
Abstract
The antiviral potential of small interfering RNAs (siRNAs) targeting rabies virus (RV) polymerase (L) and nucleoprotein (N) genes delivered through lentiviral vector was investigated. For in vitro evaluation, siRNAs expressing BHK-21 cell lines (BHK-L and BHK-N) were developed using transduction with Lenti-L and Lenti-N lentiviruses encoding siRNAs against RV-L and N genes, respectively. When these cell lines were challenged in vitro with RV Pasteur virus-11 (PV-11) strain, there was reduction in number of RV-specific foci and target gene transcripts indicating inhibitory effect on RV multiplication. For in vivo evaluation, mice were treated intracerebrally with lentiviruses and challenged with 20 LD50 of RV challenge virus standard-11 (CVS-11) strain by intramuscular route in masseter muscle. Five out of eight mice treated with Lenti-N survived indicating 62.5 % protection. The control and Lenti-L-treated mice died within 7–10 days indicating lethal nature of challenge virus and no protection. These results demonstrated that siRNA targeting RV-N could not only inhibit RV multiplication, but also conferred protection in mice against lethal RV challenge. These findings have implication on therapeutic use of siRNA targeting RV-N against RV infection.
Similar content being viewed by others


Introduction
One of the striking features of RNAi to perform sequence-specific degradation of perfectly complimentary RNA mediated through small interfering RNA (siRNA) has led to the development of a new therapeutic to target viral-associated RNAs without affecting host cellular RNAs. Animal as well as human pathogenic viruses are exceptional targets for RNAi because of their exogenous and unique nature of sequences in the host, which minimize off-target side effects. Specific inhibition of essential viral genes to prevent viral multiplication in mammalian cells can be triggered by the introduction of synthetic 21–23-nucleotide double-stranded siRNA [1, 2] or alternatively, by the transcription of siRNAs from a DNA construct driven by the RNA polymerase expression cassettes [3]. The RNAi-based antiviral approach has been investigated against several viruses ([4] for review), including human immunodeficiency virus [5, 6], hepatitis B virus [7, 8], dengue virus [9], human papilloma virus [10], poliovirus [11], hepatitis C virus [12], respiratory syncytial virus [13], severe acute respiratory syndrome coronavirus [14], and influenza virus [15]. In addition, several reports have investigated the potential of RNAi against rabies infection [16–20].
Rabies virus (RV) is an RNA virus belonging to the genus Lyssavirus under the family Rhabdoviridae. This virus is highly neurotropic and is usually transmitted from one infected animal to another animal or human by bite. The virus enters the nervous system of the new host through nerve endings at or near the bite site. RV does not establish a viremia in the infected host; however, the viral pathogenesis targets the brain using the peripheral nerves and neurons of the central nervous system (CNS) for virus replication and transport. Under natural condition, RV infection of the CNS causes only relatively mild neuropathological changes without prominent evidence of neuronal death [21]. This observation has led to the concept that the neurologic symptoms in rabies result from neuronal dysfunction rather than neuronal cell death. With promising power of RNAi to selectively eliminate target RNA in the cell without disturbing cellular function or no cytopathological changes, a strategy can be developed to inhibit the RV multiplication in CNS to block the pathogenic process and, therefore disease progression may be prevented. Among the five RV proteins, the nucleoprotein (N), phosphoprotein (P), and polymerase (L) proteins are important targets for RNAi owing to their roles in both viral transcription and replication. There are reports on in vitro evaluation of RNAi on rabies targeting viral structural genes like, N [16–20]. Further, glycoprotein (G) also remained a potential target for RNAi because RV-G is considered to be essential for virus trans-synaptic spread between neurons in vivo [22, 23]. Other targets for RNAi on rabies were nonstructural genes, like matrix (M) and phosphoprotein (P) with limited antiviral potential [24].
Previous in vitro studies have indicated that short RV-N cDNA and artificial microRNAs could interfere with RV replication [17, 24] and synthetic siRNA against RV-N could reduce RV multiplication in BHK-21 cells [16]. Our group has also evaluated siRNAs targeting RV-L and N genes for their antiviral potential delivered using adenoviral vector in vitro in BHK-21 cells and in vivo in mice to inhibit RV multiplication with moderate level of protection against lethal RV challenge [19]. In this study, lentiviral-based siRNAs delivery system was used to deliver siRNAs as shRNAs by means of U6 promoter targeting RV-L and N genes and for analyzing antiviral effects in vitro in BHK-21 cells and in vivo in mice against lethal RV challenge.
