
Abstract
In the field of medicinal chemistry there is increasing focus on identifying key proteins whose biochemical functions can firmly be linked to serious diseases. Such proteins become targets for drug or inhibitor molecules that could treat or halt the disease through therapeutic action or by blocking the protein function respectively. The protein must be targeted at the relevant biologically active site for drug or inhibitor binding to be effective. As insufficient experimental data is available to confirm the biologically active binding site for novel protein targets, researchers often rely on computational prediction methods to identify binding sites. Presented herein is a short review on structure-based computational methods that (i) predict putative binding sites and (ii) assess the druggability of predicted binding sites on protein targets. This review briefly covers the principles upon which these methods are based, where they can be accessed and their reliability in identifying the correct binding site on a protein target. Based on this review, we believe that these methods are useful in predicting putative binding sites, but as they do not account for the dynamic nature of protein–ligand binding interactions, they cannot definitively identify the correct site from a ranked list of putative sites. To overcome this shortcoming, we strongly recommend using molecular docking to predict the most likely protein–ligand binding site(s) and mode(s), followed by molecular dynamics simulations and binding thermodynamics calculations to validate the docking results. This protocol provides a valuable platform for experimental and computational efforts to design novel drugs and inhibitors that target disease-related proteins.
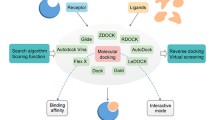
Graphical Abstract



Similar content being viewed by others


Abbreviations
- PDB:
-
Protein data bank
- SBDD:
-
Structure-based drug design
- MD:
-
Molecular dynamics
- 3D:
-
Three-dimensional
- MM-PBSA/GBSA:
-
Molecular Mechanics-Poisson Boltzmann Surface Area/Generalised Born Surface Area
- HIV:
-
Human immunodeficiency virus
- LIE:
-
Linear interaction energy
- FEP:
-
Free energy perturbation
- TI:
-
Thermodynamic integration
- ns:
-
Nanoseconds
References
Zheng, X., Gan, L., Wang, E., & Wang, J. (2012). Pocket-based drug design: exploring pocket space. The AAPS Journal, 15(1), 228–241.
Hopkins, A. L., & Groom, C. R. (2002). The druggable genome. Nature Reviews Drug Discovery, 1, 727–730.
Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23, 3–25.
Krivák, R., & Hoksza, D. (2015). Improving protein-ligand binding site prediction accuracy by classification of inner pocket points using local features. Journal of Cheminformatics, 7, 12.
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The Protein Databank. Nucleic Acids Research, 28, 235–242.
Capra, J. A., & Singh, M. (2007). Predicting functionally important residues from sequence conservation. Bioinformatics (Oxford, England), 23(15), 1875–1882.
Ghersi, D., & Sanchez, R. (2011). Beyond structural genomics: computational approaches for the identification of ligand binding sites in protein structures. Journal of Structural and Functional Genomics, 12, 109–117.
Coleman, R. G., Salzberg, A. C., & Cheng, A. C. (2006). Structure-based identification of small molecule binding sites using a free energy model. Journal of Chemical Information and Modeling, 46(6), 2631–2637.
Landon, M. R., Lancia, D. R., Yu, J., Thiel, S. C., & Vajda, S. (2007). Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. Journal of Medicinal Chemistry, 50(6), 1231–1240.
Fukunishi, Y., & Nakamura, H. (2011). Prediction of ligand-binding sites of proteins by molecular docking calculation for a random ligand library. Protein Science, 20(1), 95–106.
Heo, L., Shin, W. H., Lee, M. S., & Seok, C. (2014). GalaxySite: ligand-binding-site prediction by using molecular docking. Nucleic Acids Research, 42, W210–W214.
Komiyama, Y., Banno, M., Ueki, K., Saad, G., & Shimizu, K. (2016). Data and text mining. Automatic generation of bioinformatics tools for predicting protein—ligand binding sites. Bioinformatics (Oxford, England), 32(6), 901–907.
