
Abstract
Cuprizone (CPZ) is a neurotoxic agent that is used to induce demyelination and neurotoxicity in rats. This study aimed to investigate the protective potential of sulforaphane (SF), nuclear factor E2 related factor (Nrf-2) activator, against CPZ-induced cardiotoxicity and hepatotoxicity. Male adult Wistar rats (n = 18) were fed with a regular diet or a CPZ-contained diet (0.2%) for four weeks. The rats were divided into three groups (n = 6): negative control rats, CPZ-exposed rats, and CPZ + SF treated rats. SF was intraperitoneally administrated (2 mg/kg/day) for two weeks. The anti-inflammatory and anti-oxidative functions of SF were investigated biochemically, histologically, and immunohistochemically. CPZ increased serum levels of cardiac troponin 1 (CTn1), aspartate amino transaminase (AST), alanine amino transaminase (ALT), and alkaline phosphatase (ALP). In addition, serum levels of inflammatory interferon-gamma (IFN-γ), and pro-inflammatory interleukin 1β (IL-1β) were significantly elevated. Moreover, CPZ administration provoked oxidative stress as manifested by declined serum levels of total antioxidant capacity (TAC), as well as, stimulated lipid peroxidation and decreased catalase activities in both cardiac and hepatic tissues. SF treatment reversed all these biochemical alterations through exerting anti-oxidative and anti-inflammatory activities, and this was supported by histopathological investigations in both cardiac and hepatic tissues. This SF-triggered modulation of oxidative stress and inflammation is strongly associated with Nrf-2 activation, as evidenced by activated immunoexpression in both cardiac and hepatic tissues. This highlights the cardioprotective and hepatoprotective activities of SF via Nrf-2 activation and enhancing catalase function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cuprizone (oxalic acid bis (cyclohexylidene hydrazide) is a copper-chelating agent that is used experimentally to provoke demyelination pathology similar to that of multiple sclerosis (MS) [1]. Dietary administration of CPZ stimulates copper deficiency [2] and triggers dysfunction of the copper-dependent electron carrier “cytochrome oxidase” which is an essential component of oxidative phosphorylation [3]. Besides enhancing demyelination and glial activation, CPZ was found to cause alterations in the hepatic tissue, as demonstrated by several studies [4,5,6]. The hepatotoxic and neurotoxic potentials of CPZ could be attributed to a disturbance of cellular respiration, a key mitochondrial function [7]. It was found that a disruption in the enzymatic activities takes place prior to myelin loss during the first days or weeks of CPZ administration [5]. Concerning MS-associated cardiotoxicity, MS patients demonstrated an elevated cardiovascular risk that might be attributed to the impairment of the autonomic control of cardiovascular functions, but the underlying molecular mechanisms are not completely elucidated [8, 9]. Therefore, this study aimed to explore the influence of CPZ-induced MS on cardiovascular health, more interestingly; no study has been published so far concerning CPZ-induced cardiotoxicity.
Copper “Cu” is a vital “catalytic and structural” cofactor in several biochemical mechanisms [10]; several enzymes use Cu as a cofactor for example, superoxide dismutase-1 [11], monoamine oxidase [12], and cytochrome c oxidase assembly protein [13]. CPZ is a Cu-chelator that is toxic to mitochondria [14,15,16]. During the early stages of exposure to CPZ, a reduction in monoamine oxidase and cytochrome c oxidase in the brain and liver of mice is observed [17] in association with the development of mega-mitochondria in the liver, which is linked to metabolic disruption [15]. Therefore, CPZ is capable of suppressing the enzymatic activity of monoamine oxidase and copper-dependent enzymes such as cytochrome c oxidase [17,18,19].
Sulforaphane (SF) is a powerful phytochemical found in seeds and sprouts of cruciferous plants such as broccoli, SF is a pleiotropic agent that exhibited promising multifactorial functions against several chronic diseases, including neurodegenerative disorders and cancer [20,21,22,23,24], moreover, numerous studies showed the hepatoprotective and cardioprotective activities of SF [25,26,27,28,29]. SF is an activator of nuclear factor E2 related factor (Nrf-2) [30], which is a transcription factor involved in the adaptive response to several endogenous and exogenous stressors such as oxidative stress; SF targets the activation of the Nrf-2 pathway which regulates the expression of cytoprotective enzymes that exhibited anti-oxidative and detoxification activities [31].
Due to its electrophilic nature, SF enhances nuclear translocalization and accumulation of Nrf-2 and mediates its phosphorylation by activating different kinases including mitogen-activated protein kinase (MAPK), protein kinase C (PKC), and Akt kinase (protein kinase B or PKB), where it changes nuclear and cytoplasmic trafficking and Nrf-2 integrity and stability [32,33,34]. Furthermore, SF is considered “an indirect anti-oxidant” that is capable of exerting anti-oxidative potential through modification of Keap1 cysteine residues, activation of MAPK, phosphatidylinositol 3-kinase (PI3K), and PKC pathways, which results in the phosphorylation, nuclear accumulation, and increased transcription and stability of Nrf-2 [35].
The protective potential of SF was evidenced in several studies; however, the protective potential of SF on CPZ-induced cardiotoxicity and hepatotoxicity lacked clarification. The main target of this study is to evaluate the cardioprotective and hepatoprotective potentials of Sulforaphane (SF) in a rat model of CPZ-induced cardiotoxicity and hepatotoxicity.
Materials and Methods
Materials
DL-Sulforaphane (SF) (CAS No. 4478-93-7), and Cuprizone (CPZ) (CAS No. 370-81-0) were bought from Sigma, USA. SF was dissolved in 250 μl of Tween 80 (Sigma, St. Louis, MO, USA) and diluted to the appropriate concentration with distilled water; the final concentration of Tween 80 did not exceed 1%. Serum levels of cardiac troponin-1 (CTn1), interferon-γ (IFN-γ), and interleukin 1β (IL-1β) were estimated using commercially available enzyme-linked immunosorbent assay (ELISA) kits bought from (BioVision, USA, Catalog number: E4737; Quantikine, USA, Catalog number RIF00; and BioVision, USA, Catalog number K4796-100, respectively). Colorimetric Kits of malondialdehyde (MDA), catalase, and total antioxidants (TAC) were bought from Biodiagnostic Co., Egypt. Kits of primary Anti-Nrf-2 antibodies (GTX103322) were bought from Gene Tex Co., USA and secondary antibody HRP Envision kit (DAKO) was bought from Agilent, USA. All other chemicals used in the study were of high analytical grade.
Animals and Grouping
The experiments were performed on male adult Wistar rats (n = 18) weighing around 120 ± 20 g and aged 6 weeks from the Animal House of the National Research Center (NRC), Egypt. The animals were kept under suitable laboratory conditions throughout the period of investigation. Animals were kept with free access to food and fresh water in a room with temperatures ranging from 22 to 24 °C and a 12-h light/dark cycle. They were fed standard pellet chow, provided by the animal house at the National Research Centre (NRC), and allowed free access to water. The experiment was conducted in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978), and experimental procedures were approved by the Medical Research Ethics Committee (MREC) of the NRC, Egypt (19-313).
