multiple downstream consequences including opsonisation for phagocytosis, generation of the inflammatory mediator C5a, and formation of the membrane attack complex (MAC) [19,20,21].
Previous studies have highlighted a critical role for the classical complement cascade in physiological synaptic pruning during development. C1q and C3 fragments (C3b/iC3b) localise to and tag specific synapses for removal in the developing rodent visual system and substantial synaptic pruning defects were demonstrated in C1q knockout (KO), C3 KO, and C4 KO mice [22,23,24]. It is established that microglia are responsible for phagocytosis of synapses during developmental pruning, a process that involves interaction of complement receptor 3 (CR3; CD11b/CD18) expressed on the surface of microglia and its ligand iC3b on target synapses [25, 26].
Synaptic elimination also occurs pathologically, and is an early event in the pathogenesis of AD, occurring up to 20 years prior to the onset of cognitive dysfunction [27]. Studies in AD rodent models have implicated the classical pathway in pathological synaptic loss; C1q is deposited on synapses destined for elimination, and synaptic elimination is reduced or abolished by either C1q deletion or blocking of C1q with inhibitory antibodies in both amyloid and tau models [28, 29]. Microglia from CR3 KO mice displayed impaired synaptic engulfment triggered by local administration of oligomeric Aß [28]. Pathological synaptic pruning driven by complement is also reported in multiple sclerosis (MS) [30, 31] and in schizophrenia, where dysregulated C4 expression contributes to abnormal synaptic pruning [24, 32, 33].
While the precise mechanism of synaptic loss in AD remains conjecture, the prevailing hypothesis is that the developmental synaptic pruning process is reactivated and becomes dysregulated, resulting in inappropriate classical pathway activation and microglial phagocytosis of complement-opsonised synapses; hence, there is a pressing requirement for reliable and reproducible methods for measuring expression of C1q and other classical pathway components in fluids and pathological tissues in models and man. We have generated a panel of monoclonal antibodies against rodent C1q and developed an enzyme-linked immunosorbent assay (ELISA) that allows specific and quantitative measurement of mouse and rat C1q protein levels in serum and in brain extracts. We demonstrate that the ELISA identifies age-related changes in C1q concentration and differences in C1q levels between wild-type (WT) mice, complement KO mice and the APPNL-G-F and 3xTg mouse models of AD.
Materials and Methods
Reagents and chemicals were purchased from Thermo Fisher Scientific (Paisley, UK) unless stated otherwise. Composition of phosphate-buffered saline (PBS) is 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4. All dialysis was performed overnight at 4 °C with 12–14-kDa cut-off dialysis tubing (Medicell, London, UK). All cells were cultured at 37 °C in 5% CO2. All protein stain and Western blot images were captured using the G:BOX Chemi XX6 (Syngene, Cambridge, UK).
Animals
All procedures complied with UK Animals Scientific Procedures Act 1986 and local regulations. All animals were group-housed in environmentally enriched cages, under standard pathogen-free conditions, with a 12-h light/dark cycle, and access to food and water ad libitum. C57BL/6 (WT; Harlan, Bicester, UK), C1q KO [34], C3 KO [35], C7 KO (Jackson ImmunoResearch), Crry KO [36], APPNL-G-F [37], and 3xTg [38] mice have been described elsewhere. With the exception of the 3xTg line which is on a mixed background, all of the mouse lines were on the C57BL/6 background and all had been back-crossed onto in-house WT mice; all were maintained in the same room in the facility at the same level of containment.
All mice were humanely euthanised by increasing CO2 concentration, death was confirmed by palpation, and whole blood was collected by transcardial puncture. WT and APPNL-G-F mice were perfused with cold PBS; brains were removed, and snap-frozen. Blood samples were taken from 3-month-old male Brown Norway rats (Envigo, Bicester, UK) by tail tipping under anaesthesia. Collected mouse and rat blood was allowed to clot at room temperature for 10 min, placed on ice for 1 h, then centrifuged; serum was removed and immediately stored in aliquots at −80 °C.
Mouse brain homogenates were obtained using standard methods. In brief, frozen mouse brains (one hemisphere) were homogenised in a Dounce glass homogeniser in 1 ml of 5 mM KCl, 1 mM MgCl2, 25 mM HEPES, 120 mM NaCl, and 2 mM CaCl2 (pH 7.5), supplemented with complete mini EDTA-free protease inhibitors (Roche, Welwyn Garden City, UK), and phosphatase inhibitor cocktail V (Millipore, Watford, UK). Homogenates were passed through 80-μm nylon filters (Millipore). Samples were lysed on ice in RIPA buffer (Sigma-Aldrich, Gillingham, UK) containing protease inhibitors. The resultant lysate was centrifuged at 17,000×g for 10 min at 4 °C and supernatant collected. Total protein concentration was measured using the Pierce BCA protein assay kit.
Isolation of Mouse C1q from Mouse Serum
C1q was isolated from mouse serum via a three-step protocol on an ÄKTA pure chromatography system (GE Healthcare, Amersham, UK). Elements of the protocol were derived from published methods [39, 40]. Sterile mouse serum (TCS Biosciences, Claydon, UK) was 0.22-μm-filtered and diluted 1:1 in binding buffer (20 mM Tris, 120 mM NaCl, 20 mM EDTA, pH 7.0) to dissociate C1 complexes. Human IgG (10 mg) was immobilised on a 5-ml HiTrap NHS-Activated HP column (GE Healthcare; manufacturer’s protocol); rabbit anti-human IgG antiserum was passed over this column to saturate binding sites. The column was washed and equilibrated with 5 column volumes (CV) of binding buffer. Filtered mouse serum was applied to the column, the column was washed, and bound C1q was eluted with 3 CV of elution buffer (50 mM Tris, 1 M NaCl, 20 mM EDTA, pH 10). In order to remove contaminating mouse immunoglobulins, the eluate was applied to a 5-ml HiTrap Protein G HP column (GE Healthcare; manufacturer’s instructions), the flow-through collected, dialysed into cation exchange binding buffer (20 mM HEPES, 60 mM NaCl, 10 mM EDTA, pH 7.8), and applied to a 5-ml HiTrap SP HP column (GE Healthcare) equilibrated with 5 CV of cation exchange binding buffer. Proteins were eluted with an increasing salt gradient in cation exchange elution buffer (to 700 mM NaCl) over 20 CV. Fractions containing eluted protein were dialysed into 50 mM NaH2PO4 and 100 mM NaCl (pH 7.4), and concentrated to 0.5 ml using a 30-kDa Vivaspin 6 centrifugal concentrator (Sartorius, Epsom, UK); concentration was measured using Bradford reagent according to the manufacturer’s protocol (Sigma-Aldrich) and pure C1q-containing fractions identified by SDS-PAGE on in-house 12.5% tris-glycine gels in the presence and absence of β-mercaptoethanol and stained with Coomassie (0.25% (w/v) Coomassie Brilliant Blue R-250, 40% (v/v) methanol, 10% (v/v) acetic acid).
