
Abstract
Alpha-synuclein (aSyn) is implicated in Parkinson’s disease and several other neurodegenerative disorders. To date, the function and intracellular dynamics of aSyn are still unclear. Here, we tracked the dynamics of aSyn using photoactivatable green fluorescent protein as a reporter. We found that the availability of the aSyn N terminus modulates its shuttling into the nucleus. Interestingly, familial aSyn mutations altered the dynamics at which the protein distributes throughout the cell. Both the A30P and A53T aSyn mutations increase the speed at which the protein moves between the nucleus and cytoplasm, respectively. We also found that specific kinases potentiate the shuttling of aSyn between nucleus and cytoplasm. A mutant aSyn form that blocks S129 phosphorylation, S129A, results in the formation of cytoplasmic inclusions, suggesting phosphorylation modulates aggregation in addition to modulating aSyn intracellular dynamics. Finally, we found that the molecular chaperone HSP70 accelerates the entry of aSyn into the nuclear compartment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Misfolded and aggregated alpha-synuclein (aSyn) is the major component of intraneuronal inclusions known as Lewy bodies (LBs), the pathological hallmark of Parkinson’s disease (PD) and other synucleinopathies [1]. Despite the growing knowledge on aSyn, the normal function of the protein remains largely unclear. However, it is thought to play a role in synaptic function and plasticity, cell differentiation and vesicular trafficking [2, 3]. The subcellular distribution of aSyn is also controversial and, although it is considered a pre-synaptic protein, it has also been found to be evenly distributed throughout the cells in different cellular models and in mice [4–8].
Although the majority of PD cases are idiopathic, three missense mutations in aSyn gene (A30P, E46K and A53T), restrained in the N-terminal domain, have been identified in rare, autosomal-dominant inherited forms of PD, as well as duplications and triplications of the aSyn-containing locus [9–12]. In vitro studies revealed that the A30P mutation blocks the membrane association and inhibits the synaptic localisation of aSyn by destabilising its first helical structure [5, 13]. Conversely, A53T and E46K mutations enhance the binding to phospholipids [14].
In the normal brain, 4 % of aSyn is phosphorylated at serine 129 (S129), contrasting with 90 % of aSyn that is found to be phosphorylated in LBs. This suggests that S129 phosphorylation might interfere with the oligomerisation and aggregation process and contribute to the pathogenesis of PD [15, 16]. Among others, G protein-coupled receptor kinases (GRKs) and Polo-like kinases (PLKs) were found to phosphorylate the S129 residue of aSyn [17, 18]. Although it was proposed that S129 phosphorylation inhibits aSyn-induced regulation of tyrosine hydroxylase activity [19], the exact role of this post-translational modification (PTM) in both physiological and pathological conditions remains unclear.
Other known modifiers of aSyn aggregation are molecular chaperones, such as HSP70, which modulates the misfolding, aggregation and toxicity of aSyn in different model systems [4, 20–23]. However, the mechanisms by which HSP70 suppresses aSyn toxicity are still unclear.
The new era of time-lapse bioimaging tools combined with GFP-derived fluorescent labels enables the characterisation of protein kinetics in real time, providing invaluable insights into the molecular processes in which they are involved. Photoactivation (PA) microscopy is an emerging technique in the field of neuroscience [24] in which a non-fluorescent molecule is converted into an activated and fluorescent state by the use of an intense and brief irradiation in a selected region of the cell. This process enables the direct tracking of a protein by photo-inducing fluorescence, instead of interfering with a steady state fluorescent signal, as photobleaching (PB) methods do. The newly activated pool, obtained through the use of an ultraviolet laser, contrasts with a background of non-activated molecules and can be followed within the cells as they reach their kinetics equilibrium [25, 26] (Fig. 1a).

Strategy for nuclear and cytoplasmic photoactivation (PA) of PAGFP-labelled proteins. a PAGFP displays negligible fluorescence in the spectral range where the activated fluorescence is detected. Upon PA of a selected nuclear or cytoplasmic region with a 405 laser, PAGFP-labelled proteins become visible and the dynamics and fate of the activated molecules can be followed over time. b PAGFP constructs used in this study. We used WT, A30P, E46K, A53T and S129A aSyn variants
In order to further understand the biology of aSyn, we investigated the dynamics of aSyn between the nucleus and cytoplasm in living cells using PA microscopy (Fig. 1b). We found that the N-terminal of aSyn-wild-type (WT) determines its entry into the nuclear compartment. Moreover, aSyn shuttles between the nucleus and cytoplasm at rates which depend on mutations, phosphorylation state and on the presence of HSP70. Altogether, our novel approach provides novel insights into the biology of aSyn in living cells and may enable the development of novel strategies for therapeutic intervention in synucleinopathies.
Results
Blocking the N-terminal of aSyn Modulates Nuclear Localisation
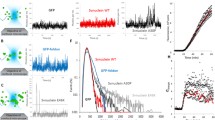
To assess whether the subcellular localisation of aSyn-WT is affected by appending different tags to either the N or C terminus, we performed immunocytochemistry analysis in cells expressing either untagged aSyn-WT or GFP, Myc- or V5-tagged versions. Both tagged and non-tagged aSyn-WT were widely distributed throughout the cell, including the nucleus (Fig. 2a–d). To further investigate the intracellular dynamics of aSyn-WT, we generated fusions with a photoactivatable green fluorescent protein (PAGFP) in order to follow the movement of a specific pool of aSyn over time at the N or C termini (aSyn-WT-PAGFP and PAGFP-aSyn-WT, respectively). H4 cells were transiently transfected with plasmids encoding aSyn-WT-PAGFP, PAGFP-aSyn-WT or PAGFP alone, as a control. As aSyn is widely distributed in the cell, we characterised the shuttling of aSyn between the nucleus and the cytoplasm. Reporter proteins were photoactivated in the nucleus or cytoplasm for 2 s using a 405-nm laser, their cellular trafficking was monitored and the fluorescence intensities quantified. After PA, the PAGFP control was quickly detected in the cytoplasm or in the nucleus after nuclear or cytoplasmic PA, respectively, reaching an equilibrium of PAGFP molecules between the two cellular compartments after 500 s. In contrast, we observed different trafficking behaviours for aSyn-WT-PAGFP and PAGFP-aSyn-WT (Figs. 2e and S1a; Videos S1, S2, S3, S4, S5 and S6). aSyn-WT-PAGFP displayed different dynamics depending on the region where PA was performed. Upon cytoplasmic PA, aSyn-WT-PAGFP molecules entered into the nucleus and after 1,000 s were evenly distributed between the two subcellular compartments. Conversely, upon nuclear PA, aSyn-WT-PAGFP was maintained in this compartment for the remainder of the time analysed (1,000 s). In contrast with the behaviour observed for photoactivated aSyn-WT-PAGFP, PAGFP-aSyn-WT was not detected in the contiguous compartment to where the PA was performed and remained in the same subcellular region (Figs. 2e and S1a). Immunoblotting analysis of nuclear and cytoplasmic extracts from cells expressing either protein confirmed the predominant localisation observed with microscopy (Fig. S2a). To further validate the observations obtained using PA, we performed fluorescence recovery after PB (FRAP) experiments in H4 cells expressing aSyn-WT-GFP, GFP-aSyn-WT or GFP alone. Upon PB of aSyn-WT-GFP in the nucleus, we found that the recovery of fluorescence in this compartment occurred after 500 s while fluorescence was not significantly recovered in the cytoplasm after PB in this region. For GFP-aSyn-WT, PB in nucleus slightly recovered fluorescence of the reporter protein after 1,000 s (about 20 % of recovery) while no cytoplasmic recovery was observed upon PB in this region (Fig. 2f; Videos S7, 8 and 9).

