
Abstract
Classic derivation of mouse embryonic stem (ES) cells from blastocysts is inefficient, strain-dependent, and requires expert skills. Over recent years, several major improvements have greatly increased the success rate for deriving mouse ES cell lines. The first improvement was the establishment of a user-friendly and reproducible medium-alternating protocol that allows isolation of ES cells from C57BL/6 transgenic mice with efficiencies of up to 75%. A recent report describes the use of this protocol in combination with leukemia inhibitory factor and pluripotin treatment, which made it possible to obtain ES cells from F1 strains with high efficiency. We report modifications of these protocols for user-friendly and reproducible derivation of mouse ES cells with efficiencies of up to 100%. Our protocol involves a long initial incubation of primary outgrowths from blastocysts with pluripotin, which results in the formation of large spherical outgrowths. These outgrowths are morphologically distinct from classical inner cell mass (ICM) outgrowths and can be easily picked and trypsinized. Pluripotin was omitted after the first trypsinization because we found that it blocks attachment of ES cells to the feeder layer and its removal facilitated formation of ES cell colonies. The newly established ES cells exhibited normal karyotypes and generated chimeras. In summary, our user-friendly modified protocol allows formation of large spherical ICM outgrowths in a robust and reliable manner. These outgrowths gave rise to ES cell lines with success rates of up to 100%.
Similar content being viewed by others



Introduction
Embryonic stem (ES) cells are capable of self renewal and differentiation into all types of embryonic and adult cells, including the germ-cell lineage. Mouse ES cells are usually isolated from the inner cell mass (ICM) of blastocysts. The first mouse ES cell lines were derived almost 30 years ago [1, 2], but the classical way for isolating mouse ES cells is very inefficient. It is highly dependent on the strain, and it requires expert skills [3]. Several major improvements have greatly increased the success rate for deriving mouse ES cell lines. These improvements include the use of a defined serum-free medium (knockout serum replacement, SR) [4], alternating between ES cell media containing SR and fetal bovine serum (FBS) [3], and the use of pharmacological compounds [5–8]. In addition, mouse and rat ES cells could be isolated and maintained in a chemically defined medium containing three (3i) or two (2i) small-molecule inhibitors and without the need for extrinsic stimuli, such as serum factors, MEFs, LIF or BMP4 [9–11]. Pluripotin, a small synthetic molecule, allows the propagation of mouse ES cells in an undifferentiated state in the absence of leukemia inhibitory factor (LIF) and mouse embryonic fibroblasts (MEFs) [12]. Finally, combining an SR/FBS medium alternation protocol with the use of MEFs, LIF and pluripotin made it possible to derive mouse ES cells from F1 strains (C57BL/6 transgenics × CD1) with high efficiency [13].
Bryja and colleagues developed a simple and user-friendly protocol that allows investigators with basic cell culture skills and standard equipment to derive ES cells [3]. However, the efficiency of their ES cell isolation protocol is only about 50% [3]. Blastocysts were plated and grown for 6 days in SR-ES cell medium. Blastocyst outgrowths were trypsinized and plated in FBS-ES cell medium and then shifted back to SR-ES cell medium the following day. After two more passages, alternating between ES cell media containing SR and FBS, ES cell lines were established and frozen. Yang and colleagues [13] modified this SR/FBS medium alternation protocol in two ways. First, blastocysts were plated and grown for 2 days in FBS-ES cell medium followed by different incubation periods (1–12 days) in SR-ES cell medium. Second, pluripotin was added to the ES cell media during the first three passages. This procedure allowed highly efficient derivation of ES cells [13].
We established a user-friendly and highly reproducible pluripotin-based ES cell derivation protocol starting from previously published protocols [3, 13]. By using our modified procedure, we were able to isolate mouse ES cell lines from wild-type C57BL/6 blastocysts as well as from seven transgenic mouse strains with efficiencies of up to 100% (Table 1). This high success rate was reproduced independently by two other researchers (Table 1). We modified the previously published protocols [3, 13] by culturing primary outgrowths from blastocysts for 12 days in pluripotin-containing SR-ES cell medium and by omitting pluripotin after the first trypsinization (Fig. 1). Prolonged cultivation of primary outgrowths in the presence of pluripotin resulted in the formation of large spheres composed of pluripotent cells that were morphologically distinct from classical ICM outgrowths (Fig. 2, compare panel a to b or c). These spheres were mostly loosely attached to their substrate and often appeared in suspension; in both cases, mouse ES cell lines could be readily isolated from them. The large spherical outgrowths were easy to pick, and after a single trypsinization step, they were readily dissociated into single cells and plated on MEFs in FBS-containing ES-cell medium. When pluripotin was present in the FBS-containing ES cell medium, most of the cells failed to attach to the MEF layer and were lost when changing the medium to SR-containing ES cell medium. In contrast, when the dissociated cells were seeded in the absence of pluripotin, they attached readily to the MEFs, and primary ES cell colonies were formed efficiently within days. The loosely attached spherical outgrowths facilitate the isolation of ES cells in several ways. First, our protocol yields large outgrowths (300–500 μm), containing several thousand cells. This provides a lot of starting material before the first trypsinization. In contrast, the outgrowths obtained according to published reports are much smaller (100 μm) [3, 13]. Second, the spherical outgrowths (obtained by our protocol) are loosely attached to the MEFs and are easy to pick and trypsinize. In contrast, the primary outgrowths obtained by the protocol of Yang and colleagues are strongly attached to the feeder layer and are difficult to pick [13]. In addition, these outgrowths are hard to dissociate and require an additional collagenase IV treatment [13]. Other differences between our ES cell procedure and previously published protocols are listed in Table 2.