The novel anti-C1q monoclonal antibodies (mAb) were also used to affinity-purify mouse C1q in a single step. The mAb (3 mg) was conjugated to a 1-ml HiTrap NHS-activated HP column (GE Healthcare; manufacturer’s protocol), pooled mouse serum was 0.22-μm-filtered and diluted 1:1 in binding buffer (10 mM Tris, 150 mM NaCl, 20 mM EDTA, pH 7.4). The mAb column was equilibrated with 5 CV of binding buffer. Filtered mouse serum was applied to the column, the column was washed in binding buffer, and bound C1q was eluted with 3 CV of elution buffer (100 mM glycine, 20 mM EDTA, pH 3). C1q-containing fractions were identified, dialysed, concentrated, quantified, and examined via SDS-PAGE as described above.
Immunisation, Generation, and Isolation of Hybridoma
C1q KO mice (8–16 weeks of age) were immunised subcutaneously with 50-μg mouse C1q in complete Freund’s adjuvant, boosted after 4 weeks with 50-μg mouse C1q in incomplete Freund’s adjuvant and then repeat boosted 1 week later. A week after the second boost, mice were tail-bled, serum-harvested, and screened for immunoreactivity against mouse C1q via direct ELISA as described below. Mice with the strongest immune response then received an intra-peritoneal boost of 50 μg C1q in PBS and were humanely sacrificed 2 days later, and spleens were removed. Splenocytes were extracted and fused with SP2/0-Ag14 mouse myeloma cells (European Collection of Animal Cell Cultures, Salisbury, UK) using an established protocol [41].
Cells from the fusion were grown in RPMI 1640 medium containing 2% penicillin/streptomycin, 2 mM glutamine, 1 mM sodium pyruvate, and 15% foetal bovine serum (FBS), supplemented with HAT (Gibco, Paisley, UK) to select hybridomas, in 96-well plates (10 plates per fusion) with C1q KO mouse peritoneal macrophages as feeder cells. After 14 days, medium was harvested from each well and screened for C1q immunoreactivity via direct ELISA as described below. Positive clones were sub-cloned three times by limiting dilution to ensure monoclonality, and ELISA was used for screening at each cloning to verify C1q specificity. Positive monoclonal hybridomas were then isotyped using an IsoStrip™ Mouse Monoclonal Antibody Isotyping Kit (Roche) and cultured in CELLine™ 1000 bioreactor flasks (VWR, Lutterworth, UK) with RPMI 1640 medium containing 2% penicillin/streptomycin, 2 mM glutamine, 1 mM sodium pyruvate, and 10% ultra-low IgG FBS (Gibco). Bioreactor flasks were harvested every 7–10 days. IgG mAb were purified using a 5-ml HiTrap Protein G HP column (GE Healthcare; manufacturer’s instructions); IgM mAb were purified using a 1-ml HiTrap Protein L column (GE Healthcare; manufacturer’s instructions). All mAb were dialysed into PBS. Selected mAb were conjugated to biotin using the EZ-Link™ Sulfo-NHS-LC-Biotin reagent (Sigma-Aldrich; manufacturer’s protocols).
Screening for Antibodies Specific to Mouse C1q
Pure mouse C1q was diluted to 0.5 μg/ml in carbonate buffer (100 mM NaHCO3, 100 mM Na2CO3, pH 9.6), dispensed (50 μl/well) into Nunc MaxiSorp™ flat-bottom 96-well plates (Invitrogen, Paisley, UK), and incubated overnight at 4 °C. Wells were blocked with 2% (w/v) bovine serum albumin (BSA) in PBS-T (100 μl/well) for 1 h at 37 °C, washed twice with PBS-T, and either serum from immunised mice (diluted 1:100 in 0.2% BSA-PBS-T) or neat hybridoma supernatant (50 μl/well) added and incubated for 90 min at 37 °C. Wells were washed again with PBS-T and then incubated with a 1:1000 dilution (50 μl/well) of peroxidase-conjugated goat anti-mouse IgG (H + L) secondary antibody (Jackson ImmunoResearch) for 30 min at 37 °C. Following two final washes, plates were developed using O-phenylenediamine dihydrochloride (SIGMAFAST™ OPD, Sigma-Aldrich). Absorbance was measured at 492 nm with a FLUOstar™ Omega Microplate Reader (BMG LABTECH).
ELISA Characterisation of Novel mAb
Anti-C1q mAb were diluted to 5 μg/ml in carbonate buffer and coated onto Nunc MaxiSorp™ flat-bottom 96-well plates (50 μl/well) for 1 h at 37 °C. Blocking and washing was performed as described above. Mouse (WT and C1q KO), rat, and human sera were loaded in a dilution series from 1 to 0.001% and incubated at 37 °C for 2 h. Bound C1q was detected using rabbit anti-human C1q polyclonal antibody (2 μg/ml; in-house) and peroxidase-conjugated donkey anti-rabbit IgG secondary antibody (1:5000; Jackson ImmunoResearch, West Grove, PA, USA). OPD development was performed as detailed above.
Western Blotting
Pure mouse C1q (1 μg) was run on in-house 12.5% tris-glycine gels in the presence and absence of β-mercaptoethanol, then transferred onto nitrocellulose 0.45-μm membrane (GE Healthcare) using a Mini Blot Module (Invitrogen). Membranes were blocked for 1 h at room temperature with 5% (w/v) BSA in PBS-T, incubated with mouse anti-C1q antiserum (1:500) or anti-C1q mAb (2 μg/ml) overnight at 4 °C and developed using peroxidase-conjugated donkey anti-mouse IgG (H + L) secondary antibody (1:5000; Jackson ImmunoResearch) followed by ECL Western blotting detection reagent (GE Healthcare).