Materials and Methods
Cells and Virus
Baby hamster kidney-21 (BHK-21) and human embryonic kidney-293T (HEK-293T) cells were grown at 37 °C under 5 % CO2 in Dulbecco’s modified minimum essential medium (DMEM, Hyclone), supplemented with 10 % fetal bovine serum (FBS, Hyclone), 1.5 g/l sodium bicarbonate, 16.8 mM HEPES (Hyclone), 1× nonessential amino acids (NEAA, Hyclone), 2.5 mg/l amphotericin B (Hyclone), and 50 mg/l of gentamicin (Sigma).
The RV Pasteur virus-11 (PV-11) strain adapted to BHK-21 was used for in vitro challenge study in BHK-21 cells. The mouse brain-adapted RV challenge virus standard-11 (CVS-11) strain was used for in vivo challenge in mice.
Experimental Animals
Swiss albino mice (3–4 weeks old of either sex) were use for in vivo experiment according to the guidelines given by Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Environment and Forests, Government of India.
Construction of RV Target Gene Reporter Plasmids
RV-L and N target gene-carrying plasmids, pDsRed-L and pDsRed-N, respectively, were constructed for expressing fusion RV-L and N protein with red fluorescent protein (RFP) at the C-terminal end. The portion of RV-L gene (636 bp)-containing siRNA target site was amplified by RT-PCR (reverse transcription PCR) with RabRfp_LF and RabRfp_LR primers (Table 1) from RV PV-11 and cloned in pDsRed-N1 vector (Clontech). Similarly, pDsRed-N was constructed by amplifying full-length (1380 bp) RV-N gene by RT-PCR using RabRfp_NF and RabRfp_NR primers (Table 1) and cloned in pDsRed-N1 vector. The expression of RFP fusion proteins (N-RFP and L-RFP) were confirmed in transfected HEK-293T cells.
Construction of siRNA Expressing Plasmids
The sense and antisense oligonucleotides (Table 1) with sense, antisense siRNAs, and loop sequences were annealed and cloned into pMCs-GFP plasmid, and recombinant plasmids were designated as shL-pMCs-GFP and shN-pMCs-GFP. The siRNAs (si-L and si-N) were expressed as shRNAs using U6 promoter. The pMCs-GFP contained enhanced green fluorescent protein (GFP) as reporter gene.
Cotransfection of Target Gene Reporter Plasmids with siRNA Expressing Plasmids
The HEK-293T cells were cotransfected with target gene reporter plasmids (pDsRed-L or pDsRed-N) expressing RV-L or N protein with C-terminal RFP fusion (L-RFP or N-RFP) and siRNA expressing plasmids (shL-pMCs-GFP and shN-pMCs-GFP), or control-scrambled siRNA expressing plasmid (shC-pMCs-GFP) using Lipofectamine 2000 reagent (Invitrogen) in a 24-well plate according to manufacturer’s instructions. At 48 h post-transfection, cells were analyzed for RFP and GFP expression using Olympus IX51 fluorescent microscope.
Production of Lentiviruses Expressing siRNAs
To produce replication-deficient lentiviruses encoding different siRNAs, lentiviruses were produced by calcium phosphate-mediated plasmids cotransfection of HEK-293T cells as per the method described earlier [25]. In brief, HEK-293T cells (80 % confluent cells in 75 cm2 flask) were cotransfected with different recombinant pMCs-GFP (either shL-pMCs-GFP, shN-pMCs-GFP or control) along with helper plasmids. Virus supernatants were collected on day 2, 3, and 4 post-transfection and concentrated using ultracentrifugation at 70,000×g for 2 h at 4 °C. The pellet was resuspended in HBSS and titrated. The lentivirus having si-L and si-N sequences were designated as Lenti-L and Lenti-N, respectively. For determination of transduction unit (TU) titer, HEK-293T cells (5 × 104 cells in a 24-well plate) were transduced with different tenfold dilutions of lentivirus stocks, and TU titer was determined based on GFP expressing cell count as per the method described earlier [25].