Wang, K., Gao, J., Shen, S., Tuszynski, J. A., Ruan, J., & Hu, G. (2013). An accurate method for prediction of protein-ligand binding site on protein surface using SVM and statistical depth function. BioMed Research International, 2013, 409658.
Ivetac, A., & McCammon, J. A. (2012). A molecular dynamics ensemble-based approach for the mapping of druggable binding sites. Methods in Molecular Biology, 819, 3–12.
Seco, J., Luque, F. J., & Barril, X. (2009). Binding site detection and druggability index from first principles. Journal of Medicinal Chemistry, 52(8), 2363–2371.
Skolnick, J., & Brylinski, M. (2009). FINDSITE: a combined evolution/structure-based approach to protein function prediction. Briefings in Bioinformatics, 10(4), 378–391.
Wass, M. N., Kelley, L. A., & Sternberg, M. J. E. (2010). 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Research, 38(Suppl 2), W469–W473.
Gao, J., Liu, Q., Kang, H., Cao, Z., & Zhu, R. (2012). Comparison of different ranking methods in protein-ligand binding site prediction. International Journal of Molecular Sciences, 13(7), 8752–8761.
Fauman, E. B., Rai, B. K., & Huang, E. S. (2011). Structure-based druggability assessment-identifying suitable targets for small molecule therapeutics. Current Opinion in Chemical Biology, 15, 463–468.
Bryliński, M., Prymula, K., Jurkowski, W., Kochańczyk, M., Stawowczyk, E., Konieczny, L., & Roterman, I. (2007). Prediction of functional sites based on the fuzzy oil drop model. PLoS Computational Biology, 3(5), 909–923.
Oda, A. (2011). Development and validation of programs for ligand-binding-pocket search. Yakugaku Zasshi, 131(10), 1429–1435.
Zhang, Z., Li, Y., Lin, B., Schroeder, M., & Huang, B. (2011). Identification of cavities on protein surface using multiple computational approaches for drug binding site prediction. Bioinformatics, 27(15), 2083–2088.
Henrich, S., Salo-Ahen, O. M. H., Huang, B., Rippmann, F., Cruciani, G., & Wade, R. C. (2010). Computational approaches to identifying and characterizing protein binding sites for ligand design. Journal of Molecular Recognition, 23(2), 209–219.
Leis, S., Schneider, S., & Zacharias, M. (2010). In silico prediction of binding sites on proteins. Current Medicinal Chemistry, 17(15), 1550–1562.
Yuan, Y., Pei, J., & Lai, L. (2013). Binding site detection and druggability prediction of protein targets for structure-based drug design. Current Pharmaceutical Design, 19(12), 2326–2333.
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2004). UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25, 1605–1612.
Oliveira, S. H. P., Ferraz, F. A. N., Honorato, R. V., Xavier-Neto, J., Sobreira, T. J. P., & de Oliveira, P. S. L. (2014). KVFinder: steered identification of protein cavities as a PyMOL plugin. BMC Bioinformatics, 15, 197.
Zhu, H., & Pisabarro, M. T. (2011). MSPocket: an orientation-independent algorithm for the detection of ligand binding pockets. Bioinformatics, 27, 351–358.
Ghersi, D., & Sanchez, R. (2009). EasyMIFs and SiteHound: a toolkit for the identification of ligand-binding sites in protein structures. Bioinformatics, 25, 3185–3186.
Laurie, A. T. R., & Jackson, R. M. (2005). Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics, 21, 1908–1916.
Schmidtke, P., & Barril, X. (2010). Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. Journal of Medicinal Chemistry, 53, 5858–5867.
Nisius, B., Sha, F., & Gohlke, H. (2012). Structure-based computational analysis of protein binding sites for function and druggability prediction. Journal of Biotechnology, 159, 123–134.
Trosset, J. Y., & Vodovar, N. (2013). Structure-based target druggability assessment. Methods in Molecular Biology, 986, 141–164.
Hussein, H. A., Borrel, A., Geneix, C., Petitjean, M., Regad, L., & Camproux, A. C. (2015). PockDrug-Server: a new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Research, 43, 436–442.
Volkamer, A., Kuhn, D., Rippmann, F., & Rarey, M. (2012). Dogsitescorer: a web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics, 28, 2074–2075.