Rat Model of CPZ-Induced Cardiotoxicity and Hepatotoxicity
CPZ-induced toxicity was induced by administrating rats a CPZ-diet prepared by carefully mixing 0.2% of CPZ into ground rodent chow for four weeks, according to Omotoso et al. [36].
Experimental Design and Treatments
The experimental period lasted for six weeks, including four weeks (30 days) for CPZ intoxication and two weeks (14 days) for treatment with SF. After an acclimatization period of two weeks, the 18 rats were randomly divided into three groups (6 rats/group) as follows:
Group (1) Healthy negative control group fed on a normal ground rodent chow.
Group (2) CPZ-exposed control group: received 0.2% CPZ-diet only daily for 30 days, and then fed on a normal ground rodent chow, and administrated the vehicle “used to dissolve SF” intraperitoneally for 14 days.
Group (3) CPZ + SF treated rats: rats received 0.2% CPZ-diet only daily for 30 days, followed by intraperitoneal (i.p.) administration of SF, at a dose of 2 mg/ kg, for 14 days.
Observation of Clinical Signs of CPZ-Induced Toxicity
Home cage observations of the experimental rats were carried out regularly (twice a week until the completion of the experiment); to evaluate clinical signs of CPZ-associated toxicity including behavioral alterations (convulsions, decreased physical activities, lethargy), morphological alterations (e.g., weight loss), and mortality. The bodyweight of rats was measured once a week using a digital balance.
Preparation of Serum and Tissue Samples
At the end of the experiment, rats were anesthetized using Thiopental sodium (50 mg/kg, i.p.) and blood samples were withdrawn. The serum was separated by centrifugation at 3000 rpm for 20 min (centrifuge 3-18KS, Germany). The rats were then sacrificed under mild anesthesia. The whole liver and heart tissues were dissected; one part was homogenized and aliquots were homogenized in 4 volumes of phosphate-buffered saline (PBS, pH 7.4), and used for biochemical investigations. The other part was weighed, fixed in 10% formal saline, and processed for histological and immunohistochemical analysis of Nrf-2.
Biochemical Investigations
Estimation of Serum Cardiac Troponin I (CTnI)
Serum CTnI was measured using ELISA, according to the manufacturer’s instructions. Results were presented as pg/ml.
Estimation of Inflammatory Marker: Interferon-gamma (IFN-γ)
Serum IFN-γ was assayed using ELISA kit, following the manufacturer's protocol. Results were presented as pg/ml protein.
Estimation of Pro-inflammatory Marker: Interleukin 1β (IL-1β)
Serum IL-1β was assayed using ELISA kit, according to the manufacturer's guidelines. Results were presented as pg/ml protein.
Serum Total Antioxidant Capacity (TAC)
The evaluation of serum TAC is performed by the reaction of antioxidants in the sample with a known amount of exogenous hydrogen peroxide (H2O2). The antioxidants in the sample eliminate a certain quantity of H2O2. The residual H2O2 is estimated by an enzymatic reaction that results in a colored product that could be measured at 505 nm [37].
Lipid Peroxidation in the Cardiac and Hepatic Tissues
Evaluating the malondialdehyde (MDA) content, in the cardiac and hepatic tissues, was used to indicate lipid peroxidation. MDA, an end-product of lipid peroxidation, was estimated according to Ohkawa et al. [38]. Briefly, 1000 µl of chromogen was added to 200 µl of tissue homogenate, and the mixture was placed in a boiling water bath for 30 min. In an acidic medium and at a high temperature (95 °C), MDA reacts with thiobarbituric acid and results in a pink-colored product that could be measured spectrophotometrically at 532 nm. The results were expressed as nmol/g tissue.
Catalase Activity in the Cardiac and Hepatic Tissues
Catalase reacts with a quantified H2O2 amount. After one minute, the catalase inhibitor ceased the reaction. Remaining H2O2 reacts with 3, 5-dichloro-2-hydroxybenzene sulfonic acid (DHBS) and, 4-aminophenazone (AAP), in the presence of peroxidase (HRP), and results in the formation of a colored product with color intensity inversely proportional to catalase activity in the sample [39].
Assessment of Liver Functions
Serum alanine amino transaminase (ALT) and serum aspartate amino transaminase (AST) were measured spectrophotometrically by quantitative colorimetric methods according to Reitman and Frankel [40]. Serum alkaline phosphatase (ALP) was measured spectrophotometrically at 510 nm [41].
Histopathological Investigation of Heart and Liver Tissues
The liver and heart tissue samples were fixed in a solution of 10% neutral buffered formalin, embedded in paraffin, sectioned at a thickness of 5 μm, and stained with hematoxylin and eosin for regular examination. The sections were analyzed using a light microscope (Leica Microsystems GmbH, Wetzlar, Germany). All standard procedures for sample fixation and staining were conducted according to Culling [42].
Immunohistochemical Analysis of Nrf-2
Sections of 5µ thickness were prepared, cardiac or hepatic samples were incubated with primary Anti-Nrf-2 antibody, at 1:500 dilutions, overnight at 4 °C. Tissue sections were washed out by PBS followed by incubation with secondary antibody HRP Envision kit (DAKO) for 20 min; washed out and incubated with diaminobenzidine (DAB) for 15 min. Tissue sections were washed by PBS then counterstained with hematoxylin, dehydrated, cleared in xylene then coverslipped for microscopic examination. Quantification of Nrf-2 was assessed by measuring the % area expression from 5 randomly chosen fields in each section and averaged using image analysis software (Image J, version 1.46a, NIH, Bethesda, MD, USA).
Statistical Analysis
All values were presented as mean ± standard error of the means (SEM), for n = 6 rats of each group. Comparisons between different groups were carried out using one-way analysis of variance (ANOVA) followed by Duncan’s multiple comparison post hoc test. The difference was considered significant when p˂0.05. The percent of the change in the value of data for the CPZ + SF group with respect to the negative control or positive control (CPZ) was calculated and presented as a percentage change. \({\text{Percentage of change }}\% \, = \,\left( {{\text{treated}}\;{\text{value}}\; - \;{\text{control}}\;{\text{value}}} \right)/{\text{control}}\;{\text{value}}\; \times \;{1}00\).
Results
Effect of CPZ Intoxication and SF Administration in Different Experimental Groups
Following dietary administration of CPZ, the clinical and behavioral signs, in addition to other toxicity signs including appearance, possible trauma, and mortality were carefully observed. No mortality rate was observed in different groups. Negative control rats and CPZ-induced rats treated with SF demonstrated normal appearance and behavior. On the other hand, CPZ-induced rats demonstrated a clear toxicological profile such as ataxia, lethargy, and decreased physical activities. In addition, CPZ-administrated rats tended to lose weight more quickly. After two weeks of CPZ administration, rats exhibited a significant decrease in body weight, as compared to control rats. However, after CPZ removal from the diet, CPZ-induced rats demonstrated a slight increase in body weight. While both groups of negative control and treated CPZ + SF group continued to gain weight throughout the experimental period, as illustrated in Fig. 1.

