
Abstract
Macroautophagy and chaperone-mediated autophagy (CMA) are two important subtypes of autophagy that play a critical role in cellular quality control under physiological and pathological conditions. Despite the marked differences between these two autophagic pathways, macroautophagy and CMA are intimately connected with each other during the autophagy-lysosomal degradation process, in particular, in the setting of neurological illness. Macroautophagy serves as a backup mechanism to removal of malfunctioning proteins (i.e., aberrant α-synuclein) from the cytoplasm when CMA is compromised, and vice versa. The molecular mechanisms underlying the conversation between macroautophagy and CMA are being clarified. Herein, we survey current overviews concentrating on the complex interactions between macroautophagy and CMA, and present therapeutic potentials through utilization and manipulation of macroautophagy-CMA crosstalk in the treatment of neurological diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autophagy is an evolutionarily conserved lysosomal degradation pathway by which cytoplasmic materials are delivered to and degraded in the lysosome [1]. It is essential for cell survival, differentiation, development, and homeostasis [2, 3]. Dysfunction of the autophagy-lysosomal pathway has been linked to a wide variety of brain pathology such as acute brain injuries [4, 5], neurodegenerative diseases (i.e., Alzheimer’s (AD), Parkinson’s (PD), and Huntington’s disease (HD)) [6–8], and brain tumors [9]. It is therefore significant to characterize the role of autophagy and its regulation in these diseases for therapeutic purpose.
Depending on the mode of cargo delivery to lysosome, autophagy can be subdivided into three subtypes: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [10]. Macroautophagy is the most universal form of autophagy characterized by the formation of autophagosome and is capable of disposing protein aggregates and damaged organelles [11, 12]. On the other hand, microautophagy is a lesser known self-eating event [13]. During the microautophagic process, cytosolic components are transported into the lysosome by direct invagination of lysosomal membrane and subsequent budding of vesicle into the lysosomal lumen [14]. In contrast, CMA is a unique pathway by which cytosolic protein aggregates are selectively transferred into lysosome for degradation [15]. Neither vesicle formation nor major changes in the lysosomal membrane occur during the CMA process [16]. Intriguingly, mounting evidences demonstrated that there is a functional relationship between macroautophagy and CMA, in particular, in the setting of neurological diseases [17, 18]. Dysfunction of either one of them can lead to a compensatory upregulation of the other. In this review, we aim to provide an integrative overview that concentrates on the interaction between macroautophagy and CMA in the mammalian systems (unless otherwise stated), and emphasize further details of the molecular mechanisms underlying the crosstalk between macroautophagy and CMA. Lastly, we will discuss therapeutic potentials by targeting macroautophagy-CMA crosstalk in the treatment of neurological diseases.
Macroautophagy and CMA: Different Paths for Intracellular Protein Turnover and Recycling
Macroautophagy: Process and Regulation
The macroautophagic degradation of long-lived proteins and damaged/superfluous organelles consists of specific steps (Fig. 1) [19, 20]. It begins with nucleation of a unique membrane structure coined as phagophore, which is followed by the sequestration of cytoplasmic constituent into the phagophore. The elongation and closure of phagophore lead to generation of a double-membrane-bound autophagosome [21]. Autophagosome can fuse with lysosome to form autolysosome, where luminal contents of the autophagosome as well as inner membrane of the autophagic vacuole are broken down by lysosomal hydrolases [22]. The resultant biomolecules, such as amino acids, sugars, and lipids, are transported back into the cytosol for reuse via permeases functioning [23].

The cellular events of macroautophagy and CMA
It is important to note that autophagy-related (Atg) proteins play an important role in autophagosome formation and macroautopahgy regulation [24–26]. For instance, the ULK1 complex, which consists of ULK1, Atg13, Atg101, and FIP200 (focal adhesion kinase family-interacting protein of 200 kD), is a vital complex in integrating incoming autophagy signals to initiate autophagosome biogenesis [27–29]. Beclin 1, the mammalian ortholog of yeast Atg6, can collaborate with other key subunits such as barkor (Beclin 1-associated autophagy-related key regulator), p150, and UVRAG (UV irradiation resistance-associated gene) to form the class III phosphatidylinositol 3-kinase (PI3K) complex that is crucial for nucleation and assembly of the initial phagophore membrane [30–33]. The elongation of pre-autophagosomal structures requires two ubiquitin-like modification systems, including Atg12-Atg5-Atg16L (Atg16-like protein) complex and LC3 (microtubule-associated protein 1 light chain 3) conjugation system [34–36]. In the first of these two ubiquitin-like conjugation reactions, the ubiquitin-like protein Atg12 is covalently tagged to Atg5 with the help of E1-like ubiquitin activating enzyme Atg7 and E2-like ubiquitin conjugating enzyme Atg10 [37–39]. The Atg12-Atg5 conjugate can then interact non-covalently with ATG16L to form the Atg12-Atg5-Atg16L tetramers, which function as a E3-like ubiquitin ligase that contributes to expansion of autophagosomal membrane by promoting LC3 lipidation [40, 41]. In the second ubiquitin-like conjugation reaction, LC3 is conjugated to phosphatidylethanolamine (PE) by E1-like Atg7 and E2-like Atg3 to form LC3-II [42, 43]. LC3-II can be specifically targeted to the elongating membrane, which aids the closure of autophagosomal membrane to form an autophagosome [44].
Multiple upstream signals, including mammalian target of rapamycin (mTOR)-dependent and -independent pathway, can regulate the process of macroautophagy [45, 46]. The serine/threonine protein kinase mTOR serves as a primary negative regulator of macroautophagy, and inactivation of mTOR promotes the macroautophagic process [47]. On the other hand, signaling pathways, such as cAMP-Epac-PLC-ε-IP3 pathway and Ca2+-calpain-G-stimulatory protein α pathway, can modulate macroautophagic response in an mTOR-independent manner [46, 48]. A more detailed regulatory mechanism of macroautophagy has been extensively described elsewhere [49–51].
CMA: Process and Regulation
CMA is another subset of autophagy that has only been described in mammalian cells, and it is distinct from macroautophagy due to its specific mechanism of cargo selection and delivery to the lysosomal lumen for disposal [52]. In this form of autophagy, protein structures are selectively delivered into lysosome individually. Heat shock cognate chaperone of 70 kDa (hsc70) plays an important role in the CMA pathway (Fig. 1) [53]. Hsc70, together with its co-chaperones (e.g., hsc90), recognizes a specific consensus pentapeptide motif KFERQ in all cytosolic proteins targeting for CMA by forming a chaperone/substrate complex [54]. Initially, the chaperone/substrate complex is associated with lysosomal membrane protein receptor LAMP-2A (lysosome-associated membrane protein type 2A) [55]. The substrate protein can then be transported into lysosomal lumen for degradation with assistance of lysosomal-hsc70 (lys-hsc70) [56, 57].
