
Abstract
Prolonged in vitro culture of human embryonic stem (hES) cells can result in chromosomal abnormalities believed to confer a selective advantage. This potential occurrence has crucial implications for the appropriate use of hES cells for research and therapeutic purposes. In view of this, time-point karyotypic evaluation to assess genetic stability is recommended as a necessary control test to be carried out during extensive ‘passaging’. Standard techniques currently used for the cytogenetic assessment of ES cells include G-banding and/or Fluorescence in situ Hybridization (FISH)-based protocols for karyotype analysis, including M-FISH and SKY. Critical for both banding and FISH techniques are the number and quality of metaphase spreads available for analysis at the microscope. Protocols for chromosome preparation from hES and human induced pluripotent stem (hiPS) cells published so far appear to differ considerably from one laboratory to another. Here we present an optimized technique, in which both the number and the quality of chromosome metaphase spreads were substantially improved when compared to current standard techniques for chromosome preparations. We believe our protocol represents a significant advancement in this line of work, and has the required attributes of simplicity and consistency to be widely accepted as a reference method for high quality, fast chromosomal analysis of human ES and iPS cells.
Similar content being viewed by others



Introduction
A euploid karyotype is one of the defining characteristics of hES cells [1]. Once established, hES cell lines are expected to be chromosomally stable. However, karyotypic abnormalities of hES cells in long term cultures have been repeatedly reported by independent laboratories [2]. Some of the recurring abnormalities, for instance trisomy 12 and trisomy 17, appear to provide a selective advantage, with cells carrying such chromosomal extra copies being able to replace the cell population in 5–10 passages [3]. The duration of culture, conditions (presence or absence of feeders) and replace-by-passaging methods (mechanical versus enzymatic) have been pointed at as possible contributing factors to chromosomal changes in hES cells [4, 5]. However, recent evidence seems to support the notion that regardless of culture conditions, some hES cell lines are inherently more inclined to karyotypic instability [6]. To which extent differences in the chromosomal complement affect their ability to differentiate, as well as their proliferative capacity, is still being investigated [7, 8]. In the meantime, while establishing and/or maintaining hES cell lines, time-point karyotypic analysis is widely recommended as one of the important steps in the quality control process [3, 9–11]. There have been a few reports underscoring an increasing interest in the possible use of microarray-based techniques—such as Comparative Genomic Hybridization (CGH), single nucleotide polymorphism (SNP) analysis and transcriptional profiling—to monitor the chromosomal stability of hES and hiPS cells in culture [12–14]. However, classical and molecular cytogenetic protocols are still considered the default approach for routine karyotypic assessment. The analysis at the microscope that classical cytogenetic protocols—such as G-banding—entail is extremely laborious and requires highly specialized training. More modern and user-friendly Fluorescence in situ Hybridization (FISH)-based protocols for karyotyping, such as Multiplex FISH (M-FISH) and Spectral Karyotyping (SKY), or simple “target analysis” protocols to detect recurring aneuplodies by means of chromosome specific probes, provide a suitable alternative, and are increasingly used by different laboratories for in-house, rapid chromosomal assessment of ES cells. Crucial for both banding and FISH techniques are the number and quality of metaphase ‘spreads’ available for analysis at the microscope. Protocols for chromosome preparation (or ‘harvesting’) from hES cells published so far appear to vary considerably from one laboratory to another. In this paper we describe an optimized technique for chromosome preparation from hES and hiPS cells, in which both the number and the quality of metaphase spreads were substantially improved when compared to current standard techniques.
Materials
Cell Culture
-
1.
hES cells or iPS cells
-
2.
1 × Phosphate Buffered Saline (PBS)
-
3.
Matrigel (BD, U.K.)
-
4.
mTeSR™1 (STEMCELL Technologies Inc, France)
-
5.
TrypLE Express 1× (Invitrogen, U.K.)
-
6.
ROCK inhibitor Y-27632 (Merck Biosciences, Germany)
-
7.