Effect of CPZ administration and SF treatment on body weight of rats in different experimental groups: Negative Control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group
Effect of SF on Cardiotoxicity Marker: CTnI in CPZ-Exposed Rats
CPZ administration enhanced a significant increase in serum CTnI level in CPZ-intoxicated rats by 94.36%, as compared to negative controls. Treatment of CPZ-induced rats with SF caused a significant reduction in CTnI levels by 56.36%, as compared to CPZ rats. These findings indicated the cardiotoxic potential of CPZ and the cardioprotective activities of SF (Fig. 2).

Effect of CPZ administration and SF treatment on serum CTnI in different experimental groups: negative control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group. Each bar represents the mean ± S.E. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group
Effect of SF on Inflammatory Marker: IFN-γ in CPZ-Exposed Rats
In CPZ-exposed rats, CPZ intake caused a significant elevation in serum IFN-γ level by 159.74%, as compared to negative controls. Treatment of CPZ-exposed rats with SF caused a significant decrease in IFN-γ levels by 45%, as compared to CPZ-induced rats. These findings indicated the anti-inflammatory potential of SF (Fig. 3).

Effect of CPZ and SF treatment on serum IFN-γ in different experimental groups: negative control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group. Each bar represents the mean ± S.E. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group
Effect of SF on Pro-inflammatory Marker: IL-1β in CPZ-Exposed Rats
In CPZ-intoxicated rats, CPZ exposure resulted in a significant increase in serum IL-1β levels by 82.68%, as compared to negative controls. Treatment of CPZ-induced rats with SF caused a significant reduction in IL-1β levels by 16.59%, as compared to CPZ-fed rats. These findings indicated the anti-inflammatory activity of SF (Fig. 4).

Effect of CPZ and SF treatment on serum IL-1β in different experimental groups: Negative Control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group. Each bar represents the mean ± S.E. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group
Effect of SF on Oxidative Stress Status in CPZ-Exposed Rats
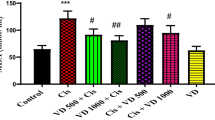
On the one hand, as compared to negative control rats, CPZ caused a significant reduction in TAC by 33.14%, cardiac catalase by 36.79% and hepatic catalase by 22.8%. Moreover, CPZ administration resulted in a significant elevation in lipid peroxidation in both cardiac and hepatic tissues by 55.87 and 58.73%, respectively. On the other hand, as compared to CPZ-exposed rats, treatment of CPZ-exposed rats with SF resulted in a significant elevation in serum TAC by 47.86%, cardiac catalase by 34%, and hepatic catalase by 40.63%, along with a significant reduction in MDA in both cardiac and hepatic tissues by 30 and 17.7%, respectively. These data demonstrated the anti-oxidative potential of SF (Fig. 5 and 6).

Effect of CPZ administration and SF treatment on serum TAC in different experimental groups: negative control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group. Each bar represents the mean ± S.E. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group

Effect of CPZ administration and SF treatment on cardiac and hepatic MDA contents and catalase activities in different experimental groups: Negative Control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group. Each bar represents the mean ± S.E. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group
Effect of SF on Enzymatic Activities of AST, ALT, and ALP
CPZ-fed rats demonstrated a significant elevation in serum AST, ALT, and ALP by 40.49, 24.63, and 51%, as compared to negative control rats. On the other side, the treatment of CPZ rats with SF caused a significant reduction of 23.83, 14, and 15.7%, as compared to CPZ-induced rats. These data demonstrated the cardioprotective and hepatoprotective activities of SF (Fig. 7).

Effect of CPZ administration and SF treatment on AST, ALT, and ALP activities in different experimental groups: negative control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day for 14 days, i.p.) group. Each bar represents the mean ± S.E. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group
Effect of SF on CPZ-Induced Histological Architecture of the Heart and Liver
Histopathological investigations of cardiac and hepatic tissues of different experimental groups are illustrated in Figs. 8 and 9.

Effect of CPZ administration and SF treatment on cardiac histoarchitecture in different experimental groups: negative control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group, (scale bar: 50 μm). Cardiac tissues of negative control rats (a) demonstrated normal cardiomyocytes with intact subcellular details (arrow). CPZ-intoxicated cardiac tissues demonstrated degenerated cardiomyocytes (dashed arrow) and infiltration of inflammatory cells (arrowhead) (b, c). CPZ + SF treated group showed almost normal histological features with well-defined cardiomyocytes (d). Preference for color: online

Effect of CPZ administration and SF treatment on hepatic histoarchitecture in different experimental groups: negative control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group, (scale bar: 200 and 50 μm). Normal control hepatic tissues exhibited normal hepatocytes with intact parenchyma (arrow) and intact hepatic vasculatures (star) (a, b). CPZ-intoxicated hepatocytes demonstrated showed moderate dilatation of hepatic vasculatures (star) and moderate periportal inflammatory cells infiltrates (black arrow), and slight hepatic degeneration (dashed arrow) (c, d). CPZ + SF treated hepatic tissues showed moderate dilatation of hepatic vasculatures were shown (star) with mild hepatocellular vacuolar degenerative alterations (dashed arrow) (e, f). Preference for color: online
Concerning the cardiac histoarchitecture, normal control samples demonstrated normal histological features of the cardiac wall with many apparent well-organized, striated and branched cardiomyocytes with intact subcellular details (arrow) and intact vasculatures (Fig. 8a). By contrast, CPZ-induced hearts exhibited focal areas of fragmented and degenerated cardiomyocytes (dashed arrow) with the mild widening of intercellular spaces accompanied by mild occasional interstitial inflammatory cells infiltrates (arrowhead) (Fig. 8b, c). Meanwhile, CPZ + SF treated hearts showed almost intact histological features without abnormal histological changes records (Fig. 8d).
Concerning the hepatic histoarchitecture, Normal control hepatic samples demonstrated normal histological features of liver parenchyma with many apparent intact well-organized hepatocytes with intact subcellular details (arrow), intact hepatic vasculatures (star), as well as hepatic sinusoids were shown without abnormal changes records (Fig. 9a, b). In contrast, CPZ-intoxicated hepatic tissues showed significant dilatation of hepatic vasculatures (star) and sinusoids accompanied by periportal inflammatory cell infiltrates (arrow), and few records of hepatocellular degenerative changes (dashed arrow) (Fig. 9c, d). On the other side, CPZ + SF treated livers showed intact subcellular details (arrow), and intact hepatic vasculatures (star) with minimal records of inflammatory cell infiltrates (Figs. 9e, f).