To validate the ELISA, pure mouse C1q (200 ng) and sera (diluted 1:10 in PBS, 10 μl loaded) from C1q KO, WT, and APPNL-G-F mice (3 and 12 months) were analysed under reducing conditions as described above. Membranes were stained with Ponceau S immediately after transfer according to the manufacturer’s instructions (Sigma-Aldrich). Following blocking, membranes were incubated with rabbit anti-C1q mAb (2 μg/ml; Abcam, Cambridge, UK) overnight at 4 °C and developed using peroxidase-conjugated donkey anti-rabbit IgG (H + L) secondary antibody (1:5000; Jackson ImmunoResearch). Densitometry was performed using GeneTools (Syngene).
Haemolytic Assays
Sheep erythrocytes were isolated from blood (TCS Biosciences) and antibody sensitized as described [42]. For assays involving mouse serum, sensitised sheep erythrocytes were additionally sensitised with 20 μg/ml mouse anti-rabbit IgG (Invitrogen). Characterisation of novel mAb in human, rat, and mouse haemolytic assays was performed according to published methods [43]. To assess the function of immunoaffinity-purified mouse C1q, a titration of purified protein (25 > 0 μg/ml) was added to 25% C1q KO mouse serum and then tested in haemolysis assays as above.
Development of a Sandwich ELISA for Quantification of Mouse C1q
The anti-C1q mAb selected for capture was diluted to 5 μg/ml in carbonate buffer and coated onto Nunc MaxiSorp™ flat-bottom 96-well plates (50 μl/well) for 1 h at 37 °C. Blocking and washing were performed as described above. Pure C1q standards in a dilution series from 2 μg/ml > 2 ng/ml were loaded in duplicate to generate a standard curve. To measure levels in serum (mouse and rat) and mouse brain homogenate, samples were analysed in triplicate (50 μl/well) at dilutions of 1:800 for serum and 0.5 mg/ml total protein for brain homogenates, respectively. Inter-assay controls were included on all plates. All samples and standards were incubated for 2 h at 37 °C then washed twice with PBS-T. The anti-C1q mAb selected for detection (biotinylated) was diluted in 0.2% (w/v) BSA-PBS-T (2 μg/ml, 50 μl/well), added, and incubated for 1 h at 37 °C. After washing, wells were incubated (1 h, 37 °C) with streptavidin-HRP (1:200; R&D Systems, Abingdon, UK) diluted in 0.2% (w/v) BSA-PBS-T (50 μl/well). After two final washes, colour was developed using OPD and absorbance measured as above. C1q concentrations in serum and brain samples were interpolated from the standard curve using Prism 5 (GraphPad, La Jolla, CA, USA).
To test assay performance, ten WT mouse serum samples (age 3 months, male n = 6, female n = 4) were analysed using the above protocol multiple times on different days and either with or without addition of 10 mM EDTA to dissociate C1. To test whether the assay could detect C1q in CSF, human CSF (hCSF) was supplemented with a known quantity of pure mouse C1q and then measured using the assay. Spike recovery was performed by adding a known amount of pure mouse C1q to WT mouse serum prior to dilution and then measuring levels in the base and supplemented serum using the assay as above. Spike recovery was calculated as a percentage of calculated recovery relative to expected recovery. Inter- and intra-assay coefficients of variability (%CV) were derived from the multiple measurements by standard methods [44].
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 5. Data points that fell 1.5× the interquartile range above or below the mean were considered outliers and excluded. All data was checked for normality with the Kolmogorov-Smirnov test. The paired t-test was used to test for differences in the same serum samples in the presence or absence of EDTA. The unpaired t-test was used to test for differences in serum C1q levels between age groups of the same complement KO genotype, and for differences in brain C1q levels between age groups of the WT and APPNL-G-F genotypes. One-way analysis of variance (ANOVA) was utilised to test for differences between age groups of the WT and APPNL-G-F genotypes, two-way ANOVA was used to test for differences between genotypes at each age. Tukey’s post hoc test and the Bonferroni post hoc test were co-implemented with one-way ANOVA and two-way ANOVA, respectively. P < 0.05 was considered significant. C1q levels are described in the results as mean ± standard deviation.
Results
Purification of Mouse C1q and Generation of mAb
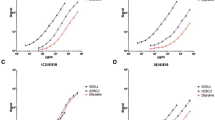
C1q was purified from mouse serum using IgG affinity chromatography followed by cation exchange (Fig. 1A); C1q eluted from cation exchange as a double peak, the first peak containing predominantly aggregated C1q (Fig. 1A, B). The second peak was aggregate-free and pure on Coomassie-stained SDS-PAGE (Fig. 1B). Two fusions were performed; from 2880 wells screened, six hybridoma clones producing mouse C1q-specific antibodies were identified, five of which were IgM mAb; two of the clones, 9H10 and 2F6, gave consistently higher ELISA signal during screening and were taken forward for full characterisation (Table 1). Species cross-reactivity of 9H10 mAb (IgG2b isotype) and 2F6 mAb (IgM isotype) was evaluated via sandwich ELISA using a polyclonal anti-human C1q for detection. 9H10 cross-reacted with mouse, rat, and human C1q (Fig. 1C); 2F6 was specific for mouse and rat C1q only and gave no signal with human C1q (Fig. 1D). C1 inhibitory activity of 9H10 and 2F6 was assessed in haemolysis assays; 9H10 did not inhibit in any of the sera tested (Fig. 1E), while 2F6 only inhibited haemolysis in rat serum (Fig. 1F). In order to determine the C1q chain specificity of the mAbs, Western blotting was performed on purified mouse C1q. 9H10 predominantly recognised the A chain of C1q (~ 27 kDa) under reducing and non-reducing conditions; 2F6 was very weak in westerns, suggesting that its epitope was conformation-dependent (Fig. 1G). Immunoaffinity chromatography on 9H10 isolated C1q from mouse serum in a single step (Fig. 1H); the purified C1q restored activity to C1q KO mouse serum, demonstrating that it was fully functional (Fig. 1I).