Generation of Homogeneous BHK-21 Cell Line Expressing Different siRNAs
The BHK-21 cells were transduced with different lentiviruses (Lenti-L or Lenti-N) with 1.0 multiplicity of infection (MOI) in the presence of polybrene (6 μg/ml). Since lentivirus backbone contained enhanced green fluorescent protein (GFP) as reporter gene, its expression indicated transduction of siRNAs expression cassette in the genome of BHK-21 cells. The GFP expressing transduced BHK-21 cells were sorted three times by fluorescent activated cell sorter (FACS) to get maximum percentage of GFP-expressing transduced BHK-21 cells. Lenti-L and lenti-N transduced homogeneous BHK-21 cells were designated as BHK-L and BHK-N, respectively. To verify the sequence integrity of siRNA expression cassette in BHK-L and BHK-N cells, the genomic DNA from cells was isolated as per the method described earlier [26], and 459-bp fragment containing siRNA expression cassette was amplified by PCR using Retro U6_F and Retro U6_R primers (Table 1), and nucleotide sequences were determined.
Transfection of siRNAs Expressing BHK-21 Cells with RV Target Gene Reporter Plasmids
The siRNAs expressing BHK-21 cell lines (BHK-L or BHK-N) were transfected with target gene expressing plasmids (pDsRed-L or pDsRed-N) in a 24-well plate using Lipofectamine 2000 reagent (Invitrogen). At 48 h post-transfection, the transfected cells were analyzed for reduction in L-RFP and N-RFP expression compared with transfected BHK-21 control cells after nuclear counter staining with DAPI.
Challenge of siRNAs Expressing BHK-21 Cells with RV
The BHK-L or BHK-N cells were infected with 0.05 MOI of RV PV-11 in 96-well plate in quadruplicate. BHK-21 cells were included in the assay as control. After 2 h of infection, the unabsorbed RV in the inoculums was removed and cell monolayer was washed three times with HBSS. At 48 h post-infection, the infected cell culture supernatant was harvested and stored at −20 °C for virus titer estimation. The infected cell monolayer was fixed with 80 % chilled acetone with denatured GFP, and there was no green fluorescence. The presence of RV in infected cells was detected by staining with anti-rabies nucleocapsid FITC-labeled antibody (BioRad) according to the manufacturer’s instructions and counterstained with DAPI (Invitrogen). The cells with green fluorescence indicated RV infection.
RV Titer Determination
The RV titer in infected cell culture supernatant was determined by fluorescent foci unit (ffu) assay. In brief, 10 μl of serial tenfold dilutions of virus supernatant, harvested at 48-h post-infection, was mixed with 2 × 102 BHK-21 cells in a flat-bottom 72-well plate in quadruplicate and incubated at 37 °C. At 48-h post-infection, infected cells were washed thrice with PBS, fixed with 80 % chilled acetone and stained with anti-rabies nucleocapsid FITC-labeled antibody (BioRad), and ffu titer was determined.
Quantitative real-time PCR (qPCR)
For quantitative determination of RV-L and N transcripts, qPCR was used. In brief, BHK-L and BHK-N cells in a 6-well plate were infected with RV PV-11 at 0.05 MOI for 24 h and total RNA was isolated using TRIzol LS reagent (Invitrogen) according to the manufacturer’s instructions. The cDNA was prepared with oligo dT primer using MMuLV-RT (Fermentas) according to the manufacturer’s protocol. The RV-L and N gene transcripts in 1:50 diluted cDNAs were quantified using gene-specific real-time primers (Table 1) using Kapa SYBR fast qPCR kit (Kapa Biosystems) according to the manufacturer’s instructions. For relative quantification, GAPDH gene transcripts were used as reference for normalization, and percent reductions in levels of target mRNA transcripts were determined as per the method described earlier [27].
Challenge of siRNAs Treated Mice with RV
Two groups of mice (n = 8 in each) were treated intracerebrally by injection with 106 Lenti-L or Lenti-N lentivirus particles at four different sites. One group of mice (n = 8) was injected with HBSS and kept as negative control. Seven days after injection with lentivirus, mice in all groups were challenged by intramuscular route in masseter muscle with 20 LD50 of virulent RV CVS-11 strain according to the NIH test for potency [28]. Mice were maintained at normal conditions and observed for 14 days. The death of animals with rabies-specific symptoms was recorded as an event at a particular day, and percent survival was calculated.
Statistical Analysis
The RV titers and level of gene transcripts were compared by statistical analysis using one-way ANOVA followed by Tukey’s test for treatment versus control. Differences between the groups with p ≤ 0.05 were considered statistically significant. All data were presented as the mean ± SEM. Comparison of survival between groups was done by Log-rank test according to Mantel–Haenszel method using GraphPad Prism version 5.01 (GraphPad).