Effect of SF on Immunohistochemical Expression of Nrf-2 in Cardiac and Hepatic Tissues
Immunohistochemical expressions of Nrf-2 in the cardiac and hepatic tissues are depicted in Fig. 10 and 11. Briefly, the normal control rats revealed a moderate immunoexpression of Nrf-2 in both heart and liver tissues (Figs. 10 and 11a), respectively. In contrast, CPZ-induced cardiac and hepatic tissues exhibited no immunoexpression of Nrf-2; indicating the inhibitory influence of CPZ on the immunoexpression of Nrf-2 (Figs. 10, 11b). On the other side, CPZ + SF treated cardiac and hepatic tissues demonstrated strong positive immunostaining cells of Nrf-2 (Figs. 10, 11c); signifying that SF upregulated the immunoexpression of Nrf-2. Figures 10 and 11d described the immunostaining area (%) of Nrf-2 immunoexpression in the heart and liver of rats from different experimental groups.

Effect of CPZ administration and SF treatment on immunostaining of Nrf-2 in the cardiac tissues of different experimental groups: negative control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group, (scale bar: 50 μm). Heart tissue of the negative control group demonstrated moderate immunoexpression of Nrf-2 with a significant increase of positive immunostaining cells (a). CPZ-intoxicated cardiac tissues showed no immunoexpression of Nrf-2 (b). CPZ + SF treated cardiac tissues exhibited strong immunoexpression of Nrf-2 with a significant increase of positive immunostaining cells (c). The immune staining area (%) of Nrf-2 in cardiac tissues is illustrated in d. Each bar represents the mean ± S.E. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group. Preference for color: online

Effect of CPZ administration and SF treatment on immunostaining of Nrf-2 in the hepatic tissues of different experimental groups: negative control, CPZ (0.2% CPZ-diet for 30 days) group, and CPZ-SF (2 mg/kg/day, for 14 days, i.p.) group, (scale bar: 50 μm). Hepatic tissue of the negative control group demonstrated moderate immunoexpression of Nrf-2 (a). CPZ-intoxicated liver tissues showed weak immunoexpression of Nrf-2 (b). CPZ + SF treated hepatic tissues exhibited strong immunoexpression of Nrf-2 with a significant increase of positive immunostaining cells (c). The immune staining area (%) of Nrf-2 in liver tissues is illustrated in d. Each bar represents the mean ± S.E. #p < 0.05 vs. negative control group, @p < 0.05 vs. CPZ positive control group. Preference for color: online
Discussion
CPZ is a well-known neurotoxicant that is commonly used to induce demyelination and oligodendroglial death by stimulating mitochondrial dysfunction and provoking oxidative stress by increasing mitochondrial generation of reactive oxygen species (ROS) [1, 43]. Herein, we investigated CPZ as a cardiotoxic and hepatotoxic chemical and examined the potential protective activities of SF against CPZ-induced cytotoxicity and hepatotoxicity via suppressing oxidative stress and modulating inflammation.
The animal weight is an essential “variable” for reliable CPZ-provoked toxicity [44]. The first effect observed by the CPZ administration was body weight loss after two weeks; the body weight of CPZ-fed rats exhibited a significant reduction of about 14% in the 3rd and 4th weeks of CPZ intake. Our results run in agreement with Akyuz and Villa [8], the loss of about 15% is a hallmark of the CPZ-induced demyelination. This long-term intoxication (four weeks) caused the morphological signs of CPZ-associated neurotoxicity in most cases accompanied by weight loss and characteristic movement disorders of the CPZ-induced animals [45, 46]. This reduction in the body weight of CPZ rats indicates reduced food intake and disruption of normal energy flow, as compared to negative controls; this might be due to the unpleasant odor or taste of CPZ and its impact on the hepatic function [47, 48]. In addition, copper chelation could be another cause of CPZ-induced weight loss, considering Cu-deficiency in rats [49]. Cessation of CPZ administration resulted in the restoration of body weight gain but at a slower rate, our results run in agreement with Babbs et al. [50] that showed recovery of weight loss following replacement of the CPZ-diet with the regular diet. On the other hand, treatment of CPZ rats with SF was able to restore body weight gain, signifying restoration of appetite upon cessation of CPZ administration. SF exhibited the potential of restoring body weight gain, ameliorating the neurotoxicity signs, and improving the clinical symptoms of CPZ intoxication. These finding showed the lowering body weight influence of CPZ, as well as, the therapeutic (gaining body weight) potential of SF. Our results highlighting the importance of body weight as a parameter of clinical appearance of toxicity.
Dietary administration of CPZ to rats is capable of initiating a cascade of oxidoinflammatory events; CPZ-provoked oxidative stress manifested as declined serum TAC levels, decreased cardiac and hepatic catalase activities, along with a significant increment in lipid peroxidation in cardiac and hepatic tissues. This oxidative potential of CPZ could be ascribed to its nature as a Cu-chelator that interferes with the activity of Cu-dependent enzymes “cuproenzymes” such as “cytochrome oxidase” that is particularly implicated in mitochondrial respiration [43]; mitochondria are the major organelles responsible for ROS propagation, which are readily scavenged by the antioxidative enzyme “catalase”. Therefore, CPZ-induced declined catalase activity promoted the oxidative impact.
CPZ-induced oxidative stress in both the hepatic and cardiac tissue, especially lipid peroxidation, might be involved in the disruption of the cellular membranes and the release of transaminases [51]; therefore, restoring these enzymatic activities to their normal levels is associated with the attenuation of the hepatotoxicity and restoration of the cardiac function. The release of intracellular ALT and AST could enhance the status of inflammation and hepatic fibrosis [52]. AST could be used as a marker of both cardiotoxicity and hepatotoxicity; CPZ administration caused significant increments in AST (40.49%), ALT (24.63%), ALP (51%), and CTn1 (94.36%), as compared to negative control rats. CPZ caused a significant increase in AST, a serum indicator of “myocardial injury” [53]; disruption of cardiomyocytes caused the leakage of AST, which normally exists in the cytoplasm of cardiomyocytes [54]. Furthermore, high serum levels of CTn1 and AST and induced ROS generation are considered key hallmarks of cardiotoxicity [51]. Elevated CTnI levels indicate irreversible cardiac damage and necrosis of cardiomyocytes [55]. This highlights the potential use of CTn1 as a highly specific and sensitive cardiac diagnostic biomarker for myocardial necrosis [56].