Isolation of pure mouse C1q and characterisation of novel monoclonal antibodies. (A) Representative ÄKTA chromatogram from the cation exchange purification of mouse C1q showing 2 peaks. (B) SDS-PAGE of cation exchange fractions (2.5 μg/lane) from peak 1 and peak 2; non-reduced (NR), or reduced with 5% β-mercaptoethanol (R), proteins were stained with Coomassie blue. Peak 1 contained aggregated mouse C1q that reduced to the individual C1q A, B, and C monomers at 31 kDa, 29 kDa, and 26 kDa; peak 2 contained pure mouse C1q with no aggregates that reduced to C1q monomers. (C) C1q sandwich ELISA using 9H10 as capture antibody and polyclonal anti-C1q as detect showing cross-reactivity with mouse, rat, and human C1q. (D) C1q sandwich ELISA using 2F6 as capture antibody and polyclonal anti-C1q as detect showing 2F6 cross-reactivity with mouse and rat C1q but not human C1q. (E, F) Classical pathway haemolytic assays showing that 9H10 had no inhibitory activity towards mouse, rat, or human C1q, while 2F6 inhibited rat C1q, but not mouse or human C1q. (G) Western blot of mouse C1q. Denatured pure C1q (1 μg/lane) under NR and R conditions and Western blotted with mouse anti-C1q antiserum (1:500), 9H10, and 2F6 (2 μg/ml). 9H10 bound the A chain in denatured C1q under both non-reducing and reducing conditions; staining with 2F6 was weak under both conditions. All membranes were exposed together, and for the same time, the images are unmodified from the original .tif files. (H) SDS-PAGE of mouse C1q isolated via IgG affinity followed by cation exchange (1) and 9H10 immunoaffinity purification (2) under both non-reducing and reducing conditions and stained with Coomassie blue. (I) Classical pathway haemolytic assay demonstrating that titration (from 25 μg/ml) of mouse C1q isolated via 9H10 immunoaffinity restored activity to C1q KO serum (25%); 116 ng/ml of mouse C1q was required to restore lysis to 50%
A quantitative sandwich ELISA was developed using 9H10 mAb as capture and biotinylated 2F6 mAb as detection. The sandwich ELISA detected C1q in both mouse and rat sera but C1q-deficient mouse serum gave no signal in the assay (Fig. 2A). The assay did not detect C1q in human serum, expected as the 2F6 mAb was not reactive against human C1q; substitution of 2F6 with a polyclonal anti-C1q antibody enabled quantification of human C1q (not shown). Using pure mouse C1q as standard, the assay had a detection limit of 2 ng/ml and a working range of 15 ng/ml to 250 ng/ml (Fig. 2B). Dissociation of the C1 complex with EDTA had no significant effect on detection of C1q (Fig. 2C). Spike recovery was 93% (Fig. 2D), which was within acceptable parameters [45]. As expected, human CSF gave no signal in the assay; however, pure mouse C1q spiked into human CSF at levels similar to those reported for C1q in human CSF (200–500 ng/ml) [46] was readily detected with recovery of 73% (Fig. 2E). Two mouse serum samples, one with high and one with low C1q levels, were measured in triplicate across 10 assay plates to calculate an inter-assay coefficient of variability (%CV): for the “high” sample, mean of means = 56.40 μg/ml, SD = 4.19 μg/ml, CV = 7.44%; for the “low” sample, mean of means = 34.71 μg/ml, SD = 4.67 μg/ml, CV = 13.36%. The overall inter-assay CV was 10.45%. Sample replicates for all cohorts were used to generate an intra-assay %CV as described [47]; the intra-assay CV was 6.14%.

Optimisation and quality testing of in-house quantitative sandwich ELISA. (A) C1q sandwich ELISA using 9H10 mAb as capture and biotinylated 2F6 mAb as detection. Standard curves were generated by titrating WT mouse, C1q KO mouse, rat, and human serum from 1% serum concentration. The assay detected C1q in mouse and rat but not human serum. (B) Pure C1q was used as standard in the assay; detection range was 10–500 ng/ml. (C) Measurement of the same WT mouse serum samples (3 months, male n = 6, female n = 4) in the presence or absence of 10 mM EDTA to dissociate the C1 complex. EDTA had no significant effect on measured C1q levels. (D) Spike recovery. Mouse C1q (50 μg/ml) was added into WT mouse serum samples (3 months, male, n = 5) and C1q levels measured. Average recovery was 93%. (E) C1q spiked into human CSF (hCSF). Mouse C1q (500 ng/ml) was spiked into hCSF samples (n = 3; hCSF/C1q) or the same volume of buffer (C1q); C1q levels were measured. Unspiked hCSF was used as a control. Average recovery was 73%
Quantification of Serum C1q in WT Mice, AD Mice, and Complement KO Mice and Rats
The concentration of C1q in serum samples from WT and APPNL-G-F mice aged 3, 6, 9, and 12 months was quantified using the sandwich ELISA (Table 2). In WT mice, serum C1q levels progressively decreased with age and were significantly decreased at 12 months compared to 3 months (Fig. 3A). APPNL-G-F mice showed a more pronounced decrease with age, significant at both 9 and 12 months relative to 3 months (Fig. 3B). These observations were replicated via Western blotting with a commercial mAb (Figs. S1A, B). Serum C1q levels were significantly decreased in APPNL-G-F mice compared to WT mice in all age groups (Fig. 3C); similar trends were observed at 4 and 12 months in the 3xTg AD mice compared to WT, reaching significance at 12 months (Fig. 3D). C1q levels in rat serum measured in the ELISA were 97.69 ± 14.31 μg/ml (n = 11; data not shown). In order to evaluate whether knocking out other complement components had any effect on serum C1q levels, C1q was measured in different complement KO mouse strains at 6 and 12 months of age (Table 2). As expected, C1q KO mouse sera gave no signal in the assay (Fig. 4A). In C3 KO mice, serum C1q levels were significantly lower compared to WT at 6 months but were not different between the two age points (Fig. 4B, E). Similarly, in C7 KO mice, C1q levels were significantly lower compared to WT at 6 and 12 months; however, in this mouse, strain levels were significantly lower at 12 months compared to 6 months (Fig. 4D, E). In Crry KO mice, C1q levels were similar to WT at 6 and 12 months and showed a small, non-significant fall at 12 months relative to 6 months (Fig. 4C, E). There were no significant differences between genders for C1q levels in any of the mouse lines (Table 2).