Results
Evaluation of Silencing Effect of siRNAs on RV Target Genes
The si-L targeting RV-L mRNA at nucleotide position (nt) 1203–1221 and si-N targeting RV-N mRNA nt 1120–1138 were cloned into pMCs-GFP as shRNA and expressed utilizing U6 RNA Pol III promoter. In plasmid cotransfection experiment, the HEK-293T cells with RFP indicated expression of RFP-tagged version of RV-L or N protein (L-RFP or N-RFP), while cells with GFP expression indicated the presence of siRNAs expressing plasmid (Fig. 1). The cells transfected with target gene reporter plasmid and siRNAs expressing plasmid indicated the reduction in RFP expression (indicating reduction of fusion transcripts of RV target gene and RFP) in comparison with control (cotransfected with negative control siRNA expressing plasmid) (Fig. 1). This confirmed that there was expression of siRNA which effectively silenced the RV target gene in a sequence-specific manner.

Evaluation of silencing effect of si-L and si-N targeting RV-L and N genes, respectively, by transient plasmid cotransfection method in HEK-293T cells. HEK-293T cells were cotransfected with target gene reporter plasmid (pDsRed-L or pDsRed-N) expressing RV-L or N protein with C-terminal RFP fusion (L-RFP or N-RFP) and siRNA expressing plasmid (shL-pMCs-GFP, shN-pMCs-GFP, or shC-pMCs-GFP) expressing different siRNAs. At 48-h post-transfection, transfected cells were analyzed for reduction of target gene expression (L-RFP or N-RFP)
Evaluation of Antiviral Effect of siRNAs In Vitro in BHK-21 Cells
Different lentiviruses, namely, Lenti-L and Lenti-N, by means of TU titer of 6.7 × 108 TU/ml and 9 × 108 TU/ml, respectively, were prepared by plasmid cotransfection method in HEK-293T cells. The BHK-21 cell lines (BHK-L or BHK-N) expressing siRNAs (si-L or si-N) were constructed by transduction using different lentiviruses (Lenti-L or Lenti-N). After sorting, the BHK-L and BHK-N cell lines indicated 86.5 and 90.7 % of GFP expressing cells, respectively, indicating homogenous nature of siRNAs expressing cell lines (Fig. 2), and siRNAs expression cassettes in these cell lines were found correct by nucleotide sequencing (data not shown). For evaluation of siRNAs to knockdown the target RV gene transcripts, BHK-L and BHK-N cells were transfected with target gene expressing plasmids (pDsRed-L or pDsRed-N). The reduction in expression of RFP tagged target gene, L-RFP, or N-RFP compared with BHK-21-transfected control cells indicated effect of siRNAs in knockdown of target gene expression (Fig. 3).

Construction and characterization of different siRNAs expressing BHK-21 cell lines. BHK-21 cells (a) were transduced using different lentiviruses (Lenti-L or Lenti-N) and analyzed under fluorescent microscope for expression of GFP. Since lentivirus backbone contained enhanced green fluorescent protein (EGFP) as reporter gene, its expression indicated transduction of siRNAs expression cassette in the genome of BHK-21 cells. The GFP-positive cells expressing siRNAs against RV-L (BHK-L) (b) and against RV-N (BHK-N) (c) were sorted and analyzed for homogenous population using flow cytometery

Qualitative evaluation of different siRNAs expressing BHK-21 cell lines to target RV-L and RV-N gene transcripts. Different homogenous siRNAs expressing BHK-21 cell lines (BHK-L or BHK-N) were transfected with target gene expressing plasmids (pDsRed-L or pDsRed-N) expressing RV-L or N genes with C-terminal fusion with red fluorescent protein (RFP). At 48-h post-transfection, cells were analyzed for reduction in L-RFP (a) and expression N-RFP (b) compared with transfected BHK-21 control cells
When siRNAs expressing cells were infected with RV, there was marked reduction in RV-specific foci in BHK-L and BHK-N cells compared with control BHK-21 cells (Fig. 4) indicating the inhibitory effect of siRNAs on RV multiplication. To estimate target gene-specific effect of siRNAs more precisely, their ability to knockdown RV gene transcripts was analyzed quantitatively using qPCR. The levels of RV-L and N gene transcripts in BHK-L cells were reduced to 41.4 and 54.1 %, respectively, compared with levels in RV-infected BHK-21 cells (Fig. 5). Similarly, levels of RV-L and N gene transcripts in BHK-N cells were also reduced to 17.5 and 15.0 %, respectively (Fig. 5). Further, to evaluate the inhibitory effect of siRNAs on RV multiplication, quantitative determination of progeny RV release from siRNAs expressing infected cells was analyzed. The RV-infected cell culture supernatants from BHK-L and BHK-N showed 7.2 ± 0.8 × 103 and 7.0 ± 1.0 × 103 ffu/ml of RV titer, respectively, while control BHK-21 cells’ supernatant showed 27.2 ± 5.0 × 103 ffu/ml of RV (Fig. 6). This indicated 73.4 % and 74.3 % reductions in RV titer with BHK-L and BHK-N, respectively, compared with control BHK-21 cells. These results were average of four independent experiments which indicated that both siRNAs, namely, si-L and si-N expressed by BHK-21 cells using U6 promoter could inhibit RV multiplication and progeny virus release in siRNAs expressing cells.