Furthermore, CPZ enhanced a significant increase in serum levels of the inflammatory marker IFN-γ (159.74%) and the pro-inflammatory marker IL-1β (82.68%), as compared to negative control rats. This increased level of the inflammatory IFN-γ might be attributed to the release of IFN-γ from the disrupted hepatocytes; IFN-γ further provoked the activation and proliferation of hepatic stellate cells (HSCs) [57]. IFN-γ is a crucial regulator of the complex process of inflammatory mediators in cardiac pathological conditions that includes myocardial infarction or myocarditis [58]. In addition, CPZ intake upregulated the expression of several genes of inflammation [59]; Il-1β is upstream of many inflammatory mechanisms [60]. Our results run in agreement with Hillis et al. [61] and Mason et al. [59] that demonstrated enhanced IL-1β expression in the CPZ-induced animals; Mason et al. [59] found that expression of IL-1β was gradually increased in the 1st week of CPZ intake, then robustly upregulated in the 3rd week till the 6th week. These biochemical results were supported by histopathological analysis of the heart and liver tissues; which demonstrated that CPZ-provoked structural alterations and induced inflammation in both tissues. Furthermore, CPZ-induced oxidative stress in both tissues was evidenced by the declined immunoexpression of Nrf-2. The disruption of Nrf-2 signaling stimulates susceptibility to oxidative and electrophilic stresses, as well as, to inflammation [62].
On the other side, treatment of CPZ-exposed rats with SF caused a significant reduction in AST, ALT, and ALP; in addition, CPZ-induced cardiac dysfunction could be reversed by SF treatment as demonstrated by decreased serum CTn1 levels. These findings run in agreement with previous studies [25, 55, 63]. This cardioprotective action of SF might be ascribed to its anti-oxidative potential and anti-lipid peroxidation activity that resulted in “membrane protection” and suppressed the cellular leakage of transaminases and CTn1, this run in agreement with several studies [28, 29, 35]. These hepatoprotective and cardioprotective actions were accredited to the up-regulation of hepatic and cardiac Nrf-2 and heme oxygenase-1 (HO-1) expression by SF [64], as evidenced by elevated Nrf-2 immunoexpression in both cardiac and hepatic tissues; suggesting that Nrf-2 is a therapeutic target in inflammation-associated conditions. Nrf-2 activated cytoprotective genetic function might act synergistically to regulate innate immune response and to inhibit the up-regulation of pro-inflammatory gene expression [65].
Nrf-2 activation restores the balance between oxidants and antioxidants and plays a key role in the expression of several antioxidant genes including catalase [62, 66, 67]. Treatment of CPZ-intoxicated rats with SF counteracted the enhanced oxidative stress in both cardiac and hepatic tissues. SF exhibited the potential to enhance the expression of detoxification or defensive enzymes via the Keap1/Nrf2/ARE signaling pathway [30]. Immunohistochemical results demonstrated Nrf-2 activation upon treatment with SF; this could be ascribed to the potential of Nrf-2 to regulate the up-regulation of detoxification genes to activate a cytoprotective response, through mediating the inactivation of oxidants and favoring the synthesis of antioxidants [68]. Moreover, the translocation of Nrf-2 to the nucleus enables its interaction with the antioxidant response element (ARE) to boost the cytoprotective “anti-oxidative” gene expression, such as HO-1 and NAD(P)H: quinone oxidoreductase 1 (NQO1), to produce catalase to exert anti-oxidative function [69]. Herein, we estimated the catalase activities and evaluated the immunostaining % of Nrf-2; as Nrf-2 controls the expression of antioxidant catalase, which is significantly influenced by redox status, as evidenced previously [70], although other antioxidant enzymes might be involved.
Nrf-2 is vital for the maintenance of redox signaling in response to stress [71]. Under stress conditions such as CPZ-induced toxicity, Nrf-2 loses its DNA binding to ARE, however, this loss of Nrf-2/ARE binding could be reversed by using “Nrf-2 agonists” such as SF [72, 73]; suggesting that Nrf-2-mediated survival pathways are reversible and responsive by using effective agonists of Nrf-2 such as SF, which act as “indirect antioxidant” with anti-inflammatory potential [30, 74]. The inhibitory potential of SF on abnormally activated inflammation is related to the activation of Nrf-2-mediated signaling pathways [75]. It was evidenced that Nrf-2 is one of the most vital defense mechanisms against oxidative and/or electrophilic stresses [71, 76]; activation of Nrf-2 signaling has conferred protection against myocardial ischemia/reperfusion (I/R) injury [77] and in isolated rat hearts [78]. It was found that SF-based Nrf-2 activation protects the myocardium from Angiotensin II-induced cardiomyopathy [79]. In contrast, the absence of Nrf-2 inhibited the expression of antioxidant genes in the myocardium, causing fibrosis and apoptosis [80, 81]; downregulation of Nrf-2 is correlated with redox-sensitive vascular dysfunction [82]. Moreover, SF is capable of interacting with cysteine residues on Kelch-like ECH-associated protein 1 (Keap1) “cysteine-rich adaptor protein”, enhancing its dissociation from Nrf-2 and enhancing the nuclear accumulation of Nrf-2 that subsequently enhances the genetic expression of phase II detoxification enzymes [83]. Therefore, the “Nrf-2 activators” could exhibit cardio-therapeutic applications where inflammation and oxidative injury play key roles [84].
The anti-oxidative and anti-inflammatory activities of SF are strongly linked to the cardioprotective and hepatoprotective activities. Experimentally, dietary SF protects against a wide variety of hepatotoxic chemicals [63, 64, 85]. SF was able to reduce ALT, AST, and ALP, decrease lipid peroxidation, and enhance TAC levels and catalase activities. Furthermore, SF administration reduced CPZ-induced oxidative stress in both cardiac and hepatic tissues. Our results run in accordance with Greco et al. [86] and Xu et al. [70] that showed that treatment of induced rats with SF increases Nrf-2-mediated antioxidant defenses; this might be explained by the ability of SF as Nrf2 regulator to control CPZ intoxication, possibly by potentiating catalase function, signifying that the re-activation of the “Nrf2-catalase signaling pathway” and enhancing the “peroxidase activity” of catalase. SF demonstrated anti-inflammatory and anti-oxidative activities as demonstrated previously [87,88,89]. SF was capable of reducing both IL-1β and IFN-γ levels, this run in agreement with several studies [90,91,92,93], signifying the role of SF as an antioxidant bioactive molecule with anti-inflammatory potential.
This study did not include negative control rats that were administrated SF alone as several studies showed the non-toxic potential of SF at a dose range of 0.5 to 12.5 mg/kg/day either orally [63, 88] or intraperitoneally [94, 95]. This step was taken as an ethical measure to decrease the number of sacrificed rats. The present study illustrated the protective role of SF against CPZ-induced cardiotoxicity and hepatotoxicity primarily via Nrf-2 activation and the accompanied anti-inflammatory and anti-oxidative functions. This study is the first to investigate the protective potential of SF against CPZ-induced cardiotoxicity and hepatotoxicity. Further studies are required to investigate the molecular mechanisms and the mechanistic pathways underlying the protective potential of SF as well as the cytotoxic potential of CPZ. However, the efficient clinical use of Nrf-2 activators to manage inflammatory diseases requires further validation to avoid the side effects of Nrf-2 activation.
Data Availability
The datasets generated during and/or analyzed during the current study are included within the manuscript.