Scheme of ES cell derivation with LIF and pluripotin. Blastocysts are flushed on day 1 and seeded in 12-well plates coated with MEFs and containing pluripotin-supplemented SR-ES cell medium (15% SR). Small spherical colonies, observed on day 6, grow rapidly to form large spherical cell aggregates that are loosely attached to the substrate on day 12. Large pluripotent cell aggregates are trypsinized with 0.25% trypsin and seeded in 96-well plates containing MEFs and FBS-ES cell medium (15% FBS) but no pluripotin. The next day, FBS-ES cell medium is replaced with SR-ES cell medium. ES cells appear after further incubation. Newly established ES cell colonies are passaged three more times, changing the medium from FBS to SR-ES and scaling up from 96-well to 24-well and then 6-well plates. On about day 24, a confluent 6-well plate of the newly established mouse ES cell line can be frozen. Modified after [3]

Formation of non-classical spherical ICM outgrowths in SR medium supplemented with pluripotin and LIF. Formation of ICM outgrowths from wild-type C57BL/6 blastocysts under different experimental conditions. a Two examples of non-classical, loosely attached, spherical ICM outgrowths formed after incubation in SR-ES medium supplemented with pluripotin and LIF. Outgrowths were plated in 12-well plates with (top) or without MEFs (bottom). b Omitting pluripotin from the SR medium in the first 2 days allows attachment of blastocysts and formation of flat ICM outgrowths, which persist after pluripotin administration. Two examples are shown. c Two examples of ICM outgrowths formed by using a previously reported protocol [13]. Blastocysts were incubated for 2 days (2 d) in FBS-ES medium supplemented with pluripotin and LIF, followed by incubation in SR-ES medium supplemented with pluripotin and LIF. DIV: days in vitro. Scale bar: 100 μm
In conclusion, our pluripotin-based protocol is user-friendly and very efficient (with success rates of up to 100%) and could make stem cell research feasible for a broader scientific community. In addition, it can generate both transgenic and littermate control ES cell lines, which allows stem cell characterization and subsequent differentiation experiments to be set up with minimal genetic variation.
Materials
Collecting Blastocysts and Morulas
-
1.
Mice (Note 1)
-
2.
Sterile dissection tools (fine scissors and forceps for dissecting out the uterus and oviduct)
-
3.
M2 medium (Sigma-Aldrich, St. Louis, MO)
-
4.
Fine forceps (Dumont #5 Standard tip Student forceps, Fine Science Tools, Heidelberg, Germany)
-
5.
A blunt 32G needle (Popper & Sons Inc., New York, Cat. No. 7400)
-
6.
A 5-ml Luer-Lok syringe
-
7.
A 23G needle
-
8.
A 1-ml syringe
-
9.
Tyrode solution (Sigma-Aldrich)
-
10.
1.5-ml tubes
-
11.
60-mm bacterial grade plates (for flushing)
-
12.
Mouth pipette (Note 2)
-
13.
Access to a stereo microscope with transmitted-light base.
-
14.
Access to a laminar air flow
Pluripotin-based ES Cell Isolation
-
1.
Mouse embryonic fibroblasts (MEFs, TgN (DR4)1 Jae strain)
-
2.
Mitomycin C (Sigma-Aldrich)
-
3.
Phosphate buffered saline (PBS) without calcium or magnesium (Gibco, Grand Island, NY)
-
4.
0.1% Gelatin (Sigma-Aldrich) (Note 3)
-
5.
MEF medium: Dulbecco’s modified Eagle’s medium (DMEM, Gibco), 10% fetal bovine serum (FBS) (Sigma-Aldrich), L-glutamine (2 mM, Gibco), Sodium puryvate (0.4 mM, Gibco), penicillin (100 U/ml, Gibco), streptomycin (100 mg/ml, Gibco).
-
6.
SR-ES cell medium: DMEM (Gibco) and F12 (Gibco) mixed in a 1:1 ratio and supplemented with 15% knock-out serum replacement (SR, Gibco, Cat. No. 10828–028), L-glutamine (2 mM, Gibco), penicillin (100 U/ml, Gibco), streptomycin (100 mg/ml, Gibco), β-mercaptoethanol (0.1 mM, Gibco), and 2,000 U/ml recombinant mouse LIF (DMBR/VIB Protein Service facility, www.dmbr.ugent.be).
-
7.
FBS-ES cell medium: Similar to SR-ES cell medium but 15% SR is replaced by 15% FBS (Hyclone, Logan, UT, Cat. No. SH30070.03E).
-
8.
Pluripotin (Cayman Chemical, Ann Arbor, MI) (Note 4)
-
9.
Mouth pipette (Note 2)
-
10.
Trypsin (0.25%) (Gibco)
-
11.
Sterile pipettes: 5 ml, 10 ml and 25 ml
-
12.
15-ml conical centrifuge tubes
-
13.
96-well culture plates (curved bottom and flat bottom)
-
14.
Culture dishes: 24 wells, 12 wells and 6 wells
-
15.
Multichannel pipettes (to pipette 30, 50, 100 and 200 μl)
-
16.
Sterile multichannel reservoirs
-
17.
Access to a laminar air flow
-
18.