Blocking the N terminus of aSyn reduces its shuttling into the nucleus. Immunofluorescence imaging of H4 cells showing the subcellular localisation of transiently transfected aSyn a N- and C-terminally tagged to Myc and V5, respectively, b tagged on both terminals with Myc (N-terminal) and V5 (C-terminal) and c untagged aSyn-WT. d Live cell imaging of aSyn N- and C-terminally tagged to GFP. e Measurements of fluorescence intensities over time in the nucleus (light grey line) and in the cytoplasm (dark grey line) of control PAGFP (dashed line) or fusion proteins of aSyn-WT with PAGFP (solid line). Values represent mean + SD of up to 15 cells analysed per condition. f Measurements of fluorescence recovery after PB over time in the nucleus (light grey line) and in the cytoplasm (dark grey line) of control GFP (dashed line) or fusion proteins of aSyn-WT with GFP (solid line). Scale bars, 10 μm
Altogether, our novel PA and FRAP experiments show, for the first time, that the movement of aSyn between the nucleus and the cytoplasm depends on the availability of the N terminus of the protein.
PD-Associated Mutations Alter the Subcellular Trafficking of aSyn
Since we established a model to study aSyn intracellular dynamics, we next investigated whether PD familial mutations (A30P, E46K and A53T) in aSyn affected its trafficking in the cell (Fig. 3a). In comparison to the aSyn-WT-PAGFP, cytoplasmic aSyn-A30P- and aSyn-E46K-PAGFP are shuttled into the nucleus in half of the time (500 s) of the WT protein (at time points 500 and 1,000 s after PA, p values between 0.0044 and <0.0001, respectively, α = 0.05; Table SIa). Because of this rapid movement into the nucleus, the fluorescence intensities in the cytoplasm and nucleus equalised earlier than in the control situation and remained in equilibrium for several minutes. In contrast to A30P and E46K mutants, aSyn-A53T-PAGFP remained in the compartment where the PA was performed, similarly to the WT protein.

Effect of aSyn mutations on its subcellular trafficking in living cells. a Fluorescence intensities after PA in the nucleus (light grey line) and in the cytoplasm (dark grey line) of aSyn-A30P-, aSyn-E46K- and aSyn-A53T PAGFP-tagged proteins (solid line) over time. Fluorescence intensities of photoactivated control aSyn-WT PAGFP fusion proteins are shown in dashed line. Values represent mean + SD of up to 15 cells analysed per condition. b Immunobloting analysis of total aSyn levels in cells expressing aSyn-WT and mutant-aSyn reporter proteins
In the fusions where PAGFP was on the N terminus of aSyn, the behaviour of A30P was similar to that of aSyn-WT. Additionally, upon cytoplasmic PA, there was a slight increase in molecules that remained on the nucleus (p value = 0.0185 at 1,000 s after nuclear or cytoplasmic PA, α = 0.05; Table SIa; Fig. 3a). In contrast, PAGFP-aSyn-E46K was translocated from the nucleus to the cytoplasm after nuclear PA (p value = 0.001 at 1,000 s after nuclear PA, α = 0.05, Table SIa). The same occurred with PAGFP-aSyn-A53T, although at a more pronounced rate, since the fluorescence intensity was higher in the cytoplasm than in the nucleus, starting at 700 s after PA (p value = 0.0013 at 1,000 s after PA, α = 0.05; Table SIa; and Fig. 3a). In order to investigate if the dynamics results were influenced by differences in the levels of WT and mutant aSyn, we performed immunobloting analysis. We verified that the levels of total aSyn did not differ between the WT and mutant forms of the protein (Fig. 3b). As expected, subcellular fractionation followed by immunobloting analysis confirmed the presence of the protein in both cytoplasmic and nuclear compartments (Fig. S2b).
Altogether, these experiments show that A30P and E46K, but not the A53T mutation, promoted a faster shuttling of aSyn with a free N terminus into the nucleus when compared with aSyn-WT. On the other hand, aSyn-A53T and aSyn-E46K with a free C terminus, but not aSyn-A30P, were delayed in the cytoplasm.
aSyn Phosphorylation by GRK5 or PLK2 Modulates Its Subcellular Trafficking
In order to assess the role of S129 aSyn phosphorylation on the intracellular dynamics of the protein, we co-expressed GRK2, GRK5, PLK2 or PLK3 kinases with aSyn-WT tagged with PAGFP on either its N- or C-terminal. The kinases tested did not significantly alter the dynamics of aSyn-WT-PAGFP. For aSyn-WT-PAGFP, co-expression with PLK2 did not alter the dynamics obtained in the absence of the kinase, but resulted in a more rapid progression towards the equilibrium fluorescence. The other kinases tested, GRK2, GRK5 and PLK3, neither did not induce significant differences on aSyn-WT-PAGFP (Table SIb; Fig. S3).