The activity of CMA can be modulated by regulating local levels of LAMP-2A and lys-hsc70 at the lysosomal compartment [54]. Inhibition of p38 mitogen activated protein kinase (MAPK) can partially suppress the activation of CMA, suggesting the involvement of this signaling pathway in the modulation of CMA [58]. However, more signaling pathways which participate in the regulation of CMA remain to be clarified.
Macroautophagy and CMA in Neurological Disorders
The autophagic-lysosomal systems, both macroautophagy and CMA, are important for cellular quality control to protect neurons from many kinds of damage and disease, such as acute injury, chronic neurodegeneration, and brain tumors. For instance, macroautophagy provides neuroprotective effects against ischemic, hemorrhagic, and traumatic brain injury [59–61]. Proper functioning of macroautophagy contributes to prevention of neurodegeneration via clearance of cytoplasmic aggregate-prone proteins and inclusions [62–64]. To date, the toxicity of causative gene products has been linked to the development of certain neurodegenerative disorder. Missense mutations in the amyloid precursor protein (APP) and presenilin genes can lead to an increase in the production of the amyloid-beta (Aβ) peptide, which can predispose to the development of AD-type brain pathology [65–69]. Pathogenic mutations in the tau gene can lead to abnormality in the function or isoform composition of this microtubule-associated protein, which contributes to the formation of abundant neurofibrillary lesions in AD and other tauopathies [70–73]. Stimulation of macroautophagy enhances the clearance of Aβ peptide and the APP-derived fragment in neurons and provides protective effects in cellular and animal models of AD [74–76]. Activation of macroautophagy can also eliminate both soluble mutant tau protein and its aggregates from the cytoplasm to promote neuronal survival in cellular and animal tauopathy models [77, 78]. PD is characterized by formation of neuronal intracellular Lewy body inclusions and the loss of dopaminergic neurons from the substantia nigra. Mutations in the α-synuclein gene (such as A53T and A30P) and the parkin gene are genetically linked to familial forms of PD [79–81]. In addition, mutations in the leucine-rich repeat kinase 2 gene (LRRK2) are clinically linked to PD [82–84]. These mutated gene products work together to promote the abnormal protein aggregation and Lewy body formation in PD [85–88]. More importantly, macroautophagy serves as a route for intracellular α-synuclein degradation to ameliorate the neurodegenerative pathology in PD models [89, 90]. In HD, an expansion of a CAG trinucleotide repeat in the interesting transcript 15 gene results to a mutant huntingtin protein with an abnormally expanded polyglutamine tract [91]. This cytotoxic polyglutamine-expanded huntingtin protein is highly expressed in neurons of the brain and can lead to the hallmark pathology of HD [92, 93]. Macroautophagy functions as a key clearance pathway for mutant huntingtin fragments that reduces intracellular huntingtin accumulation and protects cells against polyglutamine toxicity in HD models [45, 94, 63]. Similarly, activation of macroautophagy is beneficial for other so-called polyglutamine diseases, and it contributes to the degradation of mutant ataxin-3 protein that causes spinocerebellar ataxia type 3 [95–97]. Additionally, the mutant superoxide dismutase (SOD1) proteins, which are linked to familial amyotrophic lateral sclerosis, can be degraded by macroautophagy [98–100]. The impairment of the macroautophagy pathway is a crucial pathogenic mechanism of hereditary spastic paraplegia [101, 102]. Whereas, macroautophagy serves as a pro-death mechanism for brain tumor cells under certain conditions [103, 104]. Treatment with cytotoxic drugs such as arsenic trioxide and selenite triggers mitochondrial damage and initiates autophagic cell death in malignant glioma cells [103, 104].
Likewise, the signaling pathway of CMA is activated during hypoxic and ischemic stress to promote neuronal cell survival under these conditions [105]. Dysfunction of CMA causes accumulation of abnormal proteins in the pathological brain, which is involved in the pathogenesis of chronic neurodegeneration, such as PD [106], AD [107], and HD [108]. Indeed, the pathophysiological roles of CMA in the removal of aggregate-prone proteins that cause neurodegenerative diseases are being investigated. Evidence from both in vitro and in vivo studies supports that α-synuclein is a bona fide CMA substrate [109–111]. Wild-type α-synuclein, which contains a CMA-targeting motif in its sequences, can be delivered to lysosomes for its degradation via the CMA pathway [109]. Whereas, the pathogenic mutants of α-synuclein can bind to the CMA receptor LAMP-2A but fail to translocate inside the lysosomal lumen, thus impairing their own degradation along with that of other CMA substrates through this pathway [112]. Also, dopamine-modified α-synuclein can be bound to the lysosomal membrane, but it fails to translocate into lysosomes, resulting in the blockage of its degradation by CMA [113]. In the postmortem study of seven brain samples from PD patients, Alvarez-Erviti et al. reported a reduced expression of CMA proteins LAMP-2A and hsc70 in the substantia nigra pars compacta and amygdala of PD brains [114]. Downregulation of CMA activity compromises the degradation of alpha-synuclein, which underpins the Lewy body formation and PD pathogenesis [109, 115, 116]. Overexpression of LAMP-2A restores CMA activity and reduces the levels of aberrant α-synuclein and thereby ameliorates α-synuclein-induced dopaminergic neurodegeneration [106]. Although proteins like huntingtin contain CMA-targeting motifs in their sequences, the amount of these wild-type proteins degraded by CMA is negligible under normal conditions [117, 118]. However, in the case of HD, mutant huntingtin is selectively degraded by CMA through an hsc70 and LAMP-2A-dependent manner [108, 119]. Thus, functional regulation of this autophagy-lysosomal pathway provides novel means to prevent these devastating cerebral disorders.