Access to an incubator at 37°C with 5% CO2
-
19.
Access to an inverted microscope
-
20.
Access to a bench-top centrifuge
-
21.
Access to a stereo microscope with transmitted-light base
Methods
Collecting Blastocysts and Morulas
-
1.
Set up time matings: Female mice (can be superovulated) at the age of 6–16 weeks are housed with male studs. Check for copulation plugs the following morning. Plugged females are considered to be carrying 0.5 dpc (days post coitum) embryos.
-
2.
Sacrifice pregnant females to collect blastocysts (3.5 dpc) or morulas (2.5 dpc). Make a midventral incision and dissect from both sides a piece that consists of the uterus and the oviduct. A first cut is made in the uterus just before the cervix and a second cut is made between the ovary and the oviduct. Transfer the excised pieces to a 1.5-ml tube containing 500 μl M2 medium. Dissection should be done in a laminar air flow.
-
3.
In a laminar air flow, transfer the uteri to bacterial grade dishes and locate the oviduct under the stereo microscope using fine forceps. The flushing procedure differs depending on whether blastocysts (4a) or morulas (4b) are to be collected.
-
4a.
Flush blastocysts (3.5 dpc) from the uterus with a 23G needle and a 1-ml syringe filled with M2 medium. Bend the 23G needle approximately 45° and insert the needle in the end of the uterus that is closest to the oviduct. Hold the needle in place with fine forceps while pressing the plunger. Swelling of the uterus indicates successful flushing and can be monitored under the microscope.
-
4b.
Flush morulas (2.5 dpc) from the oviduct with a blunt 32G needle and a 5-ml Luer-Lok syringe filled with M2 medium. Flushing morulas from oviducts is difficult and requires technical skill. Locate the infundibulum, the funnel-shaped end of the oviduct that is nearest to the ovary. Insert the needle in the infundibulum and the first part of the oviduct and flush while holding the needle in place with fine forceps.
-
5.
Using a mouth pipette, collect all embryos and transfer them to a drop (approximately 250 μl) of M2 medium. Wash embryos two more times by transferring them twice to a fresh drop of M2 medium.
-
6.
Remove the zona pellucida from morulas by treating them with Tyrode solution (Note 5).
Pluripotin-based ES Cell Isolation (Fig. 1)
-
1.
a) Prepare gelatinized 12-well plates (1 well/embryo, Note 3) with mitomycin-C-treated MEFs (Note 6) 1 day before the embryos are collected. b) Change medium from MEF to SR-ES cells medium (2 ml/well) with pluripotin (2 μM, Note 4) 1 h before the embryos are collected (Note 7). Incubate at 37°C in 5% CO2 (Note 8).
-
2.
Plate blastocysts and morulas individually in 12-well plates using a mouth pipette. Monitor the proper distribution of embryos under a stereo microscope (inside a laminar air flow).
-
3.
Refresh the pluripotin-containing SR-ES cell medium every 2–3 days by using a mouth pipette (Note 9).
-
4.
a) Prepare a gelatinized 96-well plate (flat bottom, 1 well/outgrowth) with mitomycin-C-treated MEFs and MEF medium (200 μl/well) 1 day before the trypsinization of spherical ICM outgrowths. b) Change medium from MEF to FBS-ES cell medium without pluripotin 1 h before dissociated blastocyst outgrowths are plated. Omit pluripotin from this point on (Note 10).
-
5.
Dispense sterile PBS in a 96-well curved bottom plate (30 μl/well, 1 well/outgrowth).
-
6.
Pick the large, loosely attached spherical outgrowths between day 12 and 18 (Note 11) using a pipette with a 10-μl tip. Locate outgrowths under the stereo microscope (inside a laminar air flow) and transfer each outgrowth in approximately 5 μl medium to a separate PBS-filled well in the 96-well plate. Use a different tip for each outgrowth. If an entire row in the 96-well plate is filled (12 outgrowths), proceed to step 7. Check that all outgrowths are successfully transferred to the 96-well plate before trypsinization.
-
7.
Add 50 μl of 0.25% trypsin to each well using a multichannel pipette and incubate for 3 min at 37°C in 5% CO2 (Note 12). Trypsinize only 12 outgrowths (one row) at a time.
-
8.
Add 100 μl FBS-ES cell medium (from the flat-bottomed 96-well plate) to each well using a multichannel pipette. Dissociate outgrowths into single cells and small cell clumps by pipetting 10–15 times. Transfer dissociated cells to the 96-well mitomycin-C-treated MEF plate (flat bottom).
-
9.
Next day, change medium from FBS-ES to SR-ES cell medium.
-
10.
After 1–2 days of growth in SR-ES cell medium (refresh every day), the newly established ES cell colonies can be passaged to 24-well plates (Note 13). For that, prepare gelatinized 24-well plates (1 well/ES cell line) with mitomycin-C-treated MEFs and MEF medium (1 ml/well) 1 day in advance. Change medium from MEF to FBS-ES cell medium 1 h before the next step (trypsinization).
-
11.
Wash the ES cell colonies (from the 96-well plate) with PBS. Add 30 μl of 0.25% trypsin to each well (in the 96-well plate) and incubate for 3 min at 37°C in 5% CO2.
-
12.
Add 100 μl FBS-ES cell medium to each well, dissociate ES cell colonies into single cells by pipetting 10–15 times. Transfer dissociated cells to 24-well mitomycin-C-treated MEF plates.