Qualitative evaluation of different siRNAs expressing BHK-21 cell lines to inhibit RV multiplication. Different homogenous siRNAs expressing BHK-21 cell lines (BHK-L or BHK-N) were challenged with 0.05 MOI of RV-PV-11 strain along with BHK-21 control cells. At 48-h post-challenge, the presence of RV in infected cells was detected by staining with anti-rabies nucleocapsid FITC-labeled antibody and counterstained with DAPI. The cells with green fluorescence indicated RV-infection

Evaluation of different siRNAs expressing BHK-21 cell lines (BHK-L and BHK-N) to inhibit RV multiplication. Different homogenous siRNAs expressing cell lines, namely, BHK-L and BHK-N, were challenged with RV-PV-11 at 0.05 MOI. At 48-h post-infection, total RNA was isolated from infected cells, and cDNA was prepared using oligo dT primer. The RV-L and N gene transcripts in cDNA were quantified using RT-PCR and compared with gene transcripts in RV-infected healthy BHK-21 cells after normalization with GAPDH gene transcripts. The results presented are averages of four independent experiments (mean ± SEM). *p < 0.05 versus respective RV transcripts in control BHK-21 cells

Quantitative evaluation of inhibition of RV multiplication in different siRNAs expressing BHK-21 cell lines. Different homogenous siRNAs expressing BHK-21 cell lines (BHK-L or BHK-N) were challenged with 0.05 MOI of RV-PV-11 strain along with BHK-21 control cells. At 48-h post-challenge, the infected cell culture supernatant was harvested, and the amount of RV in the harvest was estimated using rabies fluorescent foci unit (ffu) assay. The % reduction in RV titer was determined in comparison with control. The results presented are averages of four independent experiments (mean ± SEM). *p < 0.05 versus RV titer in infected cell culture supernatant from BHK-21 cells
Evaluation of Lentiviral Delivered siRNAs for Antiviral Effect In Vivo in Mice
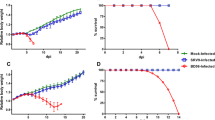
To analyze the effect of lentiviral-delivered siRNAs to inhibit RV multiplication in vivo, the lentivirus-treated mice were challenged with RV challenge virus standard-11 (CVS-11) strain according to NIH method of determining potency. All the control mice developed rabies-specific symptoms and died within 6–7 days post-challenge with median survival of 7 days. There was no protection in mice treated with Lenti-L; however, median survival days were increased to 10 days compared with control mice. Treatment of mice with Lenti-N (lentivirus expressing siRNA against RV-N) demonstrated 62.5 % protection (Fig. 7). The mice that survived after 14 days did not develop symptoms of rabies and remained healthy till the entire period of observation for 1 month. These results indicated that treatment of mice with lentivirus expressing siRNA against RV-N conferred modest protection against lethal RV challenge.