In contrast, stronger effects were observed for PAGFP-aSyn-WT. Overexpression of GRK2 did not affect the shuttling of aSyn-WT constructs between the nucleus and the cytoplasm as the values of fluorescence intensity were similar in the presence or absence of the kinase during the 1,000 s of imaging. Interestingly, GRK5 promoted the trafficking of PAGFP-aSyn-WT into the nucleus upon cytoplasmic PA, although the difference did not reach statistical significance. Nuclear PA in cells overexpressing GRK5 did not significantly alter the dynamics of PAGFP-aSyn-WT (Fig. 4a; Table SIb).

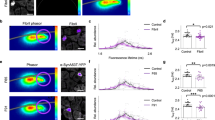
Effect of S129 phosphorylation on the subcellular dynamics of aSyn. a Fluorescence intensities after PA in the nucleus (light grey line) and in the cytoplasm (dark grey line) of PAGFP-aSyn-WT fusion protein co-expressed with GRK2, GRK5, PLK2 and PLK3 (solid line) or an empty vector (dashed lines) over time. Fluorescence intensities of photoactivated control PAGFP-aSyn-WT construct after co-transfection with an empty vector are shown by a dashed line. Values represent mean + SD of up to15 cells analysed per condition. b Immunobloting analysis of total aSyn levels in cells co-expressing aSyn-WT reporter proteins with the four tested kinases. c Fluorescence intensities after PA in the nucleus (light grey line) and in the cytoplasm (dark grey line) of PAGFP-aSyn-S129A fusion protein over time. Fluorescence intensities of photoactivated control PAGFP-aSyn-WT are shown in dashed lines. Values represent mean + SD of up to15 cells analysed per condition. d Immunobloting analysis of total aSyn levels in cells expressing PAGFP-aSyn-WT or PAGFP-aSyn-S129A. e Cytosolic inclusions in cells expressing PAGFP-aSyn-S129A. Images were taken 500 s after PA in the nucleus in order to detect the cytosolic inclusions. Scale bar, 10 μm
Co-expression of PAGFP-aSyn-WT with PLK2 promoted its shuttling to the cytoplasm upon nuclear PA (similar nuclear and cytoplasmic fluorescence levels were reached 500 s after PA, p value = 0.0141, α = 0.05; Table SIb; Fig. 4a). Upon cytoplasmic PA, PLK2 slightly accelerated the movement into the nucleus 500 s after PA (Table SIb).
Co-expression of PAGFP-aSyn-WT with PLK3 promoted its shuttling to the cytoplasm upon nuclear PA but this movement was faster in the presence of PLK2. Although the difference was already significant 500 s after PA (p value = 0.0198, α = 0.05; Table SIb), at 1,000 s the levels of fluorescence were still higher in the nucleus than in the cytoplasm. Upon cytoplasmic PA, the presence of PLK3 did not alter the dynamics of PAGFP-aSyn-WT (Fig. 4a; Table SIb). We performed immunobloting analysis to investigate if the results on the dynamics of aSyn were influenced by differences in expression in the presence and absence of the kinases tested. We verified that the levels of total aSyn were not altered in the presence of the kinases (Fig. 4b). As expected, we also confirmed that the levels of aSyn phosphorylated at S129 were increased in the presence of both kinases (Fig. S2c).
An important difference between GRK5 and PLKs was that the former induced a tendency of aSyn to traffic from the cytoplasm to the nucleus and the latter had a strong effect in promoting the trafficking of aSyn from the nucleus to the cytoplasm.
Next, we tested the dynamics of S129A aSyn, a phosphorylation-incompetent mutant, in order to further assess the effect of S129 phosphorylation in the trafficking of aSyn. No differences were observed on the dynamics of aSyn-S129A-PAGFP except for the existence of more photoactivated protein in the nucleus when compared with aSyn-WT immediately after nuclear PA (Fig. S3d; Table SIa). However, PAGFP-aSyn-S129A moved into the cytoplasm after 1,000 s of PA if PA was performed in the nucleus while the aSyn-WT did not. Moreover, PAGFP-aSyn-S129A remained in the cytoplasm if PA was performed in that compartment (p value = 0.0099, α = 0.05; Table SIa; Fig. 4c). These findings were in agreement with the fact that both aSyn-S129A fusion proteins were only marginally detected in the nuclear protein fraction, in contrast to aSyn-WT (Figs. S2a, S2d and S3e), although the total protein levels of aSyn-WT and aSyn-S129A were comparable (Fig. 4d; Fig. S3f).
Interestingly, we found that expression of either the N- or C-terminal S129A fusion proteins promoted the formation of cytosolic inclusions scattered around the nucleus (Fig. 4e; Fig. S3g).
HSP70 Modifies the Trafficking of aSyn
HSP70 modulates the accumulation of oligomeric and aggregated forms of aSyn in different model systems. Thus, we next asked whether HSP70 could interfere with the subcellular dynamics of aSyn. HSP70 did not change the intracellular dynamics of aSyn-WT-PAGFP (Table SIb; Fig. 5a). Conversely, upon cytoplasmic PA, PAGFP-aSyn-WT was shuttled into the nucleus in the presence of HSP70 within 100 s after PA (p value < 0.0001, α = 0.05; Table SIb; Fig. S1b; Fig. 5a). Although the total levels of the protein were not altered in the presence of the chaperone (Fig. 5b), the levels of aSyn were higher in the nucleus for both fusion proteins in this situation. Interestingly, HSP70 was present in the nuclear fraction only when aSyn was present (Fig. S2e). Upon nuclear PA, the tendency was for the protein to move into the cytoplasm, but not as quickly as in the former situation (p value at 100 s = 0.7836, p value at 1,000 s = 0.0014, α = 0.05; Table SIb; Fig. S1b; Fig. 5a).

Modulation of the dynamics of aSyn by HSP70. a Fluorescence intensities after PA in the nucleus (light grey) and in the cytoplasm (dark grey) of aSyn-WT PAGFP fusion proteins co-expressed with HSP70 (solid line) over time. Fluorescence intensities of photoactivated control aSyn-WT reporters after co-transfection with an empty vector are shown in dashed lined. Values are mean + SD of up to 15 cells analysed per condition. b Immunobloting analysis of total aSyn levels in cells co-expressing aSyn with HSP70 or with an empty vector. Scale bar, 10 μm
In summary, HSP70 increased the shuttling of PAGFP-aSyn-WT between the nucleus and cytoplasm.