-
13.
Next day, change medium from FBS-ES to SR-ES cell medium.
-
14.
After 1–2 days of growth in SR-ES cell medium (refresh every day), the ES cell lines can be passaged to three wells in a 24-well plate (Fig. 1). For that, repeat step 10.
-
15.
Repeat step 11 by adding 200 μl of 0.25% trypsin to each well in the 24-well plate.
-
16.
Fill a plastic pipette with 6 ml MEF medium, dissociate ES cell colonies into single cells by pipetting, and transfer the cell suspension to a 15-ml centrifuge tube. Centrifuge 5 min at 1,000 rpm. Aspirate and discard the medium.
-
17.
Resuspend the pellet in 3 ml FBS-ES cell medium and distribute in three wells of a 24-well plate (Note 14).
-
18.
Next day, change medium from FBS-ES to SR-ES cell medium.
-
19.
After 1–2 days of growth in SR-ES cell medium (refresh every day) the ES cells can be passaged to 6-well plates. For that, prepare gelatinized 6-well plates (1 well/ES cell line) with mitomycin-C-treated MEFs (similar to what is described in step 10).
-
20.
Repeat step 11 by adding 200 μl of 0.25% trypsin to each well in the 24-well plate.
-
21.
Repeat step 16.
-
22.
Resuspend the pellet in 1 ml FBS-ES cell medium and put it in a well (of a 6-well plate) that contains MEFs and 1 ml of FBS-ES cell medium.
-
23.
Next day, change medium from FBS-ES to SR-ES cell medium.
-
24.
After 1–2 days of growth in SR-ES cell medium (refresh every day), three freezings can be made from one 70% confluent well (of a 6-well plate) using standard freezing techniques (Note 15).
-
25.
Established ES cell lines can be propagated further using SR-ES medium (or other ES cell media; refresh every day). Split ES cells 1/4 or 1/5 every 2–3 days. Established ES cell lines were validated (Note 16), generated chimeras (Note 17) and displayed a normal mitotic karyotype (Note 18).
-
26.
Expected results: ES cells can be isolated from blastocysts with this protocol with an efficiency of up to 100% (Table 1) (Note 19). In comparison with previous reported protocols our method is robust and user-friendly (Note 20).
Notes
-
1.
Mice were housed in individually ventilated cages in a specific-pathogen-free animal facility. All experiments on mice were conducted according to institutional, national, and European animal regulations. Animal protocols were approved by the ethics committee of Ghent University. The generation of p120ctnKOC/+ and p120ctnKIC/+ mice, both on a C57BL/6 background, will be reported elsewhere (Pieters et al., in preparation). ROSA26 reporter (R26R) mice [14] were backcrossed to C57BL/6. The ROSA26-antiLuc reporter mice were generated by homologous recombination in G4 ES cells [15] using the ROSA26-antiLuc construct obtained from the laboratory of Dr. Dagmar Wirth [16]. B6/129 ROSA26-antiLuc F1-hybrid mice were subsequently outbred and maintained on a mixed B6/129/Swiss background. The p120ctn null allele was generated by Cre-mediated removal of a floxed region in the p120ctn gene (containing exons 3–8) [17, 18].
-
2.
A mouth pipette consists of a micropipette, a micropipette holder, tubing and an aspirator mouth piece. For detail, see Nagy et al., [19]. Micropipettes can be custom made by heating the finest part of a Pasteur pipette for a few seconds in the flame of a Bunsen burner, removing the pipette from the flame, and pulling both ends. The procedure can be repeated several times until a needle with a diameter slightly larger than a blastocyst is obtained (100–200 μm).
-
3.
All culture plates or dishes have to be coated with 0.1% gelatin. Dissolve 0.5 g in 500 ml distilled water, autoclave and store at 4°C. Before seeding the MEFs, add 0.1% gelatin to the culture plates, incubate 5 min at 37°C in 5% CO2 and aspirate the gelatin solution.
-
4.
Dissolve 5 mg pluripotin in 2.27 ml DMSO (4 mM, 2000X stock) and store aliquots at −20°C. Add pluripotin to the medium just before use (final concentration: 2 μM).
-
5.
Prepare a bacterial grade plate with several drops (250 μl) of M2 medium and Tyrode solution. Transfer up to five embryos to a drop of Tyrode solution by mouth pipetting and monitor the deterioration of the zona pellucida under the microscope. When the zona starts to deteriorate, quickly transfer the embryo to a drop of M2 medium (exposure of zona-free embryos to Tyrode solution may harm them). Remains of the zona pellucida can be removed mechanically by pipetting the embryo several times up and down using a mouth pipette. Transfer embryos freed from their zona pellucida to a fresh drop of M2 medium (make sure there are no traces of Tyrode solution left in the needle of the mouth pipette).
-
6.
Detailed protocols to isolate MEFs have been reported elsewhere [20, 21]. In brief, collect E13.5 embryos, remove the head and internal organs, and trypsinize the embryonal tissue. Dissociate the tissue into single cells and plate and grow them in MEF medium on gelatinized plates. Two embryos provide sufficient cells for one 15-cm culture dish. Confluent MEF plates can be frozen immediately (passage 0) or expanded two times before freezing (passage 2). One vial of passage-2 MEFs, frozen from a confluent 15-cm dish, can be divided over three to four gelatinized culture plates (6-, 12-, 24-, or 96-well) or over three to four gelatinized 10-cm culture dishes. After 2–3 days the MEFs reach confluency and can be inactivated by treating them with Mitomycin C (10 μg/ml) for 3 h. Two milligrams of Mitomycin C is dissolved in 2 ml sterile PBS. This 100X stock can be stored at 4°C. Mitomycin C-inactivated MEFs should be washed at least three times before use.