Abbreviations
- ALP:
-
Alkaline phosphatase
- ALT:
-
Alanine amino transaminase
- ARE:
-
Antioxidant response element
- AST:
-
Aspartate amino transaminase
- CPZ:
-
Cuprizone
- CTn1:
-
Cardiac troponin 1
- Cu:
-
Copper
- IFN-γ:
-
Inflammatory interferon-gamma
- IL-1β:
-
Interleukin 1β
- MDA:
-
Malondialdehyde
- Nrf-2:
-
Nuclear factor erythroid 2-related factor 2
- ROS:
-
Reactive oxygen species
- SF:
-
Sulforaphane
- TAC:
-
Total antioxidant capacity
References
Toomey, L. M., Papini, M., Lins, B., Wright, A. J., Warnock, A., McGonigle, T., & Fitzgerald, M. (2021). Cuprizone feed formulation influences the extent of demyelinating disease pathology. Scientific Reports, 11(1), 1–16.
Benetti, F., Ventura, M., Salmini, B., Ceola, S., Carbonera, D., Mammi, S., & Spisni, E. (2010). Cuprizone neurotoxicity, copper deficiency and neurodegeneration. Neurotoxicology, 31(5), 509–517.
Matsushima, G. K., & Morell, P. (2001). The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathology, 11(1), 107–116.
Petronilli, V., & Zoratti, M. (1990). A characterization of cuprizone-induced giant mouse liver mitochondria. Journal of Bioenergetics and Biomembranes, 22(5), 663–677.
Kipp, M., Clarner, T., Dang, J., Copray, S., & Beyer, C. (2009). The cuprizone animal model: New insights into an old story. Acta Neuropathologica, 118(6), 723–736.
Goldberg, J., Daniel, M., van Heuvel, Y., Victor, M., Beyer, C., Clarner, T., & Kipp, M. (2013). Short-term cuprizone feeding induces selective amino acid deprivation with concomitant activation of an integrated stress response in oligodendrocytes. Cellular and Molecular Neurobiology, 33(8), 1087–1098.
Arnold, S., & Beyer, C. (2009). Neuroprotection by estrogen in the brain: The mitochondrial compartment as presumed therapeutic target. Journal of Neurochemistry, 110(1), 1–11.
Akyuz, E., & Villa, C. (2020). A novel role of cardiac inwardly rectifying potassium channels explaining autonomic cardiovascular dysfunctions in a cuprizone-induced mouse model of multiple sclerosis. Autonomic Neuroscience, 225, 102647.
Mincu, R. I., Magda, L. S., Florescu, M., Velcea, A., Mihaila, S., Mihalcea, D., & Vinereanu, D. (2015). Cardiovascular dysfunction in multiple sclerosis. Maedica, 10(4), 364.
Andreini, C., Banci, L., Bertini, I., & Rosato, A. (2008). Occurrence of copper proteins through the three domains of life: A bioinformatic approach. The Journal of Proteome Research, 7(01), 209–216.
Fridovich, I. (1975). Superoxide dismutases. Annual review of biochemistry, 44(1), 147–159.
Zhang, X., & McIntire, W. S. (1996). Cloning and sequencing of a copper-containing, topa quinone-containing monoamine oxidase from human placenta. Gene, 179(2), 279–286.
Takahashi, Y., Kako, K., Kashiwabara, S. I., Takehara, A., Inada, Y., Arai, H., & Munekata, E. (2002). Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome C oxidase and embryonic development. Molecular and Cellular Biology, 22(21), 7614–7621.
Suzuki, K. (1969). Giant hepatic mitochondria: Production in mice fed with cuprizone. Science, 163(3862), 81–82.
Hoppel, C. L., & Tandler, B. (1973). Biochemical effects of cuprizone on mouse liver and heart mitochondria. Biochemical Pharmacology, 22(18), 2311–2318.
Herring, N. R., & Konradi, C. (2011). Myelin, copper, and the cuprizone model of schizophrenia. Frontiers in Bioscience (Scholar Edition), 3, 23.
Venturini, G. (1973). Enzymic activities and sodium, potassium and copper concentrations in mouse brain and liver after cuprizone treatment in vivo. Journal of Neurochemistry, 21(5), 1147–1151.
Xuan, Y., Yan, G., Peng, H., Wu, R., & Xu, H. (2014). Concurrent changes in 1H MRS metabolites and antioxidant enzymes in the brain of C57BL/6 mouse short-termly exposed to cuprizone: Possible implications for schizophrenia. Neurochemistry International, 69, 20–27.
Luo, M., Deng, M., Yu, Z., Zhang, Y., Xu, S., Hu, S., & Xu, H. (2020). Differential susceptibility and vulnerability of brain cells in C57BL/6 mouse to mitochondrial dysfunction induced by short-term cuprizone exposure. Frontiers in Neuroanatomy, 14, 30. https://doi.org/10.3389/fnana.2020.00030
Benedict, A. L., Mountney, A., Hurtado, A., Bryan, K. E., Schnaar, R. L., Dinkova-Kostova, A. T., & Talalay, P. (2012). Neuroprotective effects of sulforaphane after contusive spinal cord injury. Journal of Neurotraum. https://doi.org/10.1089/neu.2012.2474
Cuadrado, A., Manda, G., Hassan, A., Alcaraz, M. J., Barbas, C., Daiber, A., & Schmidt, H. H. (2018). Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacological reviews, 70(2), 348–383.
Nadeem, A., Ahmad, S. F., Al-Harbi, N. O., Attia, S. M., Bakheet, S. A., Ibrahim, K. E., & Alqinyah, M. (2019). Nrf2 activator, sulforaphane ameliorates autism-like symptoms through suppression of Th17 related signaling and rectification of oxidant-antioxidant imbalance in periphery and brain of BTBR T+ tf/J mice. Behavioural Brain Research, 364, 213–224.
Kamal, M. M., Akter, S., Lin, C. N., & Nazzal, S. (2020). Sulforaphane as an anticancer molecule: Mechanisms of action, synergistic effects, enhancement of drug safety, and delivery systems. Archives of Pharmacal Research, 43(4), 371–384.
Hua, D., Luo, A., Wu, Z., Huang, C., Li, S., Xu, X., & Liu, C. (2022). Sulforaphane improves cognitive dysfunction after surgery and anesthesia in mice: the role of Keap1-Nrf2 signaling. Brain Research Bulletin, 181(1), 1–11.
Nazmy, E. A., El-Khouly, O. A., Atef, H., & Said, E. (2017). Sulforaphane protects against sodium valproate–induced acute liver injury. Canadian Journal of Physiology and Pharmacology, 95(4), 420–426.
Guan, Z., Zhou, L., Zhang, Y., Chen, H., & Shao, F. (2021). Sulforaphane ameliorates the liver injury of traumatic hemorrhagic shock rats. Journal of Surgical Research, 267, 293–301.
Li, J., Xie, S., & Teng, W. (2021). Sulforaphane attenuates nonalcoholic fatty liver disease by inhibiting hepatic steatosis and apoptosis. Nutrients, 14(1), 76.