Discussion
Here, we investigated the intracellular dynamics of aSyn in living cells using photoactivatable GFP as a reporter. To control for putative effects of tagging aSyn in particular domains, we engineered fusions with PAGFP on either the N- or C-terminal of aSyn. We found that, although both aSyn fusion proteins were evenly spread throughout the cell, aSyn required a free N terminus in order to move from the cytoplasm into the nucleus.
aSyn is evenly distributed throughout the cell in different in vitro models and in mice [4–8]. The N terminus of aSyn seems to be important for membrane binding in various model organisms, ranging from yeast to rats [14, 27–29]. Although the role of aSyn in the nucleus has not been defined, it is described to interact with histones, inhibiting acetylation and enhancing chromatin binding, and promoting neurotoxicity in cellular models, in mouse nigral neurons and in Drosophila [7, 30, 31].
We also investigated whether aSyn mutations associated with familial forms of PD altered the shuttling of aSyn between the nucleus and cytoplasm. We found that (a) the A30P mutant is more prone to be located in the nucleus than the aSyn-WT; (b) the E46K mutant loses the subcellular compartmentalisation characteristic of the WT form; and (c) the A53T mutation is more prone to be located in the cytoplasm than aSyn-WT.
Until recently, aSyn was thought to be an intrinsically unfolded protein [32–34]. Nevertheless, it acquires two α-helical structures upon interaction with vesicles, contained in the residues 1–42 and from 45 to 98 [35–38]. In vitro studies showed that A30P disrupts membrane binding [5, 13], perhaps being more available to shuttle into the nucleus. Thus, it is likely that the differences in dynamics between the aSyn familial forms are related with the location and effect of the mutation on the secondary structure of aSyn protein. In addition, aSyn seems to regulate actin bundling inside the cell, and the A30P mutant affects the structure and dynamics of the actin cytoskeleton, potentiating the formation of actin foci [39]. Our results are also consistent with data showing that the A30P increases the nuclear localisation of the protein [30]. A53T is described to promote the formation of cytosolic aggregates [5, 40], which is compatible with its tendency to be localised in the cytoplasm when compared with aSyn-WT.
PTMs are known to modulate the intracellular fate of proteins, including their sub-cellular distribution. aSyn is thought to have five residues prone to phosphorylation: S87, Y125, S129, Y133 and Y136. S129 phosphorylation is the most studied, and little information exists on the kinases phosphorylating the other residues [41, 42]. In LBs, the majority of aSyn is thought to be phosphorylated on S129, contrasting with almost no phosphorylation of this residue in normal brain, However, the role of this PTM is still unclear and controversial. Phosphorylation of aSyn by GRKs inhibits its interaction with phospholipids [43]. In Drosophila, co-expression of GRK2 with aSyn leads to S129 phosphorylation and enhanced aSyn neurotoxicity [44]. Moreover, the levels of specific PLKs are increased in brains of patients with Alzheimer’s or LB disease [45].
Here, we investigated whether a selected group of kinases, GRK2, GRK5, PLK2 and PLK3, modulated the dynamics of aSyn distribution in the cell. The kinases tested only affected the dynamics of aSyn with a free C terminus, although both fusion proteins were phosphorylated in S129. This can be due to the fact that when the C-terminal of aSyn is free the protein is more prone to phosphorylation at S129, resulting in a stronger effect in its intracellular dynamics. Overall, aSyn phosphorylation by GRKs or PLKs results in different dynamics of the protein. While GRK5 potentiates the nuclear localisation of aSyn, PLKs modulate the shuttling of the protein between the nucleus and cytoplasm. In particular, PLK2 modulates the intracellular dynamics of PAGFP-aSyn-WT by increasing the movement from the nucleus to the cytoplasm at a higher rate than PLK3. Since aSyn has more residues prone to phosphorylation, the different results obtained with GRK5 and PLKs might reflect different phosphorylation patterns in residues other than S129. Due to the limited availability of antibodies these studies are still not easy to perform but as novel tools become available one might be able to discriminate between the effects of phosphorylation in different residues.
Interestingly, we also observed that nuclear PAGFP-aSyn-S129A tends to move to the cytoplasm while cytoplasmic PAGFP-aSyn-S129A remains in this subcellular compartment. This tendency might at least partially explain the cytoplasmic inclusions detected in the cells expressing this mutant aSyn and suggest that the phosphorylation status on S129 is crucial for aggregation, in agreement with recent findings in yeast, in which S129A mutant potentiates the formation of aSyn foci [46]. GRKs and PLKs modulate the dynamics of aSyn in different ways and we did not find a consistent pattern that can explain the role of S129 phosphorylation on the distribution of aSyn. One possibility is that the effects of the kinases are also due to phosphorylation of other targets in addition to aSyn. Nevertheless, we verified that the phosphorylation status of aSyn on S129 was related with the aggregation state of the protein.
Molecular chaperones, such as HSP70, hold great potential as therapeutic targets due to their ability to reverse protein aggregation and to refold or promote degradation of misfolded proteins [47]. HSP70 was shown to inhibit formation of toxic pre-fibrillar forms of aSyn [23] and to reduce its aggregation in aSyn transgenic mice [22]. In flies, it was shown that co-expression of HSP70 with aSyn-WT suppresses the loss of dopaminergic neurons, and hence, the toxicity associated with aSyn-WT overexpression [20]. A similar effect was observed in yeast and in mammalian cell models, suggesting that HSP70 inhibits aSyn toxicity by binding to the exposed hydrophobic NAC domain (non-A beta component of AD plaques; residues 61–95 of aSyn) and sequestering the protein [21, 48–50].
Here, we found that HSP70 boosted the shuttling of PAGFP-aSyn-WT to the adjacent compartment, suggesting it may assist aSyn to adopt a conformation that is more likely to cross the nuclear envelope. The selective effect with PAGFP-aSyn-WT and not with aSyn-WT-PAGFP suggests the interaction might take place through the C terminus of aSyn, which is not blocked by PAGFP in this fusion protein.
In conclusion, we showed that PD-associated mutations in aSyn, S129 phosphorylation, and HSP70 exert different effects on aSyn trafficking within the cell (Table SII; Fig. 6). While additional studies will be important to clarify the relative contribution of each condition, our goal was to demonstrate the usefulness of PA microscopy for the study of aSyn dynamics in living cells, which is not possible to achieve with other types of approaches or with untagged protein.