-
7.
Blastocysts are plated in gelatinized 12-well plates with mitomycin-C-treated MEFs (Fig. 2a, top). However, SR-ES cell medium that is supplemented with pluripotin is harmful to the MEF cells and it is not unusual that half of the MEF cells die during the first week. Nevertheless, this massive death of MEF cells does not affect the ES cell isolation efficiency. In contrast, if blastocysts were plated in uncoated 12-well plates without MEFs (Fig. 2a, bottom), they did not always form big spherical outgrowths and the success rate for isolation ES cells dropped to 40% (8 ES cell lines form 20 blastocysts). In addition, it has been reported that MEFs, unlike ES cells, do not adhere to gelatin-coated dishes in the presence of SR-ES cell medium (Gibco, data sheet Cat. No. 10828). Therefore, MEFs should always be seeded in MEF medium. This MEF medium is replaced by FBS-ES cell medium 1 h before trypsinization.
-
8.
MEFs, primary outgrowths and ES cells are grown at 37°C with 5% CO2.
-
9.
Incubation of primary outgrowths in the presence of SR-ES cell medium and pluripotin results in the formation of big spheres that do not much resemble the classical ICM outgrowths (Fig. 2, compare panel a to b or c). These spheres are mostly loosely attached to the MEFs and often appear in suspension. In contrast to classic ICM outgrowths that stick to the MEFs, the loose spherical outgrowths can be lost when the medium is aspirated. Indeed, in our hands the efficiency of initial mouse ES cell isolations (using our protocol) was not maximal due to the loss of non-attached spheres while changing medium. Therefore, loose outgrowths should be transferred to an empty well filled with SR-ES cell medium using a mouth pipette and then transferred back once the medium is refreshed. The medium of outgrowths sticking to the feeder layer can either be refreshed as such or the outgrowths are picked and transferred using a mouth pipette as mentioned above; we recommend using a mouth pipette. Culturing blastocysts the first 2 days without pluripotin resulted in the formation of flat ICM outgrowths, which stay firmly attached to the underlying feeder and trophectoderm layer after pluripotin administration (Fig. 2b). However, these flat ICM outgrowths are harder to pick and trypsinize and are less efficient (33%) in establishing ES cell lines.
-
10.
When pluripotin was present in the FBS-containing ES cell medium, most of the cells failed to attach to the MEF layer and were lost when changing the medium to SR-containing ES cell medium. In contrast, when cells dissociated after the first trypsinization were seeded in the absence of pluripotin, they readily attached to the MEFs, and primary ES cell colonies were formed efficiently within days.
-
11.
The primary outgrowth should have a diameter of about 300 μm at the time of trypsinization. Blastocyst outgrowths can be cultured for up to 18 days without losing their ability to form ES cell lines. The main advantage of a long initial cultivation in pluripotin-containing SR-ES cell medium is that it allows a large number of undifferentiated cells to develop before the first trypsinization. Apparently, the establishment of ES cell lines became more reliable once large pluripotent spheres were obtained: 79 initial outgrowths gave rise to 78 ES cell lines (99%). In addition, using a transgenic Oct4-EGFP line (EGFP driven by the Oct4-promoter), Yang and colleagues [13] showed that outgrowths from Oct4-EGFP blastocysts expressed EGFP for up to 14 days. Smaller outgrowths can also give rise to ES cell lines but they generate fewer colonies, which have to be cultured for longer times before they can be passaged.
-
12.
The outgrowths are readily dissociated into single cells in a single trypsinization step. Sequential digestion, as suggested by Yang and colleagues [13], is not required.
-
13.
If a well of the 96-well plate contains only a few ES cell colonies, it is better to let these colonies grow for a few more days, and trypsinize (step 11) and plate them again in a new well of a 96-well mitomycin-C-treated MEF plate (flat bottom, containing FBS-ES cell medium). Next day, change medium to SR-ES cell medium. Wait for ES cell colonies to appear and proceed again to step 11.
-
14.
At this stage you can decide to make a freezing (Note 15) and/or a DNA preparation. For a DNA preparation, transfer one third of the ES cells (1/3 of a 24-well) to gelatinized 24-well plates (without MEFs). After 30 min, transfer the cell suspension to a new well to get rid of most of the MEFs, which would otherwise contaminate the DNA preparation. MEFs attach faster than ES cells to gelatinized plates. Genomic DNA is prepared from confluent wells according to Laird et al. [22]. Alternatively, you can speed up the ES cell isolation by transferring ES cells from two wells (of a 24-well plate) to a 6-well plate.
-
15.
Our freezing method: ES cells (70% confluent) from a 6-well plate are trypsinized (800 μl), dissociated and centrifuged. The cell pellet is resuspended in 10% DMSO and 90% FCS (1 ml/freezing). Cells are placed on ice for 30 min and frozen at −80°C. Frozen cells are kept at −80°C for up to several weeks and in liquid nitrogen for long time storage.
-
16.