Penicillin at 10,000 units ml−1 and Streptomycin at 10 mg ml−1
-
8.
6-well culture dish(es)
-
9.
15 ml conical centrifuge tubes
-
10.
Access to a hemocytometer
-
11.
Access to a Class II microbiological cabinet
-
12.
Access to an incubator at 37°C with 5% CO2
-
13.
Access to an inverted microscope
-
14.
Access to a bench-top centrifuge
Chromosome Harvest
-
1.
Nocodazole (Sigma Aldrich, U.K., http://www.sigmaaldrich.com), 2.5 mg/ml stock solution in dimethyl sulfoxide, (DMSO) (Sigma Aldrich, U.K) (Note 1). Aliquot and store at −20°C
-
2.
“Buffered” hypotonic solution (0.4% KCl with HEPES) (Genial Genetics, U.K., http://www.genialgenetics.com)
-
3.
Carnoy’s fixative solution: 3:1 (v/v) methanol/glacial acetic acid, freshly prepared
-
4.
Pre-cleaned microscope slides (VWR International)
-
5.
1 mL disposable plastic Pasteur pipettes
-
6.
Slide storage boxes (VWR International)
-
7.
Access to a water-bath at 37°C
-
8.
Access to a phase-contrast microscope with a 10× or 20× objective
Chromosome Quality Assessment
-
1.
Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, UK)
-
2.
24XCyte mFISH probe kit (MetaSystems, Germany, http://www.metasystems-international.com)
-
3.
Access to an epifluorescent light microscope with a 63× and/or a 100× oil immersion objective equipped with a charge-coupled device (CCD) camera and appropriate operating software for digital image capturing and analysis (e.g. CytoVysion system), consisting of an Olympus BX-51 epifluorescence microscope coupled to a JAI CVM4+ CCD camera (Genetix, U.K., http://www.genetix.com)
Methods
Cell Culture
-
1.
Cells are grown in a feeder free system in a 6-well dish (Note 2).
-
2.
Two days before the chromosome harvest procedure, remove the spent medium from a well of confluent cells.
-
3.
Add 1 ml of sterile PBS, at 37°C, to wash the cells, and remove.
-
4.
Add 0.5 ml of sterile TrypLE Express, warmed at 37°C. Place the dish in a 37°C incubator, for 5 min. Using an inverted microscope, confirm that all the cells have detached from the tissue culture dish.
-
5.
Resuspend the cells in 10 ml of warm PBS, at 37°C, and transfer to a 15 ml conical centrifuge tube.
-
6.
Count the cell number using a hemocytometer cell counter.
-
7.
Centrifuge the cell suspension at 365 g for 10 min, and resuspend 1–2 × 106 cells in 2 ml of mTeSR™1 medium.
-
8.
Add ROCK inhibitor Y-27632 to a final concentration of 10 μM, and plate on a fresh well, coated in Matrigel.
-
9.
Place in the CO2 thermostatic incubator.
Chromosome Harvest and Quality Assessment
-
1.
The day after, change medium and add nocodazole at a final concentration of 0.1 μg/ml, and incubate for 16 h in the CO2 thermostatic incubator (Note 3).
-
2.
Following incubation with the mitotic synchronizing agent, trypsinize the cells as described above, collect in the culture medium, and centrifuge at 365 g, for 10 min.
-
3.
Remove the supernatant by pipetting it out of the tube, and fully resuspend the cell pellet by gently flicking the tube.
-
4.
Add 5 ml of “Buffered” hypotonic solution (Notes 4 and 5), and pipette delicately the cell suspension, to evenly disperse the cells. Incubate for 30 min at 37°C.
-
5.
Centrifuge the cells as before.
-
6.
Gently resuspend the cell pellet, and quickly add about 500 μl of cold Carnoy’s fixative solution.
-
7.
Mix by pipetting, then bring the total volume up to 10 ml with fresh cold fixative. Incubate for 30 min at room temperature.
-
8.
Centrifuge the cells as before and wash in fixative for a second time. Incubate for 10 min.