Percent survival of mice treated with different lentiviruses expressing siRNAs and challenged with lethal RV challenge virus standard-11 (CVS-11) strain. Mice (n = 8, per group) were treated intracerebrally with different lentiviruses (Lenti-L or Lenti-N) or control and challenged by the intramuscular route in masseter muscle with 20 LD50 of lethal RV CVS-11 strain. The percent survival post-challenge was monitored. Lenti-N-treated mice group differs from control or Lenti-L-treated for survival (*p < 0.05; Log-rank)
Discussion
The replication cycle of negative-sense RNA viruses, including RV, consists of two successive events, namely, transcription of the viral genes to produce viral proteins, followed by replication of virus genome using virus-encoded polymerase (L) protein. Among the five viral proteins, the nucleoprotein (N) has an important role in the formation of ribonucleoprotein-RNA complex, required as template for transcription [29]. It is observed that level of RV-N protein is very crucial for the switch from transcription to replication during virus life cycle [30, 31]. Therefore, in this study, the RV-L and N genes have been targeted through RNAi approach to evaluate their effects on RV multiplication. Previous studies, including ours, have evaluated the effect of siRNAs targeting different RV genes on RV multiplication in vitro in BHK-21 cells and in vivo in mice [16–20]. The inhibitory effect of siRNAs against RV-N was found efficient in interfering with the expression of viral genes and conferred moderate protection against lethal RV challenge in mice treated intracerebrally with adenovirus-expressing siRNAs [19]. In this present study, we selected lentiviral system for siRNAs delivery which has advantage over other siRNAs’ delivery methods, namely, long-term, continuous silencing of target gene and delivery into wide range of cells including, both dividing and nondividing cells (e.g., neurons). Apart from this, lentiviral vectors are less immunogenic, which makes them a preferred vector system for in vivo delivery [32]. These characteristics of lentiviral vector make them suitable for siRNAs delivery to nerve cells, the target cell for RV infection.
For evaluation of lentiviral-based delivery of siRNAs, in the present study, siRNAs were delivered into BHK-21 cells and evaluated for inhibition of RV multiplication using different approaches like reduction in RV-specific foci in siRNA expressing cells (for inhibition of virus multiplication), knockdown of RV target gene transcripts, and reduction in production/release of infective RV from siRNAs expressing cells. The reduction in the number of RV-infected foci in siRNAs expressing cells compared with control cells indicated inhibition in RV multiplication. This inhibition was quantified by the levels of RV gene transcripts by qPCR. Since RNAi functions by identifying and degrading mRNA that shares sequence complementary with the siRNA, qPCR analysis was used to analyze the effect of siRNAs on RV-N or L mRNA expression. The levels of RV-L and N mRNA in the siRNAs expressing and RV-infected cells were significantly reduced compared with control cells. However, the si-L was less efficient in the transcript reduction assay (Fig. 5). This is in corroboration with the observation that siRNA against N of Nipah virus was more effective than siRNA against L [33], suggesting that transcript quantity might also play a role in siRNA efficacy. In RV-infected cells among all RV transcripts, RV-N transcripts are the most abundant, and L transcripts are the least abundant [34]. Apart from transcript reduction assay, both siRNAs (si-L and si-N) were equally effective in reducing the RV target gene expression (Fig. 3), RV-specific foci (Fig. 4), and progeny RV release from siRNA expressing cells (Fig. 6). Since RV-N and L proteins have interrelated effects on virus multiplication [24], siRNAs targeting RV-L and N genes exhibited nonsignificant difference in inhibition of RV multiplication.
In the next step, the siRNAs expressing lentiviruses were analyzed for inhibition of RV multiplication in vivo in mice against virulent RV challenge. The lenti-L-treated mice showed delayed death compared with control mice, and all the lenti-L-treated mice died between 9 and 12 days post-challenge with the median survival of 10 days. The lenti-N-treated mice showed 62.5 % protection. This indicated that knockdown of RV-L gene by siRNAs could only prolong the outcome of infection. In contrast, knockdown of RV-N gene by siRNAs could not only prolong the outcome of disease but also conferred 62.5 % protection. Less effectiveness of knockdown of RV-L gene on mice protection might be due to its limited role in pathogenesis as it encodes nonstructural RNA-dependent RNA polymerase protein for virus genome replication. However, significant protection with mice treated by si-N might be attributed to the dual role of RV-N protein: as viral structural and regulatory protein during RV pathogenesis [24, 30, 31].