Crosstalk Between Macroautophagy and CMA: a Potential Target for the Treatment of Neurological Diseases
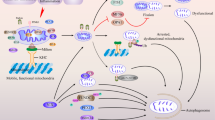
Under normal cellular settings, macroautophagy and CMA occur at a basal level, but they can be activated when cells encounter stressful stimuli [54, 120]. During nutrient deprivation, the activity of macroautophagy rapidly upregulates and reaches the maximum level around 4∼6 h postfasting but downregulates immediately thereafter [121]. If the nutritional stress continues, the CMA pathway will be enhanced. The pathway activity peaks at 12∼20 h and remains to be active in the long-term starvation [122]. Increasing evidence has indicated that macroautophagy and CMA directly communicate with each other in protein degradation to maintaining cell homeostasis (Fig. 2) [123, 124]. For instance, in a cell model of tauopathy expressing TauRD△K280, Wang and colleagues proposed that the inability of complete degradation of mutated tau protein by the CMA pathway can lead to the pro-aggregating substrate generation and tau aggregation, resulting in tau pathology. At the same time, macroautophagy is able to clear these CMA-related tau aggregates, suggesting the functional interrelationships between macroautophagy and CMA [125, 117]. With series of key findings, molecular mechanisms underlying the crosstalk between macroautophagy and CMA are beginning to be elucidated. Dissecting these interactions between macroautophagy and CMA is essential for characterizing their respective roles in physiological and pathological conditions.

Schematic illustrating the interrelations between macroautophagy and CMA. Macroautophagy and CMA communicate with each other in the process of autophagy-lysosomal degradation. CMA blockage can induce compensative upregulation of macroautophagy, and the CMA pathway compensates when macroautophagy is inhibited. Molecular directors such as hsc70, Nrf2, ubiquilin, and p53 may function as core network nodes in macroautophagy-CMA crosstalk
CMA-Blockage-Induced Macroautophagy Upregulation
CMA is a cargo-specific subtype of autophagy which dictates multiple physiological functions [126]. It contributes to amino acid recycling during prolonged starvation [122] and fulfills protein quality control functions via selective turnover of damaged or malfunctioning proteins from the cytosol [127, 128]. As mentioned above, malfunction of CMA is implicated in the pathogenesis of cerebral diseases. In PD, the pathogenic A53T and A30P α-synuclein mutants can strongly bind to the CMA receptor LAMP-2A on lysosomes. However, these mutant forms of α-synuclein cannot be translocated into the lysosomal lumen, which inhibits not only the lysosomal uptake of their own but also that of other substrates for CMA degradation [109]. More importantly, in the PC12 cell model, pathogenic α-synuclein-mutant-mediated blockage of CMA leads to compensatory activation of macroautophagy, which makes up for portions of CMA’s functions in protein degradation [109]. In the cortical neurons and differentiated SH-SY5Y cells, the dysfunction of CMA conferred by mutant A53T α-synuclein can also result in an increased activity of macroautophagy [112]. However, under this condition, the compensatory upregulation of macroautophagy secondary to CMA inhibition due to overexpression of mutant A53T α-synuclein does not contribute to an increase of macroautophagy-dependent degradation. Instead, it causes the accumulation of autophagosomes that leads to membrane destabilization of these autophagic vacuoles and the cytoplasmic release of vacuolar hydrolases, thus eventually inducing autophagic cell death of primary cortical neurons [112, 129, 130]. The molecular mechanism underlying such a compensatory response of macroautophagy owing to CMA impairment, however, is still unclear. It has been demonstrated that in the early stage after CMA blockage by RNA interference against the lysosomal membrane receptor, LAMP-2A, its macroautophagic activity decreases because of an increase in mTOR activity. However, as CMA blockage persists, the increased intracellular levels of Beclin 1, along with the reduced phosphorylation of mTOR, can work together to induce constitutive activation of the macroautophagy[131].
Under basal conditions, compensatory macroautophagy is able to help CMA-defective cell to maintain normal protein degradation and cell viability [17]. However, upon exposure to certain stressors, such as pro-oxidative and oxidative stimuli (i.e., cadmium, paraquat, and H2O2), or to UV light, CMA-defective cell undergoes more apoptotic cell death. This suggests that despite the constitutive upregulation of macroautophagy, it is unable to compensate for all CMA functions [17]. It is proposed that the persistent activation of this pathway due to CMA blockage could lead to continuous consumption of the macroautophagic machinery proteins in chronically CMA-impaired cells and thus may limit a major increase in the activity of this pathway to respond to stress [131]. Therefore, in future research, it would be important to investigate whether constitutive activation of macroautophagy can always lead to the failure of its inducible form in response to stress, and to identify the time frame needed for this switch [131]. Additionally, considering the fact that aberrant activation of macroautophagy secondary to CMA inhibition leads to autophagic cell death of cortical neurons, improving the function of CMA function is important because it can mitigate potential deleterious consequences of aberrant induction of macroautophagy in this specific context of α-synuclein overexpression [112].
CMA Compensates When Macroautophagy Is Inhibited
Macroautophagy is the bulk protein degradation pathway that plays an important role in protein quality control and maintaining cellular homeostasis of neural cells [8, 62]. A failure of macroautophagy-mediated clearance of misfolded proteins will cause the accumulation of these toxic proteins inside the affected neurons, leading to neurodegenerative disorders [132–134]. In the case of HD, activation of the kinase activity of inositol-requiring enzyme 1 mediates endoplasmic reticulum stress–induced inhibition of macroautophagic flux that causes accumulation of mutant huntingtin aggregates in cells, thus resulting in neuronal cell death [135, 136]. Martinez-Vicente et al. found that a defect in the ability of autophagic cargo recognition by autophagosomes compromises the removal of cytosolic components. As a result, there are increased levels of protein aggregates and abnormal intracellular lipid stores as well as a persistent level of dysfunctional mitochondria in HD cells [137]. Intriguingly, the impairment of macroautophagy can lead to activation of the CMA pathway under both basal and stress conditions [124]. An increase in the number of CMA translocation components (including LAMP-2A and hsc70) and CMA-competent lysosomes helps to boost the CMA activity following macroautophagy inhibition [124]. In cellular and mouse models, the HD cells are able to upregulate the activity of CMA pathway by increasing the de novo synthesis of LAMP-2A proteins and stabilizing of LAMP-2A at the lysosomal membrane followed by the blockage of macroautophagy in these cells [138]. It is proposed that cells obey different mechanisms to mediate the compensatory activation of CMA to respond to macroautophagy blockage depending on different conditions [127, 139]. The de novo synthesis of the receptor protein would be a major way to increase levels of LAMP-2A under basal conditions. In contrast, changes in the LAMP-2A protein already in the lysosomal compartment (i.e., reduced breakdown of LAMP-2A protein already at the lysosomal membrane) could be more optimal than de novo synthesis of this protein under stress conditions [124]. Therefore, it is interesting to determine whether or not there exist different mechanisms underlying compensatory upregulation of CMA secondary to macroautophagy blockage.