Bai, Y., Chen, Q., Sun, Y. P., Wang, X., Lv, L., Zhang, L. P., & Wang, X. L. (2017). Sulforaphane protection against the development of doxorubicin-induced chronic heart failure is associated with Nrf2 Upregulation. Cardiovascular Therapeutics, 35(5), e12277.
Song, L., Srilakshmi, M., Wu, Y., & Saleem, T. S. (2020). Sulforaphane attenuates isoproterenol-induced myocardial injury in mice. BioMed Research International. https://doi.org/10.1155/2020/3610285i
Wei, L., Zhang, J., Zheng, L., & Chen, Y. (2022). The functional role of sulforaphane in intestinal inflammation: A review. Food & Function. https://doi.org/10.1155/2020/3610285
Uddin, M. S., Al Mamun, A., Jakaria, M., Thangapandiyan, S., Ahmad, J., Rahman, M. A., & Aleya, L. (2020). Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Science of the Total Environment, 707, 135624.
Kubo, E., Chhunchha, B., Singh, P., Sasaki, H., & Singh, D. P. (2017). Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Scientific Reports, 7(1), 1–17.
Sun, Z., Huang, Z., & Zhang, D. D. (2009). Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS ONE, 4(8), e6588. https://doi.org/10.1371/journal.pone.0006588
Magesh, S., Chen, Y., & Hu, L. (2012). Small molecule modulators of K eap1-N rf2-ARE pathway as potential preventive and therapeutic agents. Medicinal Research Reviews, 32(4), 687–726. https://doi.org/10.1002/med.21257
Su, X., Wang, S., Zhang, H., Yang, G., Bai, Y., Liu, P., & Xin, Y. (2021). Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 through epigenetic modification. Journal of Cellular and Molecular Medicine, 25(9), 4408–4419.
Omotoso, G. O., Olajide, O. J., Gbadamosi, I. T., Adebayo, J. O., Enaibe, B. U., Akinola, O. B., & Owoyele, B. V. (2019). Cuprizone toxicity and Garcinia kola biflavonoid complex activity on hippocampal morphology and neurobehaviour. Heliyon, 5(7), e02102.
Koracevic, D., Koracevic, G., Djordjevic, V., Andrejevic, S., & Cosic, V. (2001). Method for the measurement of antioxidant activity in human fluids. Journal of Clinical Pathology, 54(5), 356–361.
Ohkawa, H., Ohishi, N., & Yagi, K. (1979). Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Analytical Biochemistry, 95(2), 351–358.
Aebi, H. (1984). Catalase in vitro. Methods in Enzymology, 105, 121–126. https://doi.org/10.1016/s0076-6879(84)05016-3
Reitman, S., & Frankel, S. (1957). A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. American Journal of Clinical Pathology, 28(1), 56–63. https://doi.org/10.1093/ajcp/28.1.56
Belfield, A., & Goldberg, D. M. (1971). Revised assay for serum phenyl phosphatase activity using 4-amino-antipyrine. Enzyme, 12(5), 561–573. https://doi.org/10.1159/000459586
Culling, C. F. A. (2013). Handbook of histopathological and histochemical techniques: Including museum techniques. Butterworth-Heinemann.
Praet, J., Guglielmetti, C., Berneman, Z., Van der Linden, A., & Ponsaerts, P. (2014). Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neuroscience and Biobehavioral Reviews, 47, 485–505. https://doi.org/10.1016/j.neubiorev.2014.10.004
Leopold, P., Schmitz, C., & Kipp, M. (2019). Animal weight is an important variable for reliable cuprizone-induced demyelination. Journal of Molecular Neuroscience: MN, 68(4), 522–528. https://doi.org/10.1007/s12031-019-01312-0
Denic, A., Johnson, A. J., Bieber, A. J., Warrington, A. E., Rodriguez, M., & Pirko, I. (2011). The relevance of animal models in multiple sclerosis research. Pathophysiology: The Official Journal of the International Society for Pathophysiology, 18(1), 21–29. https://doi.org/10.1016/j.pathophys.2010.04.004
Pandur, E., Pap, R., Varga, E., Jánosa, G., Komoly, S., Fórizs, J., & Sipos, K. (2019). Relationship of Iron Metabolism and Short-Term Cuprizone Treatment of C57BL/6 Mice. International Journal of Molecular Sciences, 20(9), 2257. https://doi.org/10.3390/ijms20092257
Ünsal, C., & Özcan, M. (2018). Neurotoxicity of cuprizone in female and male rats: Electrophysiological observations. Neurophysiology, 50(2), 108–115.
Omotoso, G. O., Ukwubile, I. I., Arietarhire, L., Sulaimon, F., & Gbadamosi, I. T. (2018). Kolaviron protects the brain in cuprizone-induced model of experimental multiple sclerosis via enhancement of intrinsic antioxidant mechanisms: Possible therapeutic applications? Pathophysiology: The Official Journal of the International Society for Pathophysiology, 25(4), 299–306. https://doi.org/10.1016/j.pathophys.2018.04.004
Taylor, C. G., Bettger, W. J., & Bray, T. M. (1988). Effect of dietary zinc or copper deficiency on the primary free radical defense system in rats. The Journal of nutrition, 118(5), 613–621. https://doi.org/10.1093/jn/118.5.613
Babbs, R. K., Beierle, J. A., Yao, E. J., Kelliher, J. C., Medeiros, A. R., Anandakumar, J., Shah, A. A., Chen, M. M., Johnson, W. E., & Bryant, C. D. (2020). The effect of the demyelinating agent cuprizone on binge-like eating of sweetened palatable food in female and male C57BL/6 substrains. Appetite. https://doi.org/10.1016/j.appet.2020.104678
Ibrahim Fouad, G., & Ahmed, K. A. (2022). Curcumin ameliorates doxorubicin-induced cardiotoxicity and hepatotoxicity via suppressing oxidative stress and modulating iNOS, NF-κB, and TNF-α in rats. Cardiovascular Toxicology, 22(2), 152–166. https://doi.org/10.1007/s12012-021-09710-w
Ibrahim Fouad, G., & Mousa, R. M. (2021). The protective potential of alpha lipoic acid on amiodarone-induced pulmonary fibrosis and hepatic injury in rats. Molecular and Cellular Biochemistry, 476(9), 3433–3448. https://doi.org/10.1007/s11010-021-04173-7
Swain, S. M., Whaley, F. S., & Ewer, M. S. (2003). Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer, 97(11), 2869–2879. https://doi.org/10.1002/cncr.11407
Haybar, H., Goudarzi, M., Mehrzadi, S., Aminzadeh, A., Khodayar, M. J., Kalantar, M., & Fatemi, I. (2019). Effect of gemfibrozil on cardiotoxicity induced by doxorubicin in male experimental rats. Biomedicine & Pharmacotherapy, 109, 530–535. https://doi.org/10.1016/j.biopha.2018.10.101