-
9.
Following a final spin, resuspend the cell pellet in 200–300 μl of fresh fixative, and drop 30–50 μl of the cell suspensions onto clean slides.
-
10.
Places the slides on a warm plate, 37°C, and allow to air-dry.
-
11.
Check the cell suspension quality by observing the slide with a phase contrast microscope, 10× objective. There should be a good number of metaphases, and no cytoplasmic halo should be visible (Note 6).
Notes
-
1.
It is important to use a very concentrated nocodazole stock solution, so that a minimal volume of DMSO is added to the cell culture, as an excess of DMSO might be toxic to the hES/hiPS cells. The stock solution may be diluted in PBS immediately prior to use, but discard any unused diluted material as nocodazole is not stable for prolonged periods when diluted in a water-based buffer.
-
2.
For our study we used two human embryonic stem cell lines—HUES-2 and HUES-10 [15]—cultured under license from the UK Stem Cell Steering Committee—and one human induced pluripotent stem cell line—iPS-DF19-9-11T.H [16]—grown as in Karlsson et al. [17]. Briefly, cells were seeded onto wells coated with Matrigel and cultured in mTeSR™1 medium with the addition of 10 μM ROCK inhibitor Y-27632. The protocol for chromosome preparation here presented works equally well with hES and hiPS cells grown on a mouse embryonic fibroblasts (MEF) feeder layer.
-
3.
Standard protocols for chromosome preparation in stem cells normally make use of Colcemid® (a synonym of demecolcine in solution), a well known and widely used mitotic spindle inhibitor. Oddly, concentrations of demecolcine used and published so far by different research groups appear to vary strikingly, the most frequently used concentration being either 0.1 μg/ml or 0.1 mg/ml. Accordingly, we set up parallel cultures of HUES-2 cells testing two different concentrations of demecolcine (0.1 μg/ml and 0.1 mg/ml), and compared them to HUES-2 cell cultures treated with nocodazole, an alternative anti-mitotic agent, at the final concentration of 0.1 μg/ml. The cultures were incubated with the mitotic agents for either 4 h or 16 h (effectively an overnight incubation). After fixation, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI), and analyzed at the microscope (Fig. 1). Ten random fields from each of the slides prepared under different conditions were collected. The efficacy of the mitotic arrest treatment was assessed dividing the total number of metaphases observed by the total number of cells analyzed (Tables 1 and 2). The statistical significance of the differences between the various treatments was measured using a 2 × 2 contingency table, with Fisher exact test. Comparison between the cultures revealed a sustained incubation (16 h) with a low dose of nocodazole (0.1 μg/ml) as the optimal mitotic arrest treatment able to provide the highest yield of metaphases (15.2%). A sustained incubation (16 h) with a low dose of demecolcine (0.1 μg/ml) provided the second highest yield of metaphases (8%).
Fig. 1 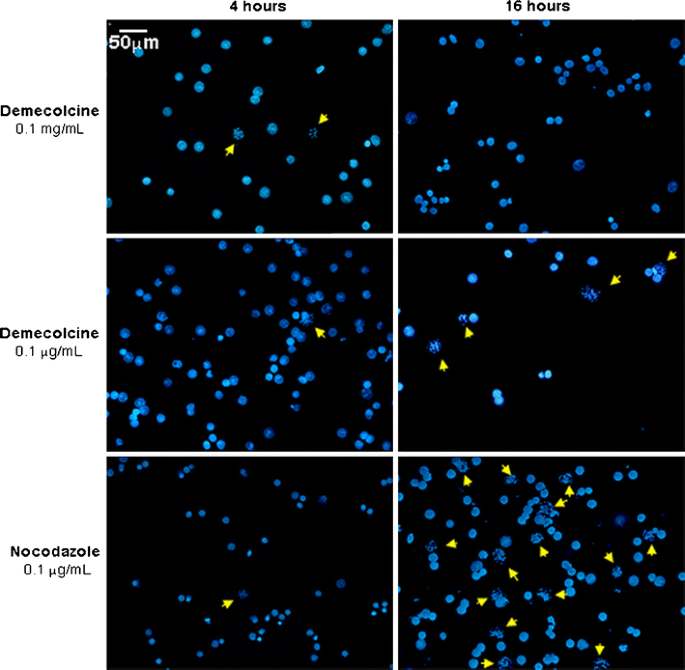
Efficiency of mitotic arrest following treatment with either demecolcine or nocodazole. Different concentrations and incubation times were compared. After fixation the cells were stained with DAPI, and analyzed at the microscope. Ten random fields from each of the slides prepared under different conditions were captured with Genus on the CytoVision system. The yellow arrows identify metaphasic cells
Table 1 Mitotic arrest efficacy Table 2 Statistical comparisons of the differences between treatments (2 × 2 contingency table, analysed with Fisher exact test, 2-tailed) -
4.