The compensative upregulation of CMA in response to macroautophagy inhibition promotes removal of oxidized proteins, which protects Atg5 −/− murine embryonic fibroblasts from oxidative-stress-mediated cell death [140, 141]. Also, evidences from an in vivo study demonstrated that a compensatory CMA due to compromised macroautophagy provides a protective effect that contributes to the function preservation of cone retinal cells in conditional Atg5 knockout in mice [142]. Notably, different with the evidence from Atg5 null mouse embryonic fibroblasts [141], enhancement of CMA pathway secondary to macroautophagy inhibition fails to prevent RALA hepatocytes from menadione-induced oxidative stress and cell death [143]. These data suggest that macroautophagy and CMA have non-redundant functions in hepatocyte to resist to oxidant stress.
Taken together, macroautophagy is able to degrade the CMA substrates; however, it cannot compensate for selective degradation of particular substrates by CMA. Comparatively, CMA is capable of degrading cytosolic proteins but fails to take care of macroautophagy-mediated degradation of expired organelles. Despite these limitations, compensatory upregulation of one of them is beneficial for cells when the other is compromised, which helps cells to resist various types of damage to maintain cell viability [17, 141].
Crosstalk Between Autophagy and Ubiquitin-Proteasome System (UPS)
The UPS is a major selective protein degradation pathway that plays important roles for eukaryotic cells [144]. UPS targets biologically non-useful proteins such as mutant, misfolded, damaged, or otherwise abnormal proteins for rapid substrate-specific proteolysis. In the CNS, UPS modulates a variety of basic cellular processes of neurons such as neuronal growth and development, synaptic function and plasticity, and neuronal survival and homeostasis [145]. Defect of the UPS has been linked to the pathogenesis of neurological disorders [146–148].
It is noteworthy that UPS and the autophagy-lysosomal system are functionally coupled in degrading excess or damaged proteins to maintain cellular homeostasis and neuronal survival [149, 150]. In a fly model of spinobulbar muscular atrophy, macroautophagy compensates for impaired UPS function in a histone deacetylase 6 (HDAC6)-dependent manner [151]. The compensatory macroautophagy due to proteasome inhibition plays an important role in controlling endoplasmic reticulum stress and reducing cell death [152, 153]. Also, in response to macroautophagy inhibition, proteasomes are activated in a compensatory manner for protein degradation [154]. Moreover, CMA and UPS collaborate to degrade the gene product of regulator of calcineurin 1, whose overexpression has been linked to Down syndrome (DS) and AD neuropathology [107]. Thus, further efforts are anticipated to explore the functional relationships between UPS and autophagy with the goal of controlling these interactions for optimizing the therapeutic approaches for cerebral diseases.
Future Direction
Macroautophagy and CMA communicate with each other extensively in the autophagy-lysosomal degradation process. In certain circumstances, one can serve as a backup mechanism when the other is defective, and vice versa. However, research in the crosstalk between macroautophagy and CMA is still in its infancy. Better knowledge of the compensatory mechanisms between these two autophagic pathways, their advantages, and their limitations is of great significance, in order to develop effective interventions to regulate them for therapeutic purposes in autophagy-associated human disorders.
In particular, elucidation of the interrelationships between macroautophagy and CMA would have great clinical significance for intervention of those devastating cerebral disorders. As described above, macroautophagy and CMA are interconnected during protein degradation process in neurons. When either one is compromised, cells initiate the other autophagic pathway in a compensatory manner. However, inappropriate or excessive compensatory activation of autophagy may lead to neuronal cell death in the pathophysiological process of cerebral disorders. In addition, compensatory activation of the autophagic pathway may promote the development of brain tumors and its resistance to anticancer therapy [155–157]. Therefore, exploring the crosstalk between these two subtypes of autophagy helps to further define their exact roles in brain pathology (i.e., if they are the primary causes or the secondary consequences of pathological alterations). As a result, this will allow further molecular insights into the progressive dysfunction of autophagic protein degradation that leads to neurological diseases. It also provides key information for better manipulation of macroautophagy and CMA in the treatment and prevention of cerebral disorders.
Therefore, a series of critical issues regarding the macroautophagy-CMA network are warranted to be addressed. First, the physiological and pathological roles of macroautophagy and CMA require further investigation [126]. To date, little is known about the regulation of basal macroautophagy and CMA under normal conditions. Whether they function as a pro-death or pro-survival program in certain cerebral disorders should be addressed. Second, although there is evidence of interrelationships between macroautophagy and CMA [158], the temporal associations, the universality, the functional consequences, and the molecular directors of the macroautophagy-CMA chat are less understood. It is worthy to note that regulation of the levels of hsc70 in the lysosomal lumen during macroautophagy blockage offers clues about possible molecular mechanisms behind the crosstalk between these two autophagic pathways [124]. In macroautophagy-deficient cells, reduced fusion of autophagosomes with lysosomes could result in less dissipation of lysosomal pH. This causes more acidic environment in these lysosomes [124]. Lower lysosomal pH confers more stability of hsc70 in the lysosomal lumen to mediate CMA [124]. Recently, Rothenberg et al. demonstrated that ubiquilin proteins can be consumed by both macroautophagy and CMA [159]. Ubiquilin can positively regulate macroautophagy by promoting the conversion of LC3-I to LC3-II and accelerating autophagosome maturation [160]. However, it is actively degraded after carrying out its function during macroautophagic process. Moreover, ubiquilin contains two pentapeptide sequences (ILKDQ and EVRFQ) known as the CMA-targeting motif, which enable its selective recognition and degradation by CMA [159]. This dual mode of consumption of ubiquilin may have important implications in the crosstalk between macroautophagy and CMA. Therefore, it will be of interest to discover how ubiquilin is selected for either of these two forms of autophagy [159]. p53, the “guardian of the cellular genome” functions in the pathophysiology of cerebral diseases [161–163], may play a role in mediating macroautophagy-CMA crosstalk. On one hand, p53 can either promote or inhibit macroautophagy, depending on its subcellular locations [164–166]. On the other hand, inhibition of macroautophagy results in increased cytosolic levels of mutant p53 proteins in non-proliferating confluent cancer cells. This allows hsc70 to recognize their KFERQ-like sequence motifs and in turn enhances the degradation of mutant p53 in a CMA-dependent manner [18]. Additionally, the Nrf2 pathway could serve as an upstream signal to regulate the activities of both macroautophagy and CMA [167]. Genetical increase of astrocyte-specific Nrf2 can attenuate the functional deficiency of macroautophagy and CMA, which promotes the autophagy-lysosomal degradation of human mutant α-synuclein in the α-synuclein mutant mouse model [167]. Future investigations should focus more on key molecules such as ubiquilin, p53, and Nrf2, which might act as a core network node in the crosstalk between macroautophagy and CMA. Finally, large-scale proteomic- and metabolomic-based studies, as well as transgenic-based approaches, should provide valuable clues on the matter of macroautophagy-CMA crosstalk.