Measurement of C1q levels in WT and APPNL-G-F mouse serum by quantitative sandwich ELISA. (A) Serum C1q levels measured in WT mice at 3, 6, 9, and 12 months. C1q levels were significantly decreased at 12 months compared to 3 months (P < 0.05). (B) Serum C1q levels measured in APPNL-G-F mice at 3, 6, 9, and 12 months. Serum C1q levels were significantly reduced at 9 and 12 months compared to 3 months (P < 0.05). (C) Serum C1q levels were significantly lower in APPNL-G-F mice than WT mice at all ages examined (* = P < 0.05, ** = P < 0.01). (D) C1q was measured in serum from male 3xTg and WT mice at 3-4 months (3xTg, n = 4; WT, n = 6) and at 12 months (3xTg, n = 5; T, n = 5). C1q levels were lower in 3xTg mice compared to WT at each age, significantly at 12 months (3-4 months, p = 0.078; 12 months, p = 0.038; t test)

Measurement of C1q levels in C1q KO, C3 KO, Crry KO, and C7 KO mouse serum by quantitative sandwich ELISA. (A) Serum C1q levels were measured in C1q KO mice; no C1q was detected at 6 or 12 months. (B-C) Measurement of serum C1q levels in B C3 KO and C Crry KO mice at 6 and 12 months of age; no significant differences in serum C1q levels were observed between 6 and 12 months (P > 0.05). (D) Measurement of serum C1q levels in C7 KO mice; a significant decrease was identified at 12 months compared to 6 months of age (P < 0.01). (E) Serum C1q levels in C3 KO, C7 KO, and C1q KO were significantly reduced relative to WT and Crry KO at 6 months. At 12 months, serum C1q was only significantly reduced in C7 KO and C1q KO mice compared to WT and Crry KO mice. There were no significant differences between WT and Crry KO mice. * = P < 0.05, ** = P < 0.01, *** = P < 0.001
Quantification of C1q in Brain Homogenates from WT and AD Mice
The ELISA was used to quantify C1q levels in brain homogenates; WT and APPNL-G-F mice aged 3 and 12 months were analysed (Table 2); brain C1q levels, expressed relative to total protein, were significantly elevated at 12 months compared to 3 months in both WT (Fig. 5A) and APPNL-G-F mice (Fig. 5B). When strains were compared, brain C1q levels were significantly reduced in APPNL-G-F mice compared to WT mice at 3 months, but not at 12 months (Fig. 5C). There were no significant differences between genders for brain C1q levels in either of the mouse lines (Table 2).

Measurement of C1q in WT and APPNL-G-F mouse whole-brain lysates by quantitative sandwich ELISA. (A) C1q levels were measured in brain lysates from WT mice at 3 and 12 months and normalised to total protein in the lysate. Brain C1q levels were significantly elevated at 12 months compared to 3 months (P < 0.01). (B) C1q levels were measured in APPNL-G-F mouse whole-brain lysates, normalised as above, at 3 and 12 months. Brain C1q levels were significantly higher at 12 months (P < 0.001). (C) Brain C1q levels were significantly lower in APPNL-G-F mice than WT mice at 3 months (P < 0.01), but were not significantly different between the lines at 12 months. Units are nanogrammes of C1q per microgramme of total brain protein
Discussion
Although long-established as hallmarks of AD and other dementias, the mechanisms of neurodegeneration and pathological synaptic loss are yet to be fully understood. There is now a substantial body of evidence that synaptic pruning, an essential developmental process, is aberrantly reactivated in the AD brain [22, 23]. In AD, it is broadly accepted that synaptic loss is the best correlate of cognitive decline [48]. Both physiological and pathological synaptic pruning are complement-dependent processes that entail activation of the classical pathway with deposition of components C1q and C3b/iC3b onto “weak” synapses, marking them for removal by microglia [28, 29]. Precisely, what C1q binds on weak synapses, and what designates synapses as “weak”, remains unknown. Many researchers are using rodent models to investigate the role of C1q in this context; however, sensitive and reproducible methods of detecting and quantifying C1q in rodent biological fluids and pathological tissues are lacking. To address this need, we generated anti-C1q mAb in C1q-KO mice using pure mouse C1q as immunogen, and developed a quantitative, sensitive, and specific sandwich ELISA from a non-competing pair of mAb that enabled measurement of C1q levels in mouse and rat serum. The sandwich ELISA passed all the standard immunoassay tests of reproducibility and reliably quantified C1q in rodent serum and brain tissue. Serum C1q levels measured in 3-month-old WT mice aligned with previous reports [49, 50].
To test the utility of the assay in experimental models, we first examined the effect of ageing on serum C1q levels in WT and AD model mice on the C57BL/6 background. We observed a progressive and significant decrease in serum C1q with age in WT mice, significant at 12 months; APPNL-G-F AD model mice also demonstrated a progressive and significant decrease in serum C1q concentrations with increasing age, more marked than in WT mice, significant at 9 and 12 months compared to 3 months. Comparison between the two strains showed decreased C1q levels in APPNL-G-F mice compared to WT at each age (Fig. 3). We observed a similar decrease in plasma C1q levels in male 3xTg AD model mice that reached significance compared to WT at 12 months despite the low sample number available for analysis. These findings contradict several reports that serum C1q levels, measured using Western blotting, a semi-quantitative and insensitive method, did not change or even increased with age in C57BL/6 mice [51, 52]. One report described a substantial increase in serum C1q levels between 2 and 12 months in C57BL/6 mice via Western blotting, supported by an unspecified ELISA technique [53]. Fonseca et al., using Western blotting and densitometry, reported no significant differences in serum C1q levels at 5 and 10 months in the Arctic48 AD model compared to WT mice [52]. The fact that we used a sensitive and specific ELISA, incorporating two mAb against mouse C1q and properly validated with appropriate quality control measure, gives us confidence that the findings we report in WT and AD model mice are correct. We show that our assay accurately measures both free C1q and C1q in the C1 complex, important in contexts where the proportions of free C1q and C1 complex might vary and enabling quantification in both serum and EDTA plasma; in the latter, Ca2+ chelation disrupts the C1 complex.