Bento, A. P., Gaulton, A., Hersey, A., Bellis, L. J., Chambers, J., Davies, M., Krüger, F. A., Light, Y., Mak, L., McGlinchey, S., Nowotka, M., Papadatos, G., Santos, R., & Overington, J. P. (2014). The ChEMBL bioactivity database: an update. Nucleic Acids Research, 42, D1083–D1090.
Halgren, T. A. (2009). Identifying and characterizing binding sites and assessing druggability. Journal of Chemical Information and Modeling, 49, 377–389.
Krasowski, A., Muthas, D., Sarkar, A., Schmitt, S., & Brenk, R. (2011). DrugPred: a structure-based approach to predict protein druggability developed using an extensive nonredundant data set. Journal of Chemical Information and Modeling, 51, 2829–2842.
Sheridan, R. P., Maiorov, V. N., Holloway, M. K., Cornell, W. D., & Gao, Y. D. (2010). Drug-like density: a method of quantifying the “bindability” of a protein target based on a very large set of pockets and drug-like ligands from the protein data bank. Journal of Chemical Information and Modeling, 50, 2029–2040.
Nayal, M., & Honig, B. (2006). On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins Structure Function and Genetics, 63, 892–906.
Cheng, A. C., Coleman, R. G., Smyth, K. T., Cao, Q., Soulard, P., Caffrey, D. R., Salzberg, A. C., & Huang, E. S. (2007). Structure-based maximal affinity model predicts small-molecule druggability. Nature Biotechnology, 25, 71–75.
Chen, K., Mizianty, M. J., Gao, J., & Kurgan, L. (2011). A critical comparative assessment of predictions of protein-binding sites for biologically relevant organic compounds. Structure, 19(5), 613–621.
Elokely, K. M., & Doerksen, R. J. (2013). Docking challenge: protein sampling and molecular docking performance. Journal of Chemical Information and Modeling, 53, 1934–1945.
Sousa, S. F., Ribeiro, A. J. M., Coimbra, J. T. S., Neves, R. P. P., Martins, S. A., Moorthy, N. S. H. N., Fernandes, P. A., & Ramos, M. J. (2013). Protein-ligand docking in the new millennium–a retrospective of 10 years in the field. Current Medicinal Chemistry, 20, 2296–2314.
Grinter, S. Z., & Zou, X. (2014). Challenges, applications, and recent advances of protein-ligand docking in structure-based drug design. Molecules, 19, 10150–10176.
Ferreira, L., dos Santos, R., Oliva, G., & Andricopulo, A. (2015). Molecular docking and structure-based drug design strategies. Molecules, 20, 13384–13421.
Chen, Y. C. (2015). Beware of docking!. Trends in Pharmacological Sciences, 36, 78–95.
Hamelberg, D., Mongan, J., & McCammon, J. A. (2004). Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. Journal of Chemical Physics, 120(24), 11919–11929.
Pierce, L. C. T., Salomon-Ferrer, R., Augusto, F., De Oliveira, C., McCammon, J. A., & Walker, R. C. (2012). Routine access to millisecond time scale events with accelerated molecular dynamics. Journal of Chemical Theory and Computation, 8(9), 2997–3002.
Durrant, J. D., & McCammon, J. A. (2011). Molecular dynamics simulations and drug discovery. BMC Biology, 9, 71.
Beveridge, D. L., & DiCapua, F. M. (1989). Free energy via molecular simulation: applications to chemical and biomolecular systems. Annual Review of Biophysics and Biophysical Chemistry, 18, 431–492.
Aldeghi, M., Heifetz, A., Bodkin, M. J., Knapp, S., & Biggin, P. C. (2016). Accurate calculation of the absolute free energy of binding for drug molecules. Chemical Science, 7(1), 207–218.
Shirts, M. R., Mobley, D. L., & Chodera, J. D. (2007). Chapter 4 Alchemical free energy calculations: ready for prime time?. Annual Reports in Computational Chemistry, 3, 41–59.