Concluding Remarks
Macroautophagy and CMA exert critical roles in maintaining neuronal homeostasis in the brain under physiological and pathological conditions. Although considerable advances have been made in our insights into the macroautophagy-CMA network, key aspects regarding the interplay of these two catabolic pathways, including the compensatory mechanisms and their benefits and limitations, remain enigmatic. Answers to these questions are particularly significant to allow better manipulation of these two important autophagy-lysosomal pathways in the treatment of autophagy-related cerebral diseases.
References
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132(1):27–42. doi:10.1016/j.cell.2007.12.018
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451(7182):1069–1075. doi:10.1038/nature06639
Mizushima N, Levine B (2010) Autophagy in mammalian development and differentiation. Nat Cell Biol 12(9):823–830. doi:10.1038/ncb0910-823
Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH (2008) Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 4(6):762–769
Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao HJ, Dai DK, Shen YW, Xu HF, Ni H, Wan L, Qin ZH, Tao LY, Zhao ZQ (2011) Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 184:54–63. doi:10.1016/j.neuroscience.2011.03.021
Sanchez-Varo R, Trujillo-Estrada L, Sanchez-Mejias E, Torres M, Baglietto-Vargas D, Moreno-Gonzalez I, De Castro V, Jimenez S, Ruano D, Vizuete M, Davila JC, Garcia-Verdugo JM, Jimenez AJ, Vitorica J, Gutierrez A (2012) Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer's mice hippocampus. Acta Neuropathol 123(1):53–70. doi:10.1007/s00401-011-0896-x
Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E (2010) Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 5(2):e9313. doi:10.1371/journal.pone.0009313
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095):885–889. doi:10.1038/nature04724
Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M, Malagnino V, Falzarano SM, Pirtoli L, Tosi P (2007) Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol 30(2):429–436
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147(4):728–741. doi:10.1016/j.cell.2011.10.026
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306(5698):990–995. doi:10.1126/science.1099993
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290(5497):1717–1721
Mijaljica D, Prescott M, Devenish RJ (2011) Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7(7):673–682
Kunz JB, Schwarz H, Mayer A (2004) Determination of four sequential stages during microautophagy in vitro. J Biol Chem 279(11):9987–9996. doi:10.1074/jbc.M307905200
Dice JF (2007) Chaperone-mediated autophagy. Autophagy 3(4):295–299
Arias E, Cuervo AM (2011) Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol 23(2):184–189. doi:10.1016/j.ceb.2010.10.009
Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM (2006) Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 103(15):5805–5810. doi:10.1073/pnas.0507436103
Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, Yuan J (2013) Chaperone-mediated autophagy degrades mutant p53. Genes Dev 27(15):1718–1730. doi:10.1101/gad.220897.113
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368(7):651–662. doi:10.1056/NEJMra1205406
Nixon RA, Yang DS (2012) Autophagy and neuronal cell death in neurological disorders. Cold Spring Harbor perspectives in biology 4 (10). doi:10.1101/cshperspect.a008839
Mizushima N (2007) Autophagy: process and function. Genes Dev 21(22):2861–2873. doi:10.1101/gad.1599207
Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8(11):931–937. doi:10.1038/nrm2245
Rubinsztein DC, Marino G, Kroemer G (2011) Autophagy and aging. Cell 146(5):682–695. doi:10.1016/j.cell.2011.07.030
Cao Y, Klionsky DJ (2007) Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 17(10):839–849. doi:10.1038/cr.2007.78
Saitoh T, Akira S (2010) Regulation of innate immune responses by autophagy-related proteins. J Cell Biol 189(6):925–935. doi:10.1083/jcb.201002021
Shpilka T, Weidberg H, Pietrokovski S, Elazar Z (2011) Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol 12(7):226. doi:10.1186/gb-2011-12-7-226
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20(7):1992–2003. doi:10.1091/mbc.E08-12-1249
Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284(18):12297–12305. doi:10.1074/jbc.M900573200
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2):132–141. doi:10.1038/ncb2152
Furuya N, Yu J, Byfield M, Pattingre S, Levine B (2005) The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 1(1):46–52
Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19(12):5360–5372. doi:10.1091/mbc.E08-01-0080
Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T (2010) Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 190(4):511–521. doi:10.1083/jcb.200911141
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9(10):1142–1151. doi:10.1038/ncb1634
Kuma A, Matsui M, Mizushima N (2007) LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 3(4):323–328
Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C, Martens S (2012) Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J 31(22):4304–4317. doi:10.1038/emboj.2012.278
Matsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F (2007) Structure of Atg5.Atg16, a complex essential for autophagy. J Biol Chem 282(9):6763–6772. doi:10.1074/jbc.M609876200
Geng J, Klionsky DJ (2008) The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 9(9):859–864. doi:10.1038/embor.2008.163
Komatsu M, Tanida I, Ueno T, Ohsumi M, Ohsumi Y, Kominami E (2001) The C-terminal region of an Apg7p/Cvt2p is required for homodimerization and is essential for its E1 activity and E1-E2 complex formation. J Biol Chem 276(13):9846–9854. doi:10.1074/jbc.M007737200
Nemoto T, Tanida I, Tanida-Miyake E, Minematsu-Ikeguchi N, Yokota M, Ohsumi M, Ueno T, Kominami E (2003) The mouse APG10 homologue, an E2-like enzyme for Apg12p conjugation, facilitates MAP-LC3 modification. J Biol Chem 278(41):39517–39526. doi:10.1074/jbc.C200334200
Kaufmann A, Beier V, Franquelim HG, Wollert T (2014) Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell 156(3):469–481. doi:10.1016/j.cell.2013.12.022
Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y (2007) The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 282(52):37298–37302. doi:10.1074/jbc.C700195200
Tanida I, Yamasaki M, Komatsu M, Ueno T (2012) The FAP motif within human ATG7, an autophagy-related E1-like enzyme, is essential for the E2-substrate reaction of LC3 lipidation. Autophagy 8(1):88–97. doi:10.4161/auto.8.1.18339
Yamada Y, Suzuki NN, Hanada T, Ichimura Y, Kumeta H, Fujioka Y, Ohsumi Y, Inagaki F (2007) The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J Biol Chem 282(11):8036–8043. doi:10.1074/jbc.M611473200
Harris H, Rubinsztein DC (2012) Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 8(2):108–117. doi:10.1038/nrneurol.2011.200
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36(6):585–595. doi:10.1038/ng1362