Singh, P., Sharma, R., McElhanon, K., Allen, C. D., Megyesi, J. K., Beneš, H., & Singh, S. P. (2015). Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radical Biology & Medicine, 86, 90–101. https://doi.org/10.1016/j.freeradbiomed.2015.05.028
Frobert, A., Valentin, J., Magnin, J. L., Riedo, E., Cook, S., & Giraud, M. N. (2015). Prognostic value of Troponin I for infarct size to improve preclinical myocardial infarction small animal models. Frontiers in Physiology, 6, 353. https://doi.org/10.3389/fphys.2015.00353
Liu, T., Wang, X., Karsdal, M. A., Leeming, D. J., & Genovese, F. (2012). Molecular serum markers of liver fibrosis. Biomarker Insights, 7, 105–117. https://doi.org/10.4137/BMI.S10009
Levick, S. P., & Goldspink, P. H. (2014). Could interferon-gamma be a therapeutic target for treating heart failure? Heart Failure Reviews, 19(2), 227–236. https://doi.org/10.1007/s10741-013-9393-8
Mason, J. L., Suzuki, K., Chaplin, D. D., & Matsushima, G. K. (2001). Interleukin-1beta promotes repair of the CNS. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 21(18), 7046–7052. https://doi.org/10.1523/JNEUROSCI.21-18-07046.2001
Shigemoto-Mogami, Y., Hoshikawa, K., Goldman, J. E., Sekino, Y., & Sato, K. (2014). Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. The Journal of neuroscience : The official journal of the Society for Neuroscience, 34(6), 2231–2243. https://doi.org/10.1523/JNEUROSCI.1619-13.2014
Hillis, J. M., Davies, J., Mundim, M. V., Al-Dalahmah, O., & Szele, F. G. (2016). Cuprizone demyelination induces a unique inflammatory response in the subventricular zone. Journal of neuroinflammation, 13(1), 190. https://doi.org/10.1186/s12974-016-0651-2
Yalniz, M., Demirel, U., Orhan, C., Bahcecioglu, I. H., Ozercan, I. H., Aygun, C., Tuzcu, M., & Sahin, K. (2012). Nadroparin sodium activates Nrf2/HO-1 pathway in acetic acid-induced colitis in rats. Inflammation, 35(3), 1213–1221. https://doi.org/10.1007/s10753-012-9431-z
Saleh, D. O., Mansour, D. F., Hashad, I. M., & Bakeer, R. M. (2019). Effects of sulforaphane on D-galactose-induced liver aging in rats: Role of keap-1/nrf-2 pathway. European Journal of Pharmacology, 855, 40–49. https://doi.org/10.1016/j.ejphar.2019.04.043
Zhao, H. D., Zhang, F., Shen, G., Li, Y. B., Li, Y. H., Jing, H. R., & Tian, X. F. (2010). Sulforaphane protects liver injury induced by intestinal ischemia reperfusion through Nrf2-ARE pathway. World journal of gastroenterology: WJG, 16(24), 3002.
Jaiswal, A. K. (2004). Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radical Biology and Medicine, 36(10), 1199–1207.
Alotaibi, M. F., Al-Joufi, F., Abou Seif, H. S., Alzoghaibi, M. A., Djouhri, L., Ahmeda, A. F., & Mahmoud, A. M. (2020). Umbelliferone inhibits spermatogenic defects and testicular injury in lead-intoxicated rats by suppressing oxidative stress and inflammation, and improving Nrf2/HO-1 signaling. Drug Design, Development and Therapy, 14, 4003.
Amaral, J. H., Rizzi, E. S., Alves-Lopes, R., Pinheiro, L. C., Tostes, R. C., & Tanus-Santos, J. E. (2019). Antioxidant and antihypertensive responses to oral nitrite involves activation of the Nrf2 pathway. Free Radical Biology & Medicine, 141, 261–268. https://doi.org/10.1016/j.freeradbiomed.2019.06.028
Hayes, J. D., & McMahon, M. (2009). NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends in Biochemical Sciences, 34(4), 176–188. https://doi.org/10.1016/j.tibs.2008.12.008
Wan, X., Liu, C., Chen, Y. B., Gu, M., Cai, Z. K., Chen, Q., & Wang, Z. (2017). Sulforaphane treatment of stress urinary incontinence via the Nrf2-ARE pathway in a rat model. Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology, 44(5), 1912–1922. https://doi.org/10.1159/000485880
Xu, J. F., Lu, J. J., Cao, Y., Wang, W., Li, H. H., Chen, J. G., Wang, F., & Wu, P. F. (2020). Sulforaphane alleviates ethanol-mediated central inhibition and reverses chronic stress-induced aggravation of acute alcoholism via targeting Nrf2-regulated catalase expression. Neuropharmacology, 176, 108235. https://doi.org/10.1016/j.neuropharm.2020.108235
Shanmugam, G., Challa, A. K., Litovsky, S. H., Devarajan, A., Wang, D., Jones, D. P., & Rajasekaran, N. S. (2019). Enhanced Keap1-Nrf2 signaling protects the myocardium from isoproterenol-induced pathological remodeling in mice. Redox Biology, 27, 101212.
Suh, J. H., Shenvi, S. V., Dixon, B. M., Liu, H., Jaiswal, A. K., Liu, R. M., & Hagen, T. M. (2004). Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proceedings of the National Academy of Sciences of the United States of America, 101(10), 3381–3386. https://doi.org/10.1073/pnas.0400282101
Thimmulappa, R. K., Mai, K. H., Srisuma, S., Kensler, T. W., Yamamoto, M., & Biswal, S. (2002). Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Research, 62(18), 5196–5203.
Joko, S., Watanabe, M., Fuda, H., Takeda, S., Furukawa, T., Hui, S. P., & Chiba, H. (2017). Comparison of chemical structures and cytoprotection abilities between direct and indirect antioxidants. Journal of Functional Foods, 35, 245–255.
Heiss, E., Herhaus, C., Klimo, K., Bartsch, H., & Gerhäuser, C. (2001). Nuclear factor κB is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. Journal of Biological Chemistry, 276(34), 32008–32015.
Satta, S., Mahmoud, A. M., Wilkinson, F. L., Yvonne Alexander, M., & White, S. J. (2017). The role of Nrf2 in cardiovascular function and disease. Oxidative Medicine and Cellular Longevity. https://doi.org/10.1155/2017/9237263
Duan, J., Guan, Y., Mu, F., Guo, C., Zhang, E., Yin, Y., & Wen, A. (2017). Protective effect of butin against ischemia/reperfusion-induced myocardial injury in diabetic mice: Involvement of the AMPK/GSK-3β/Nrf2 signaling pathway. Scientific Reports, 7(1), 1–14.
Yu, J., Wang, L., Akinyi, M., Li, Y., Duan, Z., Zhu, Y., & Fan, G. (2015). Danshensu protects isolated heart against ischemia reperfusion injury through activation of Akt/ERK1