Modifiers of aSyn intracellular dynamics. aSyn-WT shuttles into the nucleus. This is enhanced (thicker arrow) in the presence of A30P mutation or GRK5 kinase, via phosphorylation of S129. The mutants A53T and S129A, or overexpression of PLK2 and PLK3 kinases, promote the bidirectional shuttling between the nuclear and the cytoplasmic compartments. E46K mutation and HSP70 chaperone instigate a similar dynamics; however, they promote a faster shutting into the nucleus (thicker arrow)
Our data provide novel insights into the subcellular dynamics of aSyn by taking advantage of a powerful method for monitoring protein dynamics in living cells. A complete understanding of aSyn localisation, intracellular dynamics and protein–protein interactions will be crucial for understanding the normal function of aSyn and may enable the development of novel strategies for intervention in PD and other synucleinopathies.
Materials and Methods
Plasmids and Cloning Procedures
aSyn-WT-PAGFP and PAGFP-aSyn-WT constructs were generated using aSyn-GC and GN-link-aSyn pcDNA3.1 vectors [51], respectively, and verified by DNA sequencing.
In order to obtain PAGFP-aSyn-WT construct, PAGFP in C1 vector was amplified by PCR with primers 5′TAAGCTAGCATGGTGAGCAAGGGCGAGG3′ (which contains a NheI restriction site) and 5′GGACTTAAGCTTGTACAGCTCGTCCATGCC3′ (which contains a AflII restriction site and eliminates the stop codon from PAGFP). PAGFP PCR product and GN-link-aSyn were digested with NheI and AflII and ligated using T4 DNA ligase.
To obtain aSyn-WT-PAGFP, PAGFP in C1 vector was PCR amplified with the primers 5′GGGTCTAGACTATTACTTGTACAGCTCGTCCATGCC3′ (which contains a XhoI restriction site and eliminates ATG site from PAGFP) and 5′GTATCTAGACTATTACTTGTACAGCTCGTCCATGCC3′ (which contains a XbaI restriction site and the stop codon of PAGFP). PAGFP PCR product and aSyn-GC were digested with XhoI/XbaI, and ligated using T4 DNA ligase.
aSyn-A30P/E46K/A53T-PAGFP and PAGFP-aSyn-A30P/E46K/A53T constructs were generated using aSyn-WT-PAGFP and PAGFP-aSyn-WT plasmids as backbone. WT-aSyn was eliminated from aSyn-WT-PAGFP and PAGFP-aSyn-WT plasmids through NheI/XhoI and AflII/XhoI sites, respectively. PD-associated aSyn mutant forms were obtained from aSyn-BiFC plasmids [51] and were inserted in the PAGFP backbone vectors.
Mutant S129A, which mimics the constitutively unphosphorylated form of aSyn, was generated by site-directed mutagenesis from aSyn-WT constructs using primers 5′GAGGCTTATGAAATGCCTGCTGAGGAAGGGTATCAAG3′ and 5′CTTGATACCCTTCCTCAGCAGGCATTTCATAAGCCTC3′ to obtain the S129A substitution.
All constructs were generated in the pcDNA3.1 vector and verified by DNA sequencing.
The constructs for human WT-untagged aSyn (pSI-aSyn-WT), C-terminally tagged aSyn (aSyn-WT-V5 and aSyn-WT-GFP), GFP-aSyn-WT and HSP70 were a kind gift of Dr. Bradley T. Hyman and were previously described [52]. Myc-aSyn-WT-V5 and Myc-aSyn-WT have been described previously [53]. PLK and GRK encoding plasmids were a kind gift from Dr. Hilal Lashuel, Ecole polytechnique Federale de Lausanne, Switzerland.
Cell Culture, Transfections and Immunocytochemistry
H4 human neuroglioma cells were maintained under standard conditions and passaged the day before transfection [51]. Transfections with aSyn, GFP and PAGFP constructs were performed using Fugene™ 6 reagent from Roche, according to the manufacturer’s instructions. Immunocytochemistry experiments were performed as described previously for Myc-aSyn, aSyn-V5, Myc-aSyn-V5 and untagged aSyn constructs [51]. For PA experiments, cells were co-transfected with a mRFP in order to identify transfected cells. Cells were incubated for 48 h before imaging.
Live Cell Imaging
Cells were imaged using a Zeiss LSM510 META microscope with a ×63 1.4 NA oil immersion objective. aSyn-WT GFP-tagged was excited at 488 nm using an argon laser (5 % transmission) and a 505- to 550-nm band pass filter. For PA experiments, cells transfected with PAGFP constructs were first identified through a 561- to 10-nm DPSS laser (1 % transmission) to detect mRFP (561 nm) using a 575-nm long-pass filter and photoactivated using a diode laser line at 405 nm (100 % transmission) either in the cytoplasm or in the nucleus, using standard procedures for PA. PAGFP fluorescence emission was detected by excitation at 488 nm (5 % transmission) using a 505- to 550-nm band pass filter. About 500 images from each cell were taken with an interval of 2 s, and PA was performed after the second image; 15 cells per condition were analysed.
For FRAP experiments, cells transfected with GFP constructs were photobleached in the nucleus or in the cytoplasm using a diode laser line at 405 nm (100 % transmission), using standard procedures for FRAP. Recovery fluorescence of GFP constructs was detected by excitation at 488 nm (5 % transmission) using a 505- to 550-nm band pass filter. About 1,000 images from each cell were taken per second, and FRAP was performed after the second image; 15 cells per condition were analysed.
Image Analysis
The dynamics of aSyn diffusion (after PA or FRAP) was followed by analysing time-lapse series of the PAGFP or GFP reporter protein by measuring the fluorescence intensity over time, in the nucleus and in the cytoplasm, using ImageJ LSM toolbox plugin and LSM Image browser.
For PA analysis, the normalised nuclear fluorescence (NF) was obtained as the following:
The normalised cytoplasmic fluorescence (CF) was obtained as the following:
For FRAP analysis, the normalised NF was obtained as the following:
The normalised FC was obtained as the following:
Where N and C refer to nucleus and cytoplasm, respectively. t = 0 s refers to the time lapse immediately after PA or PB. These normalisations corrected for the loss of fluorescence caused by imaging both in PA and in FRAP procedures.