Several studies report that serum C1q levels increase with age in healthy human donors [54, 55]. In our recent AD plasma biomarker study, there was no difference in serum C1q levels between aged healthy controls, and individuals with MCI or AD [8]. Others have reported that C1q levels are reduced in the CSF of AD patients compared to controls and suggested that this might be an AD biomarker [56]. Of note, CSF C1q levels reported in this study were ~ 0.2–0.4 μg/ml, comfortably within the working range of our assay. We were unable to source mouse CSF for the study, so to support the capacity of our assay to measure C1q in CSF, we spiked mouse C1q into human CSF at a relevant dose and showed that it could be measured in the assay with good recovery. While we are not aware of published studies measuring C1q levels in rodent CSF, the assay detects mouse C1q in an appropriate matrix (human CSF) at the low (ng/ml) levels predicted from human data to be present in rodent CSF; these data show that the described assay offers the prospect of such studies in disease models.
Next, we explored the impact of complement gene knockouts; serum C1q was measured in C1q KO, C3 KO, C7 KO, and Crry KO mouse models (Fig. 4). All of these lines were on the C57/BL6 background, all had been back-crossed onto the in-house C57BL/6 (WT) line, and all were maintained in the same room and under identical containment; these precautions reduce but do not eliminate risk of other genetic or environmental differences impacting the inter-line comparison. As expected, serum from C1q KO mice gave no signal in the assay, confirming the specificity of the assay and the novel mAbs reported here. Serum C1q levels were reduced in C3 KO mice at 6 and 12 months, significantly in the former, compared with matched WT mice. Precisely, how the absence of C3 impacts C1q levels is unclear; however, collaboration between C1q and C3 is critical to modulation of innate and adaptive immunity in mice and men [57, 58]. Absence of C3 might thus favour immune dysregulation and increased C1q consumption. Crry is the dominant cell-associated C3 convertase inhibitor in rodents; deficiency of Crry causes systemic consumption of C3 and secondary C3 deficiency [36]. No differences in C1q levels were observed between WT and Crry KO mice at any age suggesting that secondary C3 deficiency does not have the same effect as primary C3 deficiency in the mice. In C7 KO mice, serum C1q levels were significantly reduced at 6 and 12 months compared to matched WT mice, an unexpected finding given that absence of C7 impacts only the terminal pathway and formation of MAC. MAC plays roles in the homeostatic clearance of apoptotic cells [58, 59]; we suggest that absence of MAC may lead to an increased burden of apoptotic cells during ageing that bind C1q, well described in lupus models, leading to reduced plasma C1q levels.
Finally, in order to demonstrate whether the assay could be used to measure levels of C1q in tissue extracts, we measured the protein in mouse brain homogenate (Fig. 5). The assay performed well in brain homogenates, and we observed a significant increase in brain C1q levels at 12 months compared to 3 months in WT mice, a finding that is concordant with the literature as both C1q protein and mRNA levels have been reported to increase with age in the human and mouse brain [10, 51, 60]. We also demonstrated a significant increase in brain C1q levels in aged APPNL-G-F mice, almost doubling from 3 to 12 months of age. Brain C1q levels were significantly lower in APPNL-G-F mice relative to WT mice at 3 months, but because of the extent of increase with age, they were not significantly different at 12 months. Others have reported increased C1q expression in and around areas of pathology in the APPNL-G-F model [61]. Our whole-brain measures would miss local changes in expression; dissection and separate analysis of key brain regions for measurement of such changes is an obvious next step. Elevated brain C1q levels at late disease stage in AD mouse models relative to matched WT mice have been reported in several other studies, mostly using Western blotting to semi-quantify C1q [28, 52, 60,61,62]. Although there is an abundance of immunohistochemical evidence for C1q deposition in late-stage AD brain [63, 64], we are not aware of published studies that quantitatively measure C1q protein levels in healthy and AD human brain tissue; our unpublished work suggests that the assay described here, modified to measure human C1q, could detect and quantify C1q in AD brain extracts.
Although the current work is focussed on AD models, complement-driven synaptic elimination is also reported in other neurodegenerative disorders, including MS, and in neurodevelopmental disorders such as schizophrenia. Beyond the brain, C1q is a critical factor in autoimmune and autoinflammatory diseases [65]. Hence, the availability of a novel ELISA to reliably quantify C1q levels in rodent biological fluids and tissues may have wide application in the study of models of diverse complement-driven disorders.
Data Availability
Primary data and materials described are available on reasonable request for academic use.
Code Availability
Not applicable.
References
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V et al (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genet 41(10):1088–1093. https://doi.org/10.1038/ng.440
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D et al (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genet 41(10):1094–1099. https://doi.org/10.1038/ng.439
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC et al (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature Genet 45(12):1452–1458. https://doi.org/10.1038/ng.2802
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M et al (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nature Genet 51(3):414–430. https://doi.org/10.1038/s41588-019-0358-2
Hakobyan S, Harding K, Aiyaz M, Hye A, Dobson R, Baird A, Liu B, Harris CL et al (2016) Complement biomarkers as predictors of disease progression in Alzheimer’s disease. J Alz Dis 54(2):707–716. https://doi.org/10.3233/JAD-160420
Hu WT, Watts KD, Tailor P, Nguyen TP, Howell JC, Lee RC, Seyfried NT, Gearing M et al (2016) CSF complement 3 and factor H are staging biomarkers in Alzheimer’s disease. Acta Neuropathol Comm 4(14). https://doi.org/10.1186/s40478-016-0277-8
Morgan AR, Touchard S, O’Hagan C, Sims R, Majounie E, Escott-Price V, Jones L, Williams J et al (2017) The correlation between inflammatory biomarkers and polygenic risk score in Alzheimer’s disease. J Alz Dis 56(1):25–36. https://doi.org/10.3233/JAD-160889