The newly established ES cell lines could be maintained in a morphologically undifferentiated state for over 10 passages (Fig. 3a), they expressed the transcription factor Oct-4 (Fig. 3b), and they readily formed embryoid bodies (EBs, Fig. 3c). Those EBs could be differentiated into various cell types (Fig. 4d), including neurectoderm (Nestin), mesoderm (Smooth muscle actin), skeletal muscle (α-Dystrobrevin), beating cardiomyocytes (Troponin), and endothelial and hematopoietic cell types (data not shown). The procedure for forming embryoid bodies was similar to that described before [23, 24]. In brief, ES cells were feeder-depleted and passaged once on gelatinized plates. For random differentiation into EBs, ES cells were plated in different dilutions unto bacterial grade Petri-dishes in differentiation medium (IMDM medium (Invitrogen) supplemented with 15% FBS (Hyclone), 5% PFMHII (Invitrogen), 2 mM L-glutamine (Invitrogen), 0.4 mM MTG (Sigma), 50 μg/ml ascorbic acid (Sigma) and penicillin (100 U/ml)-streptomycin (100 μg/ml) (Invitrogen). EBs were allowed to form in these dishes for 10 days. The EBs were then plated unto gelatinized culture dishes and allowed to attach and spread out for 10 days in differentiation medium. The staining procedure for ES cells and plated EBs involved fixation with 4% paraformaldehyde, permeabilization with 0.2% TX-100 and incubation at room temperature for 2 h with primary antibody and 1 h with secondary antibody (Molecular Probes, Eugene, OR). The following primary antibodies were used: mouse monoclonal anti-Oct-4 (1/100, Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal anti-Nestin (1/100, Becton Dickinson, New York), mouse mocolonal anti-Smooth muscle actin (SMA, 1/100, Sigma), mouse monoclonal anti-Troponin (1/100, Thermo Scientific, Waltham, MA), rabbit polyclonal anti-α-Dystrobrevin (1/200, H-300, Santa Cruz). Confocal microscopy was performed using a Leica TCS SP5 confocal scan head attached to a Leica DM IRE2 inverted microscope and a PC running Leica AF software version 2.5.
Fig. 3 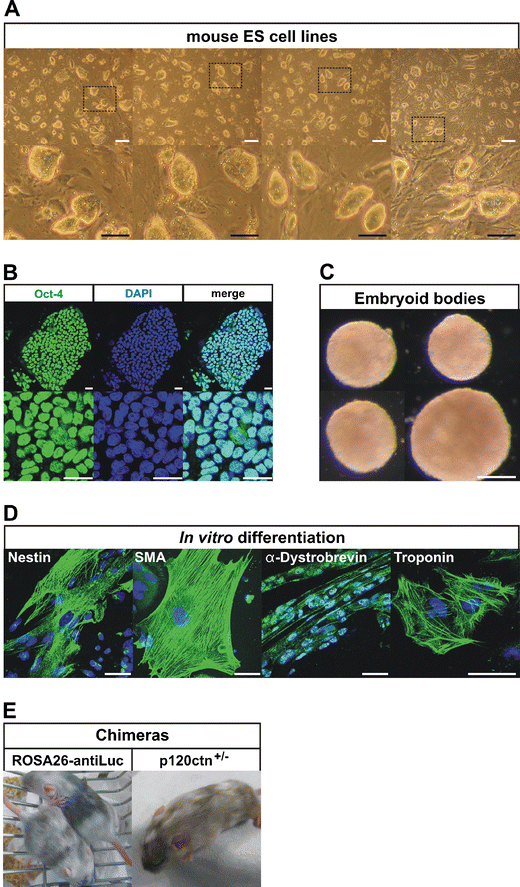
Properties of ES cell lines derived from pluripotin-treated primary outgrowths. a Morphology of newly established mouse ES cell lines. White scale bars: 200 μm, black scale bars: 100 μm. b ES cells are positive for stem cell marker Oct-4. Scale bars: 25 μm. c ES cells form embryoid bodies (EBs) after 10 days of culture in suspension. Scale bars: 200 μm. d Marker analysis after random in vitro differentiation of ES cells. EBs (from c) were plated on gelatinized culture dishes and allowed to attach and spread out for another 10 days in order to differentiate into various cell types, including neurectoderm (Nestin), mesoderm (Smooth muscle actin), skeletal muscle (α-Dystrobrevin) and beating cardiomyocytes (Troponin). Scale bars: 50 μm. e Chimeras generated from ROSA26-AntiLuc and p120ctn+/− ES cells
Fig. 4 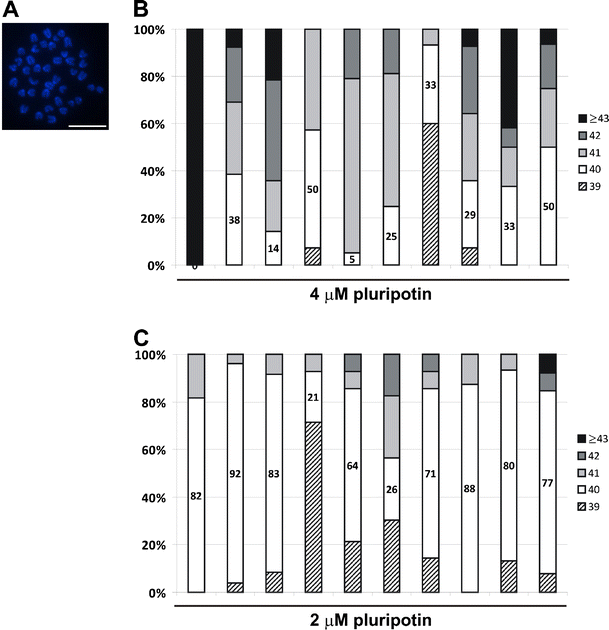
Chromosomal analysis of pluripotin-derived ES cells a Fluorescent image of a DAPI-stained mitotic spread from a mouse ES cell line, depicting 40 acrocentric chromosomes. Scale bar: 5 μm. b–c Graphs depicting the percentage of mitotic spreads that contain 39, 40, 41, 42 and 43 or more chromosomes. Mitotic spreads from 10 ES cell lines derived with 4 μM (b) or 2 μM (c) pluripotin were analyzed. An ES cell line is considered normal if 70% or more of its spreads contain 40 chromosomes. All ES cell lines derived with 4 μM pluripotin showed chromosomal instability, whereas most ES cell lines derived with 2 μM pluripotin exhibited a normal karyotype
-
17.