With the aim of optimizing the quality of metaphase spreading, we tested two different hypotonic treatments. We set as first parameter for our quality analysis the average number of chromosome overlaps per metaphase. A high number of overlaps makes chromosome identification laborious and can ultimately hinder high-quality karyotypic analysis. The average number of chromosome overlaps was measured and compared in HUES-2 cells treated with either demecolcine or nocodazole, both at 0.1 μg/ml for 16 h, and harvested with either a standard hypotonic solution (0.075 M KCl) or a buffered hypotonic solution (0.4% KCl with HEPES), obtained commercially. We randomly acquired and analyzed twenty-five metaphases from each of the four different harvesting conditions (Fig. 2). We initially scored the number of overlaps between chromosomes per metaphase (Table 3). In some cases, where the metaphase quality was poor and it was impossible to distinguish individual chromosomes, we assigned an arbitrary maximum overlap number of 23, meaning that all the chromosomes were involved in at least one overlapping event. When the different harvests were compared, the number of overlaps in chromosomes harvested with the buffered hypotonic solution—following either nocodazole or demecolcine treatment—was significantly lower than in chromosomes harvested with the standard hypotonic solution (p < 0.001, Student’s T-test for independent samples). In contrast, neither the difference between nocodazole and demecolcine harvests after buffered hypotonic (p = 0.335) nor the difference between nocodazole and demecolcine harvests after standard hypotonic (p = 0.47) was significant. The pH of the cell preparations resuspended in the standard hypotonic solution was slightly acidic (pH 6.5–6.7), similar to the pH of the spent tissue culture medium, while the buffered hypotonic solution maintained a pH of 7.4. The chromosome suspensions obtained by “acidified” KCl were consistently of poor quality, in comparison to the preparation obtained with the buffered hypotonic treatment. However, the addition of a PBS wash, before resuspending the cells in hypotonic, lead to a slight improvement in the KCl-prepared metaphase quality, but decreased their overall number.
Fig. 2 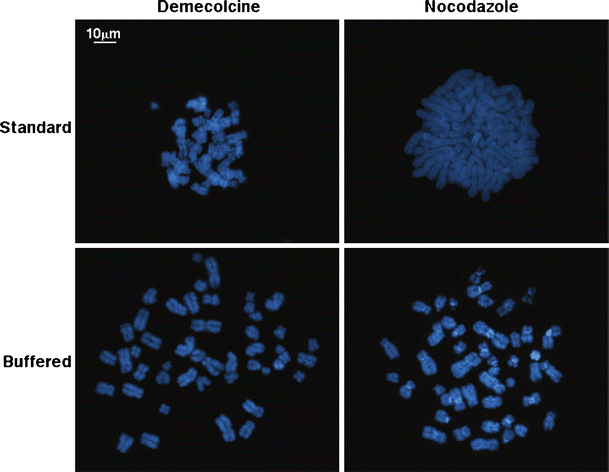
Assessment of chromosome overlaps following different mitotic and hypotonic treatments. The number of chromosome overlaps per metaphase was compared in four different ‘harvest’ conditions. Where the metaphase quality was poor, we assigned an arbitrary number of 23 overlaps per metaphase, meaning that all the chromosomes were involved in at least one overlapping event
Table 3 Metaphase spread quality assessment: chromosome overlaps -
5.