The results from previous studies showed that synthetic siRNA [16] and artificial microRNA (miRNA) [17] against RV-N could reduce RV titer in cells. These studies supported that RV-N mRNA could be a good target for intervention to inhibit RV replication. In our previous study using adenoviral delivery, the siRNAs targeting RV-L and N genes demonstrated low (33.3 %) and moderate (66.6 %) levels of protection, respectively, in mice treated with adenoviruses expressing siRNAs [19]. In this study using lentiviral delivery, the siRNA targeting RV-L inhibited RV multiplication in vitro in BHK-21 cells but did not protect mice against lethal RV challenge. However, the siRNA targeting RV-N could not only inhibit RV multiplication in vitro in BHK-21 cells, but also demonstrated consistent moderate level of protection in treated mice against lethal RV challenge. These reports indicated significant implication on the use of viral vector-based siRNA delivery as an antiviral approach against RV infection. Further, the potential of siRNA targeting RV-N for therapeutic use against RV infection can be explored after careful optimization of dose and time of therapeutic administration either alone or in combination with siRNAs targeting other RV genes.
References
Elbashir, S. M., Harborth, J., Lendeckel, W., Yalcin, A., Weber, K., & Tuschl, T. (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498.
Paul, C. P., Good, P. D., Winer, I., & Engelke, D. R. (2002). Effective expression of small interfering RNA in human cells. Nature Biotechnology, 20, 505–508.
Brummelkamp, T. R., Bernards, R., & Agami, R. (2002). A system for stable expression of short interfering RNAs in mammalian cells. Science, 296, 550–553.
Ma, Y., Chan, C. Y., & He, M. L. (2007). RNA interference and antiviral therapy. World Journal of Gastroenterology, 13, 5169–5179.
Boden, D., Pusch, O., Silbermann, R., Lee, F., Tucker, L., & Ramratnam, B. (2004). Enhanced gene silencing of HIV-1 specific siRNA using microRNA designed hairpins. Nucleic Acids Research, 32, 1154–1158.
Kumar, P., Ban, H. S., Kim, S. S., Wu, H., Pearson, T., Greiner, D. L., et al. (2008). T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell, 134, 577–586.
Song, E., Lee, S. K., Wang, J., Ince, N., Ouyang, N., Min, J., et al. (2003). RNA interference targeting Fas protects mice from fulminant hepatitis. Nature Medicine, 9, 347–351.
Morrissey, D. V., Lockridge, J. A., Shaw, L., Blanchard, K., Jensen, K., Breen, W., et al. (2005). Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nature Biotechnology, 23, 1002–1007.
Padwad, Y. S., Mishra, K. P., Jain, M., Chanda, S., Karan, D., & Ganju, L. (2009). RNA interference mediated silencing of Hsp60 gene in human monocytic myeloma cell line U937 revealed decreased dengue virus multiplication. Immunobiology, 214, 422–429.
Niu, X. Y., Peng, Z. L., Duan, W. Q., Wang, H., & Wang, P. (2006). Inhibition of HPV 16 E6 oncogene expression by RNA interference in vitro and in vivo. International Journal of Gynecological Cancer, 16, 743–751.
Saleh, M. C., Van Rij, R. P., & Andino, R. (2004). RNA silencing in viral infections: insights from poliovirus. Virus Research, 102, 11–17.
Lupberger, J., Brino, L., & Baumert, T. F. (2008). RNAi: a powerful tool to unravel hepatitis C virus–host interactions within the infectious life cycle. Journal of Hepatology, 48, 523–525.
Bitko, V., Musiyenko, A., Shulyayeva, O., & Barik, S. (2005). Inhibition of respiratory viruses by nasally administered siRNA. Nature Medicine, 11, 50–55.
Li, B. J., Tang, Q., Cheng, D., Qin, C., Xie, F. Y., Wei, Q., et al. (2005). Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nature Medicine, 11, 944–951.
Zhang, W., Wang, C. Y., Yang, S. T., Qin, C., Hu, J. L., & Xia, X. Z. (2009). Inhibition of highly pathogenic avian influenza virus H5N1 replication by the small interfering RNA targeting polymerase A gene. Biochemical and Biophysical Research Communications, 390, 421–426.
Brandao, P. E., Castilho, J. G., Fahl, W., Carnieli, P, Jr, Oliveira, R., Macedo, C. I., et al. (2007). Short-interfering RNAs as antivirals against rabies. Brazilian Journal of Infectious Diseases, 11, 224–225.
Israsena, N., Supavonwong, P., Ratanasetyuth, N., Khawplod, P., & Hemachudha, T. (2009). Inhibition of rabies virus replication by multiple artificial microRNAs. Antiviral Research, 84, 76–83.