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC (2008) Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4(5):295–305. doi:10.1038/nchembio.79
Pyo JO, Nah J, Jung YK (2012) Molecules and their functions in autophagy. Exp Mol Med 44(2):73–80. doi:10.3858/emm.2012.44.2.029
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16(1):46–56. doi:10.1038/cdd.2008.110
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. doi:10.1146/annurev-genet-102808-114910
Mehrpour M, Esclatine A, Beau I, Codogno P (2010) Overview of macroautophagy regulation in mammalian cells. Cell Res 20(7):748–762. doi:10.1038/cr.2010.82
Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL (2008) The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 4(7):851–865
Kaushik S, Cuervo AM (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22(8):407–417. doi:10.1016/j.tcb.2012.05.006
Chiang HL, Terlecky SR, Plant CP, Dice JF (1989) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246(4928):382–385
Li W, Yang Q, Mao Z (2011) Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mole Life Sci: CMLS 68(5):749–763. doi:10.1007/s00018-010-0565-6
Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28(18):5747–5763. doi:10.1128/MCB.02070-07
Agarraberes FA, Terlecky SR, Dice JF (1997) An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 137(4):825–834
Cuervo AM, Dice JF, Knecht E (1997) A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem 272(9):5606–5615
Finn PF, Mesires NT, Vine M, Dice JF (2005) Effects of small molecules on chaperone-mediated autophagy. Autophagy 1(3):141–145
Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, Wang G, Chen Z (2013) Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 9(9):1321–1333. doi:10.4161/auto.25132
Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G (2012) Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience 213:144–153. doi:10.1016/j.neuroscience.2012.03.055
Viscomi MT, D'Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F, Fanelli F, Maccarrone M, Moreno S, Cecconi F, Molinari M (2012) Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy 8(2):222–235. doi:10.4161/auto.8.2.18599
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441(7095):880–884. doi:10.1038/nature04723
Yamamoto A, Cremona ML, Rothman JE (2006) Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol 172(5):719–731. doi:10.1083/jcb.200510065
Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M (2012) Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain : J Neurol 135(Pt 7):2169–2177. doi:10.1093/brain/aws143
Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S (1996) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383(6602):710–713. doi:10.1038/383710a0
Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ (1997) Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med 3(1):67–72
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391(6665):387–390. doi:10.1038/34910
Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4(1):97–100
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. doi:10.1146/annurev.neuro.24.1.1121
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33(1):95–130
Ribe EM, Perez M, Puig B, Gich I, Lim F, Cuadrado M, Sesma T, Catena S, Sanchez B, Nieto M, Gomez-Ramos P, Moran MA, Cabodevilla F, Samaranch L, Ortiz L, Perez A, Ferrer I, Avila J, Gomez-Isla T (2005) Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol Dis 20(3):814–822. doi:10.1016/j.nbd.2005.05.027
Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293(5534):1487–1491. doi:10.1126/science.1058189
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS ONE 5(4):e9979. doi:10.1371/journal.pone.0009979
Tian Y, Bustos V, Flajolet M, Greengard P (2011) A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J: Off Publ Fed Am Soc Exp Biol 25(6):1934–1942. doi:10.1096/fj.10-175158
Jiang T, Yu JT, Zhu XC, Tan MS, Wang HF, Cao L, Zhang QQ, Shi JQ, Gao L, Qin H, Zhang YD, Tan L (2014) Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer's disease. Pharmacol Res : Off J Ital Pharmacol Soc 81:54–63. doi:10.1016/j.phrs.2014.02.008
Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, Solano RM, Gomez A, Perucho J, Cuervo AM, Garcia de Yebenes J, Mena MA (2010) Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis 39(3):423–438. doi:10.1016/j.nbd.2010.05.014
Kruger U, Wang Y, Kumar S, Mandelkow EM (2012) Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging 33(10):2291–2305. doi:10.1016/j.neurobiolaging.2011.11.009
Conway KA, Harper JD, Lansbury PT (1998) Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med 4(11):1318–1320. doi:10.1038/3311
Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A, French Parkinson's Disease Genetics Study G, European Consortium on Genetic Susceptibility in Parkinson's D (2000) Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 342(21):1560–1567. doi:10.1056/NEJM200005253422103
Periquet M, Latouche M, Lohmann E, Rawal N, De Michele G, Ricard S, Teive H, Fraix V, Vidailhet M, Nicholl D, Barone P, Wood NW, Raskin S, Deleuze JF, Agid Y, Durr A, Brice A, French Parkinson's Disease Genetics Study G, European Consortium on Genetic Susceptibility in Parkinson's D (2003) Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain : a journal of neurology 126(Pt 6):1271–1278
Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, Lynch T, Bhatia KP, Gasser T, Lees AJ, Wood NW, International LC (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case–control study. The Lancet Neurology 7(7):583–590. doi:10.1016/S1474-4422(08)70117-0
Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, Bacon JA, Bardien S, Bozi M, Brice A, Brighina L, Van Broeckhoven C, Carr J, Chartier-Harlin MC, Dardiotis E, Dickson DW, Diehl NN, Elbaz A, Ferrarese C, Ferraris A, Fiske B, Gibson JM, Gibson R, Hadjigeorgiou GM, Hattori N, Ioannidis JP, Jasinska-Myga B, Jeon BS, Kim YJ, Klein C, Kruger R, Kyratzi E, Lesage S, Lin CH, Lynch T, Maraganore DM, Mellick GD, Mutez E, Nilsson C, Opala G, Park SS, Puschmann A, Quattrone A, Sharma M, Silburn PA, Sohn YH, Stefanis L, Tadic V, Theuns J, Tomiyama H, Uitti RJ, Valente EM, van de Loo S, Vassilatis DK, Vilarino-Guell C, White LR, Wirdefeldt K, Wszolek ZK, Wu RM, Farrer MJ, Genetic Epidemiology Of Parkinson's Disease C (2011) Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case–control study. The Lancet Neurology 10(10):898–908. doi:10.1016/S1474-4422(11)70175-2
West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 102(46):16842–16847. doi:10.1073/pnas.0507360102
Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron 64(6):807–827. doi:10.1016/j.neuron.2009.11.006
Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ (2001) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson's disease. Science 293(5528):263–269. doi:10.1126/science.1060627
Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA (2005) Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A 102(51):18676–18681. doi:10.1073/pnas.0508052102
Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM (2001) Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med 7(10):1144–1150. doi:10.1038/nm1001-1144
Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278(27):25009–25013. doi:10.1074/jbc.M300227200
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci : Off J Soc Neurosci 29(43):13578–13588. doi:10.1523/JNEUROSCI.4390-09.2009
The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72(6):971–983
Sharp AH, Loev SJ, Schilling G, Li SH, Li XJ, Bao J, Wagster MV, Kotzuk JA, Steiner JP, Lo A et al (1995) Widespread expression of Huntington's disease gene (IT15) protein product. Neuron 14(5):1065–1074
Schilling G, Sharp AH, Loev SJ, Wagster MV, Li SH, Stine OC, Ross CA (1995) Expression of the Huntington's disease (IT15) protein product in HD patients. Hum Mol Genet 4(8):1365–1371
Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O'Kane CJ, Schreiber SL, Rubinsztein DC (2007) Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol 3(6):331–338. doi:10.1038/nchembio883
Yu X, Munoz-Alarcon A, Ajayi A, Webling KE, Steinhof A, Langel U, Strom AL (2013) Inhibition of autophagy via p53-mediated disruption of ULK1 in a SCA7 polyglutamine disease model. J Mole Neurosci : MN 50(3):586–599. doi:10.1007/s12031-013-0012-x
Nascimento-Ferreira I, Nobrega C, Vasconcelos-Ferreira A, Onofre I, Albuquerque D, Aveleira C, Hirai H, Deglon N, Pereira de Almeida L (2013) Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado-Joseph disease. Brain:J Neurol 136(Pt 7):2173–2188. doi:10.1093/brain/awt144
Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC (2010) Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain : J Neurol 133(Pt 1):93–104. doi:10.1093/brain/awp292
Kabuta T, Suzuki Y, Wada K (2006) Degradation of amyotrophic lateral sclerosis-linked mutant Cu, Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem 281(41):30524–30533. doi:10.1074/jbc.M603337200
Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH (2009) XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 23(19):2294–2306. doi:10.1101/gad.1830709
Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, Galbiati M, Fontana E, Marino M, Carra S, Bendotti C, De Biasi S, Poletti A (2010) The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 19(17):3440–3456. doi:10.1093/hmg/ddq257
Oz-Levi D, Ben-Zeev B, Ruzzo EK, Hitomi Y, Gelman A, Pelak K, Anikster Y, Reznik-Wolf H, Bar-Joseph I, Olender T, Alkelai A, Weiss M, Ben-Asher E, Ge D, Shianna KV, Elazar Z, Goldstein DB, Pras E, Lancet D (2012) Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet 91(6):1065–1072. doi:10.1016/j.ajhg.2012.09.015
Vantaggiato C, Crimella C, Airoldi G, Polishchuk R, Bonato S, Brighina E, Scarlato M, Musumeci O, Toscano A, Martinuzzi A, Santorelli FM, Ballabio A, Bresolin N, Clementi E, Bassi MT (2013) Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain: J Neurol 136(Pt 10):3119–3139. doi:10.1093/brain/awt227
Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S (2005) Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 24(6):980–991. doi:10.1038/sj.onc.1208095
Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, Choi KS (2007) Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res 67(13):6314–6324. doi:10.1158/0008-5472.CAN-06-4217
Dohi E, Tanaka S, Seki T, Miyagi T, Hide I, Takahashi T, Matsumoto M, Sakai N (2012) Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem Int 60(4):431–442. doi:10.1016/j.neuint.2012.01.020
Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, Kirik D, Stefanis L (2013) Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain: J Neurol 136(Pt 7):2130–2146. doi:10.1093/brain/awt131
Liu H, Wang P, Song W, Sun X (2009) Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J: Off Publ Fed Am Soc Exp Biol 23(10):3383–3392. doi:10.1096/fj.09-134296
Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH (2012) The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 7(10):e46834. doi:10.1371/journal.pone.0046834
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305(5688):1292–1295. doi:10.1126/science.1101738
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L (2008) Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283(35):23542–23556. doi:10.1074/jbc.M801992200
Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA (2010) Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem 285(18):13621–13629. doi:10.1074/jbc.M109.074617
Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L (2009) Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 4(5):e5515. doi:10.1371/journal.pone.0005515
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM (2008) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118(2):777–788. doi:10.1172/JCI32806
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH (2010) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 67(12):1464–1472. doi:10.1001/archneurol.2010.198
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16(4):394–406. doi:10.1038/nn.3350
Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper JM (2013) Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson's disease. Cell Death Dis 4:e545. doi:10.1038/cddis.2013.73
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18(21):4153–4170. doi:10.1093/hmg/ddp367
Koga H, Cuervo AM (2011) Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis 43(1):29–37. doi:10.1016/j.nbd.2010.07.006
Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, Miyazaki H, Matsumoto G, Kino Y, Nagai Y, Nukina N (2010) Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol 28(3):256–263. doi:10.1038/nbt.1608
Boya P, Reggiori F, Codogno P (2013) Emerging regulation and functions of autophagy. Nat Cell Biol 15(7):713–720. doi:10.1038/ncb2788
Massey AC, Kaushik S, Cuervo AM (2006) Lysosomal chat maintains the balance. Autophagy 2(4):325–327
Cuervo AM, Knecht E, Terlecky SR, Dice JF (1995) Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol 269(5 Pt 1):C1200–C1208
Orenstein SJ, Cuervo AM (2010) Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol 21(7):719–726. doi:10.1016/j.semcdb.2010.02.005
Kaushik S, Massey AC, Mizushima N, Cuervo AM (2008) Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 19(5):2179–2192. doi:10.1091/mbc.E07-11-1155
Wang Y, Martinez-Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E (2010) Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy 6(1):182–183
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24(1):92–104. doi:10.1038/cr.2013.153
Kiffin R, Christian C, Knecht E, Cuervo AM (2004) Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 15(11):4829–4840. doi:10.1091/mbc.E04-06-0477
Zhang C, Cuervo AM (2008) Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med 14(9):959–965. doi:10.1038/nm.1851
Kroemer G, Jaattela M (2005) Lysosomes and autophagy in cell death control. Nat Rev Cancer 5(11):886–897. doi:10.1038/nrc1738
Bains M, Florez-McClure ML, Heidenreich KA (2009) Insulin-like growth factor-I prevents the accumulation of autophagic vesicles and cell death in Purkinje neurons by increasing the rate of autophagosome-to-lysosome fusion and degradation. J Biol Chem 284(30):20398–20407. doi:10.1074/jbc.M109.011791