Morgan AR, Touchard S, Leckey C, O’Hagan C, Nevado-Holgado AJ, NIMA Consortium, Barkhof F, Bertram L, Blin O et al (2019) Inflammatory biomarkers in Alzheimer’s disease plasma. Alz Dement 15(6):776–787. https://doi.org/10.1016/j.jalz.2019.03.007
Rasmussen KL, Nordestgaard BG, Frikke-Schmidt R, Nielsen SF (2018) An updated Alzheimer hypothesis: complement C3 and risk of Alzheimer’s disease—A cohort study of 95,442 individuals. Alz Dement 14(12):1589–1601. https://doi.org/10.1016/j.jalz.2018.07.223
Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, Cotman CW (2012) Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflamm 9(179):1–18. https://doi.org/10.1186/1742-2094-9-179
Ishii T, Haga S (1984) Immuno-electron-microscopic localization of complements in amyloid fibrils of senile plaques. Acta Neuropathol 63(4):296–300. https://doi.org/10.1007/BF00687336
Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L et al (1992) Complement activation by β-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 89(21):10016–10020. https://doi.org/10.1073/pnas.89.21.10016
Veerhuis R, Van Breemen MJ, Hoozemans JM, Morbin M, Ouladhadj J, Tagliavini F, Eikelenboom P et al (2003) Amyloid β plaque-associated proteins C1q and SAP enhance the Aβ1-42 peptide-induced cytokine secretion by adult human microglia in vitro. Acta Neuropathol 105(2):135–144. https://doi.org/10.1007/s00401-002-0624-7
Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci U S A 99(16):10837–10842. https://doi.org/10.1073/pnas.162350199
Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA (2008) Complement C3 deficiency leads to accelerated amyloid β plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci 28(25):6333–6341. https://doi.org/10.1523/JNEUROSCI.0829-08.2008
Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA (2017) Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Science Trans Med 9(392). https://doi.org/10.1126/scitranslmed.aaf6295
Reid KBM, Porter RR (1976) Subunit composition and structure of subcomponent C1q of the first component of human complement. Biochem J 155(1):19–23. https://doi.org/10.1042/bj1550019
Müller-Eberhard HJ, Polley MJ, Calcott MA (1967) Formation and functional significance of a molecular complex derived from the second and the fourth component of human complement. J Exp Med 125(2):359–380
Müller-Eberhard HJ, Dalmasso A, Calcott M (1966) The reaction mechanism of beta-1C-globulin (C’3) in immune hemolysis. J Exp Med 123(1):33–54. https://doi.org/10.1084/jem.132.4.775
Cochrane C, Müller-Eberhard H (1968) The derivation of two distinct anaphylatoxin activities from the third and fifth components of human complement. J Exp Med 127:371–386
Serna M, Giles JL, Morgan BP, Bubeck D (2016) Structural basis of complement membrane attack complex formation. Nature Comm 7:1–7. https://doi.org/10.1038/ncomms10587
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131(6):1164–1178. https://doi.org/10.1016/j.cell.2007.10.036
Bialas AR, Stevens B (2013) TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nature Neurosci 16(12):1773–1782. https://doi.org/10.1038/nn.3560
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J et al (2016) Schizophrenia risk from complex variation of complement component 4. Nature 530(7589):177–183. https://doi.org/10.1038/nature16549
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA et al (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333(6048):1456–1458. https://doi.org/10.1126/science.1202529
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME et al (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74(4):691–705. https://doi.org/10.1016/j.neuron.2012.03.026
Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27(10):1372–1384. https://doi.org/10.1016/j.neurobiolaging.2005.09.012
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352(6286):712–716. https://doi.org/10.1126/science.aad8373
Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V et al (2018) Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron 100(6):1322–1336. https://doi.org/10.1016/j.neuron.2018.10.014
Michailidou I, Willems JG, Kooi EJ, van Eden C, Gold SM, Geurts JJ, Baas F, Huitinga I et al (2015) Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann Neurol 77(6):1007–1026. https://doi.org/10.1002/ana.24398
Watkins LM, Neal JW, Loveless S, Michailidou I, Ramaglia V, Rees MI, Reynolds R, Robertson NP et al (2016) Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflamm 13(1):1–13. https://doi.org/10.1186/s12974-016-0611-x
Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, Fu T, Worringer K et al (2019) Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nature Neurosci 22(3):374–385. https://doi.org/10.1038/s41593-018-0334-7
Comer AL, Jinadasa T, Sriram B, Phadke RA, Kretsge LN, Nguyen TPH, Antognetti G, Gilbert JP et al (2020) Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLoS Biol 18(1):e3000604. https://doi.org/10.1371/journal.pbio.3000604
Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP et al (1998) Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nature Genet 19(1):56–59. https://doi.org/10.1038/ng0598-56
Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC (1995) Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A 92(25):11490–11494. https://doi.org/10.1073/pnas.92.25.11490
Ruseva MM, Hughes TR, Donev RM, Sivasankar B, Pickering MC, Wu X, Harris CL, Morgan BP (2009) Crry deficiency in complement sufficient mice: C3 consumption occurs without associated renal injury. Mol Immunol 46(5):803–811. https://doi.org/10.1016/j.molimm.2008.09.003
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC (2014) Single app knock-in mouse models of Alzheimer’s disease. Nature Neurosci 17(5):661–663. https://doi.org/10.1038/nn.3697
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP et al (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 39(3):409–421. https://doi.org/10.1016/s0896-6273(03)00434-3
Wing MG, Seilly DJ, Bridgman DJ, Harrison RA (1993) Rapid isolation and biochemical characterization of rat C1 and C1q. Mol Immunol 30(5):433–440. https://doi.org/10.1016/0161-5890(93)90111-N
Györffy BA, Kun J, Török G, Bulyáki É, Borhegyi Z, Gulyássy P, Kis V, Szocsics P et al (2018) Local apoptotic-like mechanisms underlie complement mediated synaptic pruning. Proc Natl Acad Sci U S A 115(24):6303–6308. https://doi.org/10.1073/pnas.1722613115
Harris CL, Lublin DM, Morgan BP (2002) Efficient generation of monoclonal antibodies for specific protein domains using recombinant immunoglobulin fusion proteins: Pitfalls and solutions. J Immunol Methods 268(2):245–258. https://doi.org/10.1016/S0022-1759(02)00207-7