Chimeras were generated from different pluripotin-derived ES cells (Fig. 3e). ROSA-antiLuc ES cell lines generated weak chimeras, probably because these ES cells exhibited significant chromosomal instability as they were derived with 4 μM pluripotin (Note 18). p120ctn+/− ES cells derived with 2 μM pluripotin generated strong chimeras. Chimeras were generated by diploid aggregation. Briefly, embryos were collected on day E2.5 from superovulated Swiss female mice. Zona pellucida of embryos was removed by treatment with acid Tyrode’s solution (Note 5, Sigma). ES cell colonies were briefly treated with 0.25% trypsin-EDTA (Invitrogen) to form loosely connected clumps of 7–10 cells. Each zona-free embryo was aggregated with a clump of 7–10 cells inside depression wells made in a 35-mm plastic dish (VWR International) with an aggregation needle (BLS Ltd, Hungary). They were cultured overnight in microdrops of KSOM with amino acids (Biognost) under mineral oil (Sigma) at 37°C in 95% air/5% CO2. The next day, blastocysts were transferred into the uteri of 2.5-dpc pseudopregnant Swiss females previously mated with vasectomized males. Chimeras were identified at birth by the presence of black eyes and later by agouti coat pigmentation.
-
18.
Normal mitotic karyotype of mouse ES cells consists of 40 acrocentric chromosomes (Fig. 4a). Chromosomal analysis was performed on 10 ES cell lines derived with 4 μM pluripotin. Chromosomal instability was observed in all of them (Fig. 4b). Thus, these lines are only suitable for in vitro analysis. In contrast, ES-cell lines derived with 2 μM pluripotin during the blastocyst outgrowth phase were generated with high efficiency (100%) and these lines had a normal karyotype (Fig. 4c). In addition, strong chimeras could be derived from these ES cell lines. Yang and colleagues [13] derived ES cells with 3–5 μM pluripotin. Their ES cells showed some chromosomal abnormalities and were not proven to be germline competent [13]. By lowering the pluripotin concentration from 4 to 2 μM, we obtained ES cells that generated strong chimeras and displayed a normal karyotype. To make mitotic spreads for counting chromosomes, ES cells were passaged to gelatinized six-well plates. Nearly confluent ES cell cultures were grown for 2 h with demecolcine (50 ng/ml, Sigma), washed with PBS, and trypsinized. Resuspended ES cells were incubated 15–20 min in warm KCl solution (75 mM) and washed four times with freshly-made cold fixative consisting of a 3:1 mixture of methanol and glacial acetic acid. ES cells were resuspended in 500 μl fixative and dispersed on three wet slides (80 μl/slide) (SuperFrost Plus microscope slides, Klinipath). Air-dried slides were stained with DAPI and 20 individual spreads were counted. Spreads with 38 chromosomes or less were discarded. An ES cell line is considered normal if 70% or more of its spreads contain 40 chromosomes.
-
19.
Isolation of ES cells from morulas turned out to be much less efficient (Table 3) due to decreased formation of primary outgrowths (32% compared to 95%).
Table 3 The derivation efficiency of ES cells from morulas -
20.
Differences between our protocol and that of Yang et al. [13] are listed in Table 2. We also compared the performance of our modified mouse ES cell derivation protocol with other protocols, including the one published by Yang and colleagues [13]. From the nine wild-type C57BL/6 blastocysts that we processed with Yang’s protocol, only five formed small ICM outgrowths (Fig. 2c; top, DIV 10). Most outgrowths spontaneously dissociated after 10 days of culture (Fig. 2c; bottom, DIV 10) and no ES cell lines could be derived from them. In contrast, all six wild-type C57BL/6 blastocysts processed according to our modified protocol formed large spherical ICM outgrowths after 10 days of culture (Fig. 2a, DIV 10). ES cell lines could be established for all six outgrowths (Table 1). Remarkably, the two pluripotin-based protocols gave rise to morphologically different ICM outgrowths. This discrepancy between our results and those of Yang and colleagues [13] might have been due to the use of different ES cell media in which blastocysts are seeded. On the first 2 days, Yang and colleagues [13] used FBS-ES cell medium (Table 2), which might contain factors that stimulate attachment of blastocyst outgrowths to the substrate in the presence of pluripotin. These factors are probably not present in the chemically defined SR-ES cell medium that we used. On the other hand, seeding blastocysts in SR-ES cell medium alone allows attachment of the outgrowths to the MEFs and the formation of flat ICM outgrowths (Fig. 2b). Our data indicate that pluripotin, at least in SR-ES cell medium, blocks the attachment of outgrowths to the MEFs. Such a block might promote growth and result in the formation of bigger but loosely attached outgrowths.