The average chromosome length was set as the second parameter for metaphase spread quality. Increased chromosome length means improved chromosomal morphology and, most importantly, improved longitudinal resolution for karyotyping purposes. For our analysis we chose chromosome 1—the longest chromosome—to reduce the% error during measurements. The length of the two chromosome 1 homologues was measured in μm using the image analysis package MetaMorph v7.6 (Molecular Devices Inc, Danaher Corporation, U.S.A.) in twenty-five randomly captured metaphases from chromosome harvests prepared using the buffered hypotonic, which had given the best results in the previous assay (Fig. 3). The average length of chromosome 1 in the demecolcine 16 h/buffered hypotonic harvest was found to be 20% lower than the average length of chromosome 1 in the nocodazole 16 h/buffered hypotonic solution harvest (Student t-test for independent samples showing a two tailed p value of 0.01).
Fig. 3 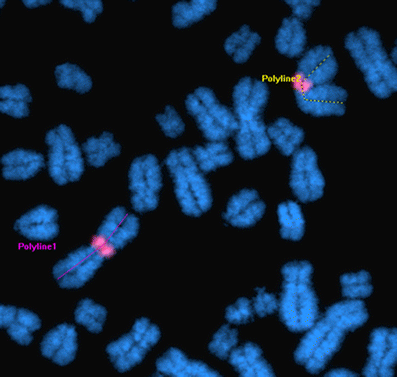
Chromosome length as a parameter for metaphase spread quality for karyotype analysis. The average length of chromosome 1 (here with the centromeric region marked in red) was measured using the image analysis package MetaMorph v7.6 (example above), and compared between the nocodazole 16 h/buffered hypotonic harvest and the demecolcine 16 h/buffered hypotonic harvest
-
6.
Having established—on the basis of the number and quality of metaphase spreads recovered- the ‘0.1 μg/ml nocodazole 16 h/buffered hypotonic solution’ to be the best protocol combination for chromosome harvests of hES cells, we proceeded to further confirm its suitability for FISH-based karyotyping techniques by analyzing by M-FISH chromosome spreads obtained from the three different cell lines used in this study, namely HUES-2 (passage 40), HUES-10 (passage 37) and iPS-DF19-9-11T.H (passage 29) (Fig. 4). M-FISH was performed as recommended by the 24XCyte mFISH probe kit manufacturer. The high standard and improved speed of the M-FISH analysis together confirmed the newly identified mitotic arrest and hypotonic conditions as optimal.
Fig. 4 
Twenty-four color karyotyping of hES cells (HUES-2 and HUES-10) and iPS-DF19-9-11T.H by M-FISH. The high standard and improved speed of the M-FISH analysis have together confirmed the newly identified optimal mitotic arrest and hypotonic conditions to provide a significant technical breakthrough for chromosomal analysis of hES and hiPS cells. While HUES-10 (passage 37) and iPS-DF19-9-11T.H (passage 29) presented a normal karyotype, M-FISH analysis on HUES-2 at passage 40 revealed, as well as chromosome 12 partial trisomy, a couple of structural abnormalities to include a translocation involving an extra copy of chromosome 1q and chromosome 18, and an unbalanced translocation involving chromosomes 17 and 22




References
Hoffman, L. M., & Carpenter, M. K. (2005). Characterization and culture of human embryonic stem cells. Nature Biotechnology, 23, 699–708.
Baker, D. E., Harrison, N. J., Maltby, E., Smith, K., Moore, H. D., et al. (2007). Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nature Biotechnology, 25, 207–215.
Meisner, L. F., & Johnson, J. A. (2008). Protocols for cytogenetic studies of human embryonic stem cells. Methods, 45, 133–141.