Massey AC, Follenzi A, Kiffin R, Zhang C, Cuervo AM (2008) Early cellular changes after blockage of chaperone-mediated autophagy. Autophagy 4(4):442–456
Viscomi MT, D'Amelio M (2012) The “Janus-faced role” of autophagy in neuronal sickness: focus on neurodegeneration. Mol Neurobiol 46(2):513–521. doi:10.1007/s12035-012-8296-3
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci : Off J Soc Neurosci 28(27):6926–6937. doi:10.1523/JNEUROSCI.0800-08.2008
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118(6):2190–2199. doi:10.1172/JCI33585
Lee H, Noh JY, Oh Y, Kim Y, Chang JW, Chung CW, Lee ST, Kim M, Ryu H, Jung YK (2012) IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet 21(1):101–114. doi:10.1093/hmg/ddr445
Reijonen S, Putkonen N, Norremolle A, Lindholm D, Korhonen L (2008) Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp Cell Res 314(5):950–960. doi:10.1016/j.yexcr.2007.12.025
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 13(5):567–576. doi:10.1038/nn.2528
Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM (2011) Constitutive upregulation of chaperone-mediated autophagy in Huntington's disease. J Neurosci : Off J Soc Neurosci 31(50):18492–18505. doi:10.1523/JNEUROSCI.3219-11.2011
Cuervo AM, Dice JF (2000) Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci 113(Pt 24):4441–4450
Singh R, Czaja MJ (2008) Compensatory mechanisms and the type of injury determine the fate of cells with impaired macroautophagy. Autophagy 4(4):516–518
Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T, Xiang Y, Cuervo AM, Czaja MJ (2008) Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem 283(8):4766–4777. doi:10.1074/jbc.M706666200
Rodriguez-Muela N, Koga H, Garcia-Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, Boya P (2013) Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 12(3):478–488. doi:10.1111/acel.12072
Wang Y, Singh R, Xiang Y, Czaja MJ (2010) Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 52(1):266–277. doi:10.1002/hep.23645
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479. doi:10.1146/annurev.biochem.67.1.425
Yi JJ, Ehlers MD (2007) Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev 59(1):14–39. doi:10.1124/pr.59.1.4
Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ (2008) Impaired ubiquitin-proteasome system activity in the synapses of Huntington's disease mice. J Cell Biol 180(6):1177–1189. doi:10.1083/jcb.200709080
de Pril R, Fischer DF, Maat-Schieman ML, Hobo B, de Vos RA, Brunt ER, Hol EM, Roos RA, van Leeuwen FW (2004) Accumulation of aberrant ubiquitin induces aggregate formation and cell death in polyglutamine diseases. Hum Mol Genet 13(16):1803–1813. doi:10.1093/hmg/ddh188
Liu CL, Martone ME, Hu BR (2004) Protein ubiquitination in postsynaptic densities after transient cerebral ischemia. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow and Metab 24(11):1219–1225. doi:10.1097/01.WCB.0000136706.77918.21
Schreiber A, Peter M (2014) Substrate recognition in selective autophagy and the ubiquitin-proteasome system. Biochim Biophys Acta 1843(1):163–181. doi:10.1016/j.bbamcr.2013.03.019
Nedelsky NB, Todd PK, Taylor JP (2008) Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta 1782(12):691–699. doi:10.1016/j.bbadis.2008.10.002
Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447(7146):859–863. doi:10.1038/nature05853
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM (2007) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171(2):513–524. doi:10.2353/ajpath.2007.070188
Zhu K, Dunner K Jr, McConkey DJ (2010) Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29(3):451–462. doi:10.1038/onc.2009.343
Wang XJ, Yu J, Wong SH, Cheng AS, Chan FK, Ng SS, Cho CH, Sung JJ, Wu WK (2013) A novel crosstalk between two major protein degradation systems: regulation of proteasomal activity by autophagy. Autophagy 9(10):1500–1508. doi:10.4161/auto.25573
Quintavalle C, Di Costanzo S, Zanca C, Tasset I, Fraldi A, Incoronato M, Mirabelli P, Monti M, Ballabio A, Pucci P, Cuervo AM, Condorelli G (2014) Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J Cell Physiol 229(10):1359–1368. doi:10.1002/jcp.24569
Xiao C, Yang BF, Asadi N, Beguinot F, Hao C (2002) Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-FLIP and PED/PEA-15 in glioma cells. J Biol Chem 277(28):25020–25025. doi:10.1074/jbc.M202946200
Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, Aghi MK (2012) Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 72(7):1773–1783. doi:10.1158/0008-5472.CAN-11-3831
Yang F, Yang YP, Mao CJ, Liu L, Zheng HF, Hu LF, Liu CF (2013) Crosstalk between the proteasome system and autophagy in the clearance of alpha-synuclein. Acta Pharmacol Sin 34(5):674–680. doi:10.1038/aps.2013.29
Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA, Monteiro MJ (2010) Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet 19(16):3219–3232. doi:10.1093/hmg/ddq231
N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ (2009) PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 10(2):173–179. doi:10.1038/embor.2008.238
Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV (2003) Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med 9(3):338–342. doi:10.1038/nm826
Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A (2005) p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron 47(1):29–41. doi:10.1016/j.neuron.2005.06.005
Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavanee W, Vogelstein B (1992) Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 355(6363):846–847. doi:10.1038/355846a0
Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G (2008) A dual role of p53 in the control of autophagy. Autophagy 4(6):810–814
Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G (2010) Autophagy regulation by p53. Curr Opin Cell Biol 22(2):181–185. doi:10.1016/j.ceb.2009.12.001
Zhang XD, Wang Y, Wang Y, Zhang X, Han R, Wu JC, Liang ZQ, Gu ZL, Han F, Fukunaga K, Qin ZH (2009) p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 5(3):339–350
Gan L, Vargas MR, Johnson DA, Johnson JA (2012) Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci: Off J Soc Neurosci 32(49):17775–17787. doi:10.1523/JNEUROSCI.3049-12.2012
Acknowledgments of Funding
This work was supported by National Natural Science Foundation of China (grant 81171096, 81371433, 81271273, and 81371369), Research Fund for the Doctoral Program of Higher Education of China (20120101120030), Zhejiang Provincial Medical Science and Technology Planning Project (grant 2012RCA030 and 2013KYA088), Zhejiang Provincial Education Planning Project (Y201226287), and Zhejiang Provincial Natural Science Foundation of China (grant Y13H090007 and LY13H090002).
Conflict of interest
No potential conflicts of interest were disclosed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Haijian Wu and Sheng Chen provided equal contribution to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Wu, H., Chen, S., Ammar, AB. et al. Crosstalk Between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol Neurobiol 52, 1284–1296 (2015). https://doi.org/10.1007/s12035-014-8933-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8933-0