Srinivasan, J., Cheatham, T. E., Cieplak, P., Kollman, P. A., & Case, D. A. (1998). Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. Journal of the American Chemical Society, 120(37), 9401–9409.
Kollman, P. A., Massova, I., Reyes, C., Kuhn, B., Huo, S., Chong, L., Lee, M., Lee, T., Duan, Y., Wang, W., Donini, O., Cieplak, P., Srinivasan, J., Case, D. A., & Cheatham, T. E. (2000). Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Accounts of Chemical Research, 33(12), 889–897.
Aqvist, J., Medina, C., & Samuelsson, J. E. (1994). A new method for predicting binding affinity in computer-aided drug design. Protein Engineering, 7, 385–391.
Lill, M. A., & Thompson, J. J. (2011). Solvent interaction energy calculations on molecular dynamics trajectories: increasing the efficiency using systematic frame selection. Journal of Chemical Information and Modeling, 51(10), 2680–2689.
Genheden, S., & Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opinion on Drug Discovery, 10(5), 449–461.
Srivastava, H. K., Chourasia, M., Kumar, D., & Sastry, G. N. (2011). Comparison of computational methods to model DNA minor groove binders. Journal of Chemical Information and Modeling, 51(3), 558–571.
Srivastava, H. K., & Sastry, G. N. (2013). Efficient estimation of MMGBSA-based BEs for DNA and aromatic furan amidino derivatives. Journal of Biomolecular Structure & Dynamics, 31(5), 522–537.
Homeyer, N., & Gohlke, H. (2012). Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Molecular Informatics, 31(2), 114–122.
Hou, T., Wang, J., Li, Y., & Wang, W. (2010). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. the accuracy of binding free energy calculations based on molecular dynamics simulations. Journal of Chemical Information and Modeling, 51, 69–82.
Genheden, S., Mikulskis, P., Hu, L., Kongsted, J., Söderhjelm, P., & Ryde, U. (2011). Accurate predictions of nonpolar solvation free energies require explicit consideration of binding-site hydration. Journal of the American Chemical Society, 133(33), 13081–13092.
Genheden, S., & Ryde, U. (2010). How to obtain statistically converged MM/GBSA results. Journal of Computational Chemistry, 31, 837–846.
Genheden, S., & Ryde, U. (2012). Will molecular dynamics simulations of proteins ever reach equilibrium?. Physical Chemistry Chemical Physics, 14, 8662–8677.
Henriksen, N. M., Hayatshahi, H. S., Davis, D. R., & Cheatham, T. E. (2014). Structural and energetic analysis of 2-aminobenzimidazole inhibitors in complex with the hepatitis C virus IRES RNA using molecular dynamics simulations. Journal of Chemical Information and Modeling, 54(6), 1758–1772.
Sun, H., Li, Y., Tian, S., Xu, L., & Hou, T. (2014). Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Physical Chemistry Chemical Physics, 16, 16719–16729.
Srivastava, H. K., & Sastry, G. N. (2012). Molecular dynamics investigation on a series of HIV protease inhibitors: assessing the performance of MM-PBSA and MM-GBSA approaches. Journal of Chemical Information and Modeling, 52(11), 3088–3098.
Chetty, S., & Soliman, M. E. S. (2015). Possible allosteric binding site on Gyrase B , a key target for novel anti-TB drugs : homology modelling and binding site identification using molecular dynamics simulation and binding free energy calculations. Medicinal Chemistry Research, 24, 2055–2074.
Acknowledgments
The authors would like to acknowledge The School of Health Sciences, University of KwaZulu-Natal, Westville campus for providing the infrastructure for this work. Neal K. Broomhead would like to acknowledge the National Research Foundation of South Africa for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no financial or intellectual conflicts of interest.
Rights and permissions
About this article

Cite this article
Broomhead, N.K., Soliman, M.E. Can We Rely on Computational Predictions To Correctly Identify Ligand Binding Sites on Novel Protein Drug Targets? Assessment of Binding Site Prediction Methods and a Protocol for Validation of Predicted Binding Sites. Cell Biochem Biophys 75, 15–23 (2017). https://doi.org/10.1007/s12013-016-0769-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-016-0769-y