Sonwane, A. A., Dahiya, S. S., Saini, M., Chaturvedi, V. K., Singh, R. P., & Gupta, P. K. (2012). Inhibition of rabies virus multiplication by siRNA delivered through adenoviral vector in vitro in BHK-21 cells and in vivo in mice. Research in Veterinary Science, 93, 498–503.
Gupta, P. K., Sonwane, A. A., Singh, N. K., Meshram, C. D., Dahiya, S. S., Pawar, S. S., et al. (2012). Intracerebral delivery of small interfering RNAs (siRNAs) using adenoviral vector protects mice against lethal peripheral rabies challenge. Virus Research, 163, 11–18.
Yang, Y. J., Zhao, P. S., Zhang, T., Wang, H. L., Liang, H. R., Zhao, L. L., et al. (2012). Small interfering RNAs targeting the rabies virus nucleoprotein gene. Virus Research, 169, 169–174.
Iwasaki, Y., & Tobita, M. (2002). Pathology. In A. C. Jackson & W. H. Wunner (Eds.), Rabies (pp. 283–306). San Diego: Academic Press.
Etessami, R., Conzelmann, K. K., Fadai-Ghothi, B., Natelson, B., Tsiang, H., & Ceccaldi, P. E. (2000). Spread and pathogenic characteristics of a G-deficient rabies virus recombinant: an in vitro and in vivo study. Journal of General Virology, 81, 2147–2153.
Yan, X., Mohankumar, P. S., Dietzschold, B., Schnell, M. J., & Fu, Z. F. (2002). The rabies virus glycoprotein determines the distribution of different rabies virus strains in the brain. Journal of NeuroVirology., 8, 345–352.
Wunner, W. H., Pallatroni, C., & Curtis, P. J. (2004). Selection of genetic inhibitors of rabies virus. Archives of Virology, 149, 1653–1662.
Tiscornia, G., Singer, O., & Verma, I. M. (2006). Production and purification of lentiviral vectors. Nature Protocols, 1, 241–245.
Wang, T. Y., Wang, L., Zhang, J. H., & Dong, W. H. (2011). A simplified universal genomic DNA extraction protocol suitable for PCR. Genetics and Molecular Research, 10, 519–525.
Deng, L., Li, G., Xi, L., Yin, A., Gao, Y., You, W., et al. (2009). Hepatitis B virus inhibition in mice by lentiviral vector mediated short hairpin RNA. BMC Gastroenterology, 9, 73–83.
Wilbur, L. A., & Aubert, F. A. (1996). The NIH test for potency. In F. X. Meslin, M. M. Kaplan, & H. Koprowski (Eds.), Laboratory Techniques in Rabies (pp. 360–368). Geneva: WHO.
Barik, S. (2004). Control of nonsegmented negative-strand RNA virus replication by siRNA. Virus Research, 102, 27–35.
Finke, S., & Conzelmann, K. K. (2005). Replication strategies of rabies virus. Virus Research, 111, 120–131.
Finke, S., Mueller-Waldeck, R., & Conzelmann, K. K. (2003). Rabies virus matrix protein regulates the balance of virus transcription and replication. Journal of General Virology, 84, 1613–1621.
Sliva, K., & Schnierle, B. S. (2010). Selective gene silencing by viral delivery of short hairpin RNA. Virology Journal, 7, 248.
Mungall, B. A., Schopman, N. C., Lambeth, L. S., & Doran, T. J. (2008). Inhibition of Henipavirus infection by RNA interference. Antiviral Research, 80, 324–331.
Kissi, B., Tordo, N., & Bourhy, H. (1995). Genetic polymorphism in the rabies virus nucleoprotein gene. Virology, 209, 526–537.
Acknowledgments
The authors thank the Director, Indian Veterinary Research Institute (IVRI) for providing necessary facilities to carry out this research. This study was supported by a project “Evaluation of anti-rabies effect of small interfering RNA (siRNA) delivered through viral vector” Grant No. BT/PR9941/AGR/36/22/2007 from Department of Biotechnology (DBT), Government of India.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Singh, N.K., Meshram, C.D., Sonwane, A.A. et al. Protection of Mice Against Lethal Rabies Virus Challenge Using Short Interfering RNAs (siRNAs) Delivered Through Lentiviral Vector. Mol Biotechnol 56, 91–101 (2014). https://doi.org/10.1007/s12033-013-9685-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-013-9685-1