Statistical Analysis
The numerical results are given as mean of NF or CF ± SD of up to 15 independent experiments.
The significance of the difference between the experimental and the control values of fluorescence was evaluated at three time points, 100, 500 and 1,000 s, in the nuclear compartment using 95 % confidence intervals (α = 0.05) through single comparisons by the two-tailed unpaired Student’s t test followed by a Fisher’s exact test to compare variances between the control and experimental groups.
Immunoblot Analysis
H4 total protein extracts were obtained 48-h post-transfection using standard procedures. Briefly, cells were washed twice in PBS and lysed in NP40 buffer (glycerol, 10 %; HEPES, 20 mM (pH 7.9); KCl, 10 mM; EDTA, 1 mM; NP40, 0.2 %; and DTT, 1 mM) containing protease and phosphatase inhibitors cocktail (1 tablet/10 ml; Roche Diagnostics). After centrifugation at 16,000×g for 20 min at 4 °C, supernatants were collected (cytoplasmic extract). The pellet was resuspended in NaCl buffer (glycerol, 20 %; HEPES, 20 mM (pH 7.9); KCl, 10 mM; EDTA, 1 mM; NaCl, 400 mM; and DTT, 1 mM) containing protease and phosphatase inhibitors cocktail tablets and then centrifuged again. After centrifugation, the supernatant corresponds to the nuclear extract. Protein concentration was determined using the BCA protein assay and 20 μg of protein lysates were resolved in 12 % SDS-PAGE. Resolved proteins were transferred to nitrocellulose membranes. After quick washing in Tris-buffered saline and 0.1 % Tween 20 (TBS-T), membranes were blocked in 5 % non-fat dry milk in TBS for 1 h and then incubated with primary antibodies in 5 % BSA in TBS overnight at 4 °C. The primary antibodies used were mouse anti-aSyn, 1:1,000 (BD Transduction); mouse anti-GAPDH, 1:4,000 (Ambion); rabbit anti-HSP70, 1:1,000 (Assay Designs); and goat anti-LamininB C20, 1:500 (Santa Cruz Biotechnology). The membrane was then washed three times for 10 min each in TBS-T at room temperature and probed with ECL™ IgG horseradish peroxidase-conjugated (HRP) anti-mouse, anti-rabbit (GE Healthcare) or IgG HRP-conjugated anti-goat (Santa Cruz Biotechnology) secondary antibodies (1:10,000) for 1 h at room temperature. The membrane was then washed four times for 15 min each with TBS-T, and the signal was detected with an ECL chemiluminescence kit (Millipore Immobilon Western Chemiluminescent HRP Substrate).
References
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388(6645):839–840. doi:10.1038/42166
Schneider BL, Seehus CR, Capowski EE, Aebischer P, Zhang SC, Svendsen CN (2007) Over-expression of alpha-synuclein in human neural progenitors leads to specific changes in fate and differentiation. Hum Mol Genet 16(6):651–666
Crews L, Mizuno H, Desplats P, Rockenstein E, Adame A, Patrick C, Winner B, Winkler J, Masliah E (2008) Alpha-synuclein alters Notch-1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J Neurosci 28(16):4250–4260. doi:10.1523/JNEUROSCI.0066-08.2008
Klucken J, Outeiro TF, Nguyen P, McLean PJ, Hyman BT (2006) Detection of novel intracellular alpha-synuclein oligomeric species by fluorescence lifetime imaging. FASEB J 20(12):2050–2057. doi:10.1096/fj.05-5422com
Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH (2004) Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci 24(30):6715–6723. doi:10.1523/JNEUROSCI.1594-04.2004
Vivacqua G, Casini A, Vaccaro R, Fornai F, Yu S, D’Este L (2011) Different sub-cellular localization of alpha-synuclein in the C57BL\6J mouse’s central nervous system by two novel monoclonal antibodies. J Chem Neuroanat 41(2):97–110. doi:10.1016/j.jchemneu.2010.12.003
Goers J, Manning-Bog AB, McCormack AL, Millett IS, Doniach S, Di Monte DA, Uversky VN, Fink AL (2003) Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 42(28):8465–8471. doi:10.1021/bi0341152
Unni VK, Weissman TA, Rockenstein E, Masliah E, McLean PJ, Hyman BT (2010) In vivo imaging of alpha-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS One 5(5):e10589. doi:10.1371/journal.pone.0010589
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) Alpha-synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18(2):106–108
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55(2):164–173
Ulmer TS, Bax A (2005) Comparison of structure and dynamics of micelle-bound human alpha-synuclein and Parkinson disease variants. J Biol Chem 280(52):43179–43187. doi:10.1074/jbc.M507624200
Bodner CR, Maltsev AS, Dobson CM, Bax A (2010) Differential phospholipid binding of alpha-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry 49(5):862–871. doi:10.1021/bi901723p
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T (2002) Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4(2):160–164
Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281(40):29739–29752. doi:10.1074/jbc.M600933200
Pronin AN, Morris AJ, Surguchov A, Benovic JL (2000) Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem 275(34):26515–26522
Inglis KJ, Chereau D, Brigham EF, Chiou SS, Schobel S, Frigon NL, Yu M, Caccavello RJ, Nelson S, Motter R, Wright S, Chian D, Santiago P, Soriano F, Ramos C, Powell K, Goldstein JM, Babcock M, Yednock T, Bard F, Basi GS, Sham H, Chilcote TJ, McConlogue L, Griswold-Prenner I, Anderson JP (2009) Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J Biol Chem 284(5):2598–2602
Lou H, Montoya SE, Alerte TN, Wang J, Wu J, Peng X, Hong CS, Friedrich EE, Mader SA, Pedersen CJ, Marcus BS, McCormack AL, Di Monte DA, Daubner SC, Perez RG (2010) Serine 129 phosphorylation reduces the ability of alpha-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J Biol Chem 285(23):17648–17661. doi:10.1074/jbc.M110.100867
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295(5556):865–868. doi:10.1126/science.1067389
Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN (2005) Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J Mol Biol 351(5):1081–1100. doi:10.1016/j.jmb.2005.06.060
Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ (2004) Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem 279(24):25497–25502. doi:10.1074/jbc.M400255200