Caisander, G., Park, H., Frej, K., Lindqvist, J., Bergh, C., et al. (2006). Chromosomal integrity maintained in five human embryonic stem cell lines after prolonged in vitro culture. Chromosome Research, 14, 131–137.
Grandela, C., & Wolvetang, E. (2007). hESC adaptation, selection and stability. Stem Cell Reviews, 3, 183–191.
Catalina, P., Montes, R., Ligero, G., Sanchez, L., de la Cueva, T., et al. (2008). Human ESCs predisposition to karyotypic instability: is a matter of culture adaptation or differential vulnerability among hESC lines due to inherent properties? Molecular Cancer, 7, 76.
Yang, S., Lin, G., Tan, Y. Q., Deng, L. Y., Yuan, D., et al. (2010). Differences between karyotypically normal and abnormal human embryonic stem cells. Cell Proliferation, 43, 195–206.
Imreh, M. P., Gertow, K., Cedervall, J., Unger, C., Holmberg, K., et al. (2006). In vitro culture conditions favoring selection of chromosomal abnormalities in human ES cells. Journal of Cellular Biochemistry, 99, 508–516.
The International Stem Cell Banking Initiative. (2009). Consensus guidance for banking and supply of human embryonic stem cell lines for research purposes. Stem Cell Reviews, 5, 301–314.
Catalina, P., Cobo, F., Cortes, J. L., Nieto, A. I., Cabrera, C., et al. (2007). Conventional and molecular cytogenetic diagnostic methods in stem cell research: a concise review. Cell Biology International, 31, 861–869.
Nieto, A., Cobo, F., Barroso-Deljesus, A., Barnie, A. H., Catalina, P., et al. (2006). Embryonic stem cell bank: a work proposal. Stem Cell Reviews, 2, 117–126.
Elliott, A. M., Hohenstein Elliott, K. A., & Kammesheidt, A. (2010). High resolution array-CGH characterization of human stem cells using a stem cell focused microarray. Molecular Biotechnology, 46, 234–242.
Narva, E., Autio, R., Rahkonen, N., Kong, L., Harrison, N., et al. (2010). High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nature Biotechnology, 28, 371–377.
Mayshar, Y., Ben-David, U., Lavon, N., Biancotti, J. C., Yakir, B., et al. (2010). Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell, 7, 521–531.
Cowan, C. A., Klimanskaya, I., McMahon, J., Atienza, J., Witmyer, J., et al. (2004). Derivation of embryonic stem-cell lines from human blastocysts. The New England Journal of Medicine, 350, 1353–1356.
Yu, J., Hu, K., Smuga-Otto, K., Tian, S., Stewart, R., et al. (2009). Human induced pluripotent stem cells free of vector and transgene sequences. Science, 324, 797–801.
Karlsson, K. R., Cowley, S., Martinez, F. O., Shaw, M., Minger, S. L., et al. (2008). Homogeneous monocytes and macrophages from human embryonic stem cells following coculture-free differentiation in M-CSF and IL-3. Experimental Hematology, 36, 1167–1175.
Acknowledgements
We thank Prof. William James and Dr. Sally Cowley (William Dunn School of Pathology, University of Oxford) for their assistance in setting up the hESc cultures, and the kind gift of the HUES-2 cells. We also thank Dr. Keith Morris (Molecular Cytogenetics and Microscopy Core, Wellcome Trust Centre for Human Genetics) for help with chromosome measurements. M.A.M is a recipient of the PGSD NSERC and EPA Cephalosporin studentships. S.K is a recipient of the Clarendon Fund/Keble Sloane Robinson Scholarship. This work was supported by The Wellcome Trust [075491/Z/04].
Conflict of interest
The authors declare no potential conflicts of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Daniela Moralli and Mohammed Yusuf have contributed equally to this paper.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Moralli, D., Yusuf, M., Mandegar, M.A. et al. An Improved Technique for Chromosomal Analysis of Human ES and iPS Cells. Stem Cell Rev and Rep 7, 471–477 (2011). https://doi.org/10.1007/s12015-010-9224-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-010-9224-4