Zelek WM, Harris CL, Morgan BP (2018) Extracting the barbs from complement assays: Identification and optimisation of a safe substitute for traditional buffers. Immunobiol 223(12):744–749. https://doi.org/10.1016/j.imbio.2018.07.016
Zelek WM, Taylor PR, Morgan BP (2019) Development and characterization of novel anti-C5 monoclonal antibodies capable of inhibiting complement in multiple species. Immunology 157(4):283–295. https://doi.org/10.1111/imm.13083
Reed GF, Lynn F, Meade BD (2002) Use of coefficient of variation in assessing variability of quantitative assays. Clin Diag Lab Immunol 9(6):1235–1239. https://doi.org/10.1128/CDLI.9.6.1235
Andreasson U, Perret-Liaudet A, van Waalwijk van Doorn LJ, Blennow K, Chiasserini D, Engelborghs S, Fladby T, Genc S et al (2015) A practical guide to immunoassay method validation. Front Neurol 6(179):1–8. https://doi.org/10.3389/fneur.2015.00179
Hanneman SK, Cox CD, Green KE, Kang DH (2011) Estimating intra- and inter-assay variability in salivary cortisol. Biol Res Nurs 13(3):243–250. https://doi.org/10.1177/1099800411404061
Sandholm K, Persson B, Skattum L, Eggertsen G, Nyman D, Gunnarsson I, Svenungson E, Nilsson B et al (2019) Evaluation of a novel immunoassay for quantification of C1q for clinical diagnostic use. Front Immunol 10:7. https://doi.org/10.3389/fimmu.2019.00007
Barthet G, Mulle C (2020) Presynaptic failure in Alzheimer’s disease. Prog Neurobiol. Nov;194:101801. https://doi.org/10.1016/j.pneurobio.2020.101801
Cortes-Hernandez J, Fossati-Jimack L, Petry F, Loos M, Izui S, Walport MJ, Cook HT, Botto M (2004) Restoration of C1q levels by bone marrow transplantation attenuates autoimmune disease associated with C1q deficiency in mice. Eur J Immunol 34(12):3713–3722. https://doi.org/10.1002/eji.200425616
Yabumoto C, Akazawa H, Yamamoto R, Yano M, Kudo-Sakamoto Y, Sumida T, Kamo T, Yagi H et al (2015) Angiotensin II receptor blockade promotes repair of skeletal muscle through down-regulation of aging-promoting C1q expression. Sci Rep 5:14453. https://doi.org/10.1038/srep14453
Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH et al (2013) A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33(33):13460–13474. https://doi.org/10.1523/JNEUROSCI.1333-13.2013
Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, Tenner AJ (2017) Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflamm 14(1):1–15. https://doi.org/10.1186/s12974-017-0814-9
Naito AT, Sumida T, Nomura S, Liu ML, Higo T, Nakagawa A, Okada K, Sakai T et al (2012) Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell 149(6):1298–1313. https://doi.org/10.1016/j.cell.2012.03.047
Yonemasu K, Kitajima H, Tanabe S, Ochi T, Shinkai H (1978) Effect of age on C1q and C3 levels in human serum and their presence in colostrum. Immunology 35(3):523–530. https://doi.org/10.1093/clind/15.6.1049
Watanabe S, Sato K, Hasegawa N, Kurihara T, Matsutani K, Sanada K, Hamaoka T, Fujita S et al (2015) Serum C1q as a novel biomarker of sarcopenia in older adults. FASEB J 29(3):1003–1010. https://doi.org/10.1096/fj.14-262154
Smyth MD, Cribbs DH, Tenner AJ, Shankle WR, Dick M, Kesslak JP, Cotman CW (1994) Decreased levels of C1q in cerebrospinal fluid of living Alzheimer patients correlate with disease state. Neurobiol Aging 15(5):609–614. https://doi.org/10.1016/0197-4580(94)00055-7
Pekkarinen PT, Heikkilä N, Kisand K, Peterson P, Botto M, Daha MR, Drouet C, Isaac L et al (2015) Dysregulation of adaptive immune responses in complement C3-deficient patients. Eur J Immunol 45(3):915–921. https://doi.org/10.1002/eji.201444948
Scott D, Botto M (2016) The paradoxical roles of C1q and C3 in autoimmunity. Immunobiol 221(6):719–725. https://doi.org/10.1016/j.imbio.2015.05.001
Cole DS, Morgan BP (2003) Beyond lysis: How complement influences cell fate. Clin Sci 104(5):455–466. https://doi.org/10.1042/CS20020362
Reichwald J, Danner S, Wiederhold KH, Staufenbiel M (2009) Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflamm 6(35):1–12. https://doi.org/10.1186/1742-2094-6-35
Chen WT, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, Qian X, Laláková J et al (2020) Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 182(4):976–991. https://doi.org/10.1016/j.cell.2020.06.038
Benoit ME, Hernandez MX, Dinh ML, Benavente F, Vasquez O, Tenner AJ et al (2013) C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models are essential for the C1q-mediated protection against amyloid- β neurotoxicity. J Biol Chem 288(1):654–665. https://doi.org/10.1074/jbc.M112.400168
Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ (1996) Localization and cell association of C1q in Alzheimer’s disease brain. Exp Neurol 138(1):22–32. https://doi.org/10.1006/exnr.1996.0043
Veerhuis R, van der Valk P, Janssen I, Zhan SS, Van Nostrand WE, Eikelenboom P (1995) Complement activation in amyloid plaques in Alzheimer’s disease brains does not proceed further than C3. Virch Archiv 426(6):603–610. https://doi.org/10.1007/BF00192116
Thielens NM, Tedesco F, Bohlson SS, Gaboriaud C, Tenner AJ (2017) C1q: A fresh look upon an old molecule. Mol Immunol 89:73–83. https://doi.org/10.1016/j.molimm.2017.05.025
Acknowledgements
The work was supported by the UK Dementia Research Institute. We thank Neil Evans for technical and animal support, Dr. Wioleta Zelek for help with ELISA optimisation, and Dr. Tim Hughes for access to 3xTg samples.
Funding
This work was supported by a Programme Grant from the UK Dementia Research Institute, part funded by the UK Medical Research Council.
Author information
Authors and Affiliations
Contributions
RB led the assay development work; BPM designed and supervised the project; all other authors contributed to aspects of the experimental work. RB, MT, and SC drafted the manuscript; BPM finalised the submission.
Corresponding author
Ethics declarations
Ethics Approval
No human subjects were involved in this research so informed consent is not relevant. Animals use was under the auspices of UK Home Office Licence numbers PF4167C0A and P8159A562.
Consent to Participate
No human subjects were involved in this research, so consent to participate is not relevant.
Consent for Publication
All authors have approved this manuscript and consented to its submission for publication.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PPTX 1321 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Byrne, R.A.J., Torvell, M., Daskoulidou, N. et al. Novel Monoclonal Antibodies Against Mouse C1q: Characterisation and Development of a Quantitative ELISA for Mouse C1q. Mol Neurobiol 58, 4323–4336 (2021). https://doi.org/10.1007/s12035-021-02419-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02419-5