Dedmon MM, Christodoulou J, Wilson MR, Dobson CM (2005) Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. J Biol Chem 280(15):14733–14740. doi:10.1074/jbc.M413024200
Roy S, Yang G, Tang Y, Scott DA (2012) A simple photoactivation and image analysis module for visualizing and analyzing axonal transport with high temporal resolution. Nat Protoc 7(1):62–68. doi:10.1038/nprot.2011.428
Lippincott-Schwartz J, Altan-Bonnet N, Patterson GH (2003) Photobleaching and photoactivation: following protein dynamics in living cells. Nat Cell Biol Suppl:S7–14
Patterson GH, Lippincott-Schwartz J (2002) A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297(5588):1873–1877
Vamvaca K, Volles MJ, Lansbury PT Jr (2009) The first N-terminal amino acids of alpha-synuclein are essential for alpha-helical structure formation in vitro and membrane binding in yeast. J Mol Biol 389(2):413–424. doi:10.1016/j.jmb.2009.03.021
Yang ML, Hasadsri L, Woods WS, George JM (2010) Dynamic transport and localization of alpha-synuclein in primary hippocampal neurons. Mol Neurodegener 5(1):9. doi:10.1186/1750-1326-5-9
Specht CG, Tigaret CM, Rast GF, Thalhammer A, Rudhard Y, Schoepfer R (2005) Subcellular localisation of recombinant alpha- and gamma-synuclein. Mol Cell Neurosci 28(2):326–334. doi:10.1016/j.mcn.2004.09.017
Kontopoulos E, Parvin JD, Feany MB (2006) Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 15(20):3012–3023
Siddiqui A, Chinta SJ, Mallajosyula JK, Rajagopolan S, Hanson I, Rane A, Andersen JK (2012) Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson’s disease. Free Radic Biol Med. doi:10.1016/j.freeradbiomed.2012.05.024
Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT Jr (1996) NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35(43):13709–13715. doi:10.1021/bi961799n
Bartels T, Choi JG, Selkoe DJ (2011) Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477(7362):107–110. doi:10.1038/nature10324
Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ (2011) A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 108(43):17797–17802. doi:10.1073/pnas.1113260108
Zhu M, Fink AL (2003) Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem 278(19):16873–16877. doi:10.1074/jbc.M210136200
Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC (2003) A broken alpha-helix in folded alpha-synuclein. J Biol Chem 278(17):15313–15318. doi:10.1074/jbc.M213128200
Perrin RJ, Woods WS, Clayton DF, George JM (2000) Interaction of human alpha-synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J Biol Chem 275(44):34393–34398. doi:10.1074/jbc.M004851200
Ulmer TS, Bax A, Cole NB, Nussbaum RL (2005) Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem 280(10):9595–9603. doi:10.1074/jbc.M411805200
Sousa VL, Bellani S, Giannandrea M, Yousuf M, Valtorta F, Meldolesi J, Chieregatti E (2009) {alpha}-synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol Biol Cell 20(16):3725–3739. doi:10.1091/mbc.E08-03-0302
Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT Jr (2002) Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 322(5):1089–1102
Oueslati A, Paleologou KE, Schneider BL, Aebischer P, Lashuel HA (2012) Mimicking phosphorylation at serine 87 inhibits the aggregation of human alpha-synuclein and protects against its toxicity in a rat model of Parkinson’s disease. J Neurosci 32(5):1536–1544. doi:10.1523/JNEUROSCI.3784-11.2012
Hejjaoui M, Butterfield S, Fauvet B, Vercruysse F, Cui J, Dikiy I, Prudent M, Olschewski D, Zhang Y, Eliezer D, Lashuel HA (2012) Elucidating the role of C-terminal post-translational modifications using protein semisynthesis strategies: alpha-synuclein phosphorylation at tyrosine 125. J Am Chem Soc 134(11):5196–5210. doi:10.1021/ja210866j
Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C (2000) Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J Biol Chem 275(1):390–397
Chen L, Feany MB (2005) Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci 8(5):657–663
Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, Olschewski D, Yin G, Zweckstetter M, Masliah E, Kahle PJ, Hirling H, Lashuel HA (2010) Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem 285(4):2807–2822. doi:10.1074/jbc.M109.081950
Fiske M, Valtierra S, Solvang K, Zorniak M, White M, Herrera S, Konnikova A, Brezinsky R, Debburman S (2011) Contribution of alanine-76 and serine phosphorylation in alpha-synuclein membrane association and aggregation in yeasts. Park Dis 2011:392180. doi:10.4061/2011/392180
Witt SN (2010) Hsp70 molecular chaperones and Parkinson’s disease. Biopolymers 93(3):218–228. doi:10.1002/bip.21302
Zhou Y, Gu G, Goodlett DR, Zhang T, Pan C, Montine TJ, Montine KS, Aebersold RH, Zhang J (2004) Analysis of alpha-synuclein-associated proteins by quantitative proteomics. J Biol Chem 279(37):39155–39164. doi:10.1074/jbc.M405456200
Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VM (2003) Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42(28):8530–8540. doi:10.1021/bi027363r
Lee JC, Langen R, Hummel PA, Gray HB, Winkler JR (2004) Alpha-synuclein structures from fluorescence energy-transfer kinetics: implications for the role of the protein in Parkinson’s disease. Proc Natl Acad Sci U S A 101(47):16466–16471
Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ (2008) Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One 3(4):e1867. doi:10.1371/journal.pone.0001867
McLean PJ, Kawamata H, Hyman BT (2001) Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104(3):901–912
Outeiro TF, Klucken J, Bercury K, Tetzlaff J, Putcha P, Oliveira LM, Quintas A, McLean PJ, Hyman BT (2009) Dopamine-induced conformational changes in alpha-synuclein. PLoS One 4(9):e6906. doi:10.1371/journal.pone.0006906
Acknowledgments
We are grateful to José Rino and António Temudo from the Bioimaging Unit, Instituto de Medicina Molecular, Lisbon, Portugal, for the valuable support with imaging optimisation. This work was supported by Fundação para a Ciência e Tecnologia (PTDC/SAU-NEU/105215/2008). SG was supported by AXA Research Fund and by Fundação Ciência e Tecnologia (grant No. SFRH/BD/79337/2011). TFO was supported by an FP7 Marie Curie International Reintegration Grant (Neurofold) and by an EMBO Installation Grant.