References
Martin, G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proceedings of the National Academy of Sciences of the United States of America, 78, 7634–7638.
Evans, M. J., & Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature, 292, 154–156.
Bryja, V., Bonilla, S., Cajanek, L., et al. (2006). An efficient method for the derivation of mouse embryonic stem cells. Stem Cells, 24, 844–849.
Cheng, J., Dutra, A., Takesono, A., Garrett-Beal, L., & Schwartzberg, P. L. (2004). Improved generation of C57BL/6J mouse embryonic stem cells in a defined serum-free media. Genesis, 39, 100–104.
Buehr, M., & Smith, A. (2003). Genesis of embryonic stem cells. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 358, 1397–1402. discussion 1402.
Lodge, P., McWhir, J., Gallagher, E., & Sang, H. (2005). Increased gp130 signaling in combination with inhibition of the MEK/ERK pathway facilitates embryonic stem cell isolation from normally refractory murine CBA blastocysts. Cloning and Stem Cells, 7, 2–7.
Qi, X., Li, T. G., Hao, J., et al. (2004). BMP4 supports self-renewal of embryonic stem cells by inhibiting mitogen-activated protein kinase pathways. Proceedings of the National Academy of Sciences of the United States of America, 101, 6027–6032.
Umehara, H., Kimura, T., Ohtsuka, S., et al. (2007). Efficient derivation of embryonic stem cells by inhibition of glycogen synthase kinase-3. Stem Cells, 25, 2705–2711.
Ying, Q. L., Wray, J., Nichols, J., et al. (2008). The ground state of embryonic stem cell self-renewal. Nature, 453, 519–523.
Li, P., Tong, C., Mehrian-Shai, R., et al. (2008). Germline competent embryonic stem cells derived from rat blastocysts. Cell, 135, 1299–1310.
Buehr, M., Meek, S., Blair, K., et al. (2008). Capture of authentic embryonic stem cells from rat blastocysts. Cell, 135, 1287–1298.
Chen, S., Do, J. T., Zhang, Q., et al. (2006). Self-renewal of embryonic stem cells by a small molecule. Proceedings of the National Academy of Sciences of the United States of America, 103, 17266–17271.
Yang, W., Wei, W., Shi, C., et al. (2009). Pluripotin combined with leukemia inhibitory factor greatly promotes the derivation of embryonic stem cell lines from refractory strains. Stem Cells, 27, 383–389.
Soriano, P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genetics, 21, 70–71.
George, S. H., Gertsenstein, M., Vintersten, K., et al. (2007). Developmental and adult phenotyping directly from mutant embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America, 104, 4455–4460.
Sandhu, U., Cebula, M., Behme, S., et al. (2010). Strict control of transgene expression in a mouse model for sensitive biological applications based on RMCE compatible ES cells. Nucleic Acids Research, 39, e1.
Betz, U. A., Vosshenrich, C. A., Rajewsky, K., & Muller, W. (1996). Bypass of lethality with mosaic mice generated by Cre-loxP-mediated recombination. Current Biology, 6, 1307–1316.
Davis, M. A., & Reynolds, A. B. (2006). Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Developmental Cell, 10, 21–31.
Nagy, A., Gertsenstein, M., Vintersten, K., & Behringer, R. (2003). Manipulating the mouse embryo: A laboratory manual (3rd ed.). New York: Cold Spring Harbor Laboratory Press.
Hogan, B., Beddington, R., Constantini, F., & Lacy, E. (1994). Manipulating the mouse embryo: A laboratory manual (2nd ed.). New York: Cold Spring Harbor Laboratory Press.
Bryja, V., Bonilla, S., & Arenas, E. (2006). Derivation of mouse embryonic stem cells. Nature Protocols, 1, 2082–2087.
Laird, P. W., Zijderveld, A., Linders, K., Rudnicki, M. A., Jaenisch, R., & Berns, A. (1991). Simplified mammalian DNA isolation procedure. Nucleic Acids Research, 19, 4293.
Bibel, M., Richter, J., Lacroix, E., & Barde, Y. A. (2007). Generation of a defined and uniform population of CNS progenitors and neurons from mouse embryonic stem cells. Nature Protocols, 2, 1034–1043.
Bibel, M., Richter, J., Schrenk, K., et al. (2004). Differentiation of mouse embryonic stem cells into a defined neuronal lineage. Nature Neuroscience, 7, 1003–1009.
Acknowledgements
This work was supported by grants from the Queen Elisabeth Medical Foundation (G.S.K.E.), Belgium, Research Foundation (F.W.O.) - Flanders, and from the Geconcerteerde Onderzoeksacties of Ghent University. Tim Pieters and Lieven Haenebalcke have been supported by the Instituut voor de Aanmoediging van Innovatie door Wetenschap en Technologie in Vlaanderen (IWT). We acknowledge Dr. Amin Bredan for critical reading of the manuscript, and the members of our research group for valuable discussions.
Conflict of Interest
The authors declare no potential conflicts of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pieters, T., Haenebalcke, L., Hochepied, T. et al. Efficient and User-Friendly Pluripotin-based Derivation of Mouse Embryonic Stem Cells. Stem Cell Rev and Rep 8, 768–778 (2012). https://doi.org/10.1007/s12015-011-9323-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-011-9323-x