Conflict of Interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Fig. S1
Subcellular dynamics of aSyn-WT. a Distribution of PAGFP, aSyn-WT-PAGFP or PAGFP-aSyn-WT in H4 cells before (−2 s), during (0 s) and after (10, 100, 250, 500, 750 and 1,000 s) nuclear and cytoplasmic PA. b Distribution of PAGFP-aSyn-WT co-expressed with HSP70 in H4 cells before (−2 s), during (0 s) and after (10, 100, 250, 500, 750 and 1,000 s) nuclear or cytoplasmic PA. Scale bars, 10 μm. (JPEG 245 kb)
{kind=link}
Fig. S2
Immunobloting analysis of nuclear and cytosolic protein extracts from cells expressing a aSyn-WT reporter proteins. b A30P, E46K and A53T aSyn reporter proteins. c PAGFP-aSyn-WT with GRK2, GRK5, PLK2 and PLK3. d PAGFP-aSyn-S129A. e aSyn-WT in the presence or absence of HSP70. N nuclear fraction, C cytoplasmic fraction, T total fraction. (JPEG 111 kb)
{kind=link}
Fig. S3
Effect of S129 phosphorylation status on the subcellular dynamics of aSyn-WT-PAGFP. a Fluorescence intensities after PA in the nucleus (light grey lines) and in the cytoplasm (dark grey lines) of aSyn-WT-PAGFP fusion proteins co-expressed with GRK2, GRK5, PLK2 and PLK3 (solid lines) over time. Fluorescence intensities of photoactivated control aSyn-WT-PAGFP constructs after co-transfection with an empty vector are shown in dashed lines. Values are mean + SD of 15 cells analysed per condition. b Immunobloting analysis of total aSyn levels in cells expressing aSyn-WT-PAGFP or aSyn-S129A-PAGFP. c Immunobloting analysis of aSyn-WT-PAGFP construct co-expressed with GRK2, GRK5, PLK2 and PLK3. N nuclear fraction, C cytoplasmic fraction, T total fraction. d Fluorescence intensities after PA in the nucleus (light grey) and in the cytoplasm (dark grey) of aSyn-S129A-PAGFP fusion proteins (solid lines) over time. Fluorescence intensities of photoactivated control aSyn-WT-PAGFP fusion proteins are shown in dashed lines. Values are mean + SD of up to 15 cells analysed for each condition. e Immunobloting analysis of cells expressing aSyn-S129A-PAGFP. N nuclear fraction, C cytoplasmic fraction, T total fraction. f Immunobloting analysis of total aSyn levels in cells expressing aSyn-WT-PAGFP or aSyn-S129A-PAGFP. g Cytosolic inclusions in cells expressing aSyn-S129A-PAGFP. Images were taken 500 s after PA in the nucleus in order to increase the contrast of the cytosolic inclusions. Scale bar, 10 μm. (JPEG 189 kb)
{kind=link}
Video S1
Fluorescence dynamics of PAGFP upon nuclear PA. PAGFP was expressed in cells and displays negligible fluorescence. However, after PA in the nucleus, PAGFP becomes fluorescent and spreads into the cytoplasm reaching complete fluorescence homogeneity within the cell in less than 500 s. Scale bar, 10 μm. (MPG 107 kb)
Video S2
Fluorescence dynamics of aSyn-WT-PAGFP upon nuclear PA. aSyn-WT-PAGFP was expressed in cells and displays negligible fluorescence. After PA in the nucleus, aSyn-WT-PAGFP molecules become fluorescent and are maintained in the nucleus over time (1,000 seconds). Scale bar, 10 μm. (MPG 245 kb)
Fluorescence dynamics of PAGFP-aSyn-WT upon nuclear PA. PAGFP-aSyn-WT was expressed in cells and displays negligible fluorescence. After PA in the nucleus, PAGFP-aSyn-WT molecules become fluorescent and are maintained in the nucleus over time (1,000 s). Scale bar, 10 μm. (MPG 84.6 kb)
Fluorescence dynamics of PAGFP upon cytoplasmic PA. PAGFP was expressed in cells and displays negligible fluorescence. After PA in the cytoplasm, PAGFP becomes fluorescent and spreads into the nucleus reaching complete fluorescence homogeneity in the cell in less than 500 s. Scale bar, 10 μm. (MPG 187 kb)
Fluorescence dynamics of aSyn-WT-PAGFP upon cytoplasmic PA. aSyn-WT-PAGFP was expressed in cells and displays negligible fluorescence. However, after PA in the cytoplasm, aSyn-WT-PAGFP molecules become fluorescent and move into the nucleus over time (1,000 s). Scale bar, 10 μm. (MPG 143 kb)
Video S6
Fluorescence dynamics of PAGFP-aSyn-WT upon cytoplasmic PA. PAGFP-aSyn-WT was expressed in cells and displays negligible fluorescence. However, after PA in the cytoplasm, PAGFP-aSyn-WT-tagged molecules become fluorescent and are maintained in the cytoplasm over time (1,000 s). Scale bar, 10 μm. (MPG 128 kb)
GFP fluorescence recovery after nuclear and cytoplasmic PB. After nuclear (left cell) and cytoplasmic (right cell) PB of cells expressing GFP, the nucleus (left cell) and the cytoplasm (right cell) recover the fluorescence in less than 500 s. Scale bar, 10 μm. (MPG 129 kb)
Video S8
aSyn-WT-GFP fluorescence recovery after nuclear and cytoplasmic PB. After nuclear PB (bottom cells), the nucleus recovers the fluorescence along the time (1,000 s). After cytoplasmic PB (top cells), the cytoplasm does not recover the fluorescence. Scale bar, 10 μm. (MPG 151 kb)
GFP-aSyn-WT fluorescence recovery after nuclear and cytoplasmic PB. After nuclear (right cell) or cytoplasmic (left cell) PB, neither the nucleus (right cell) nor the cytoplasm (left cell) recover the fluorescence along the time (1,000 s). Scale bar, 10 μm. (MPG 104 kb)
Table SI
(DOCX 629 kb)
Table SII
(DOCX 26.3 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Gonçalves, S., Outeiro, T.F. Assessing the Subcellular Dynamics of Alpha-synuclein Using Photoactivation Microscopy. Mol Neurobiol 47, 1081–1092 (2013). https://doi.org/10.1007/s12035-013-8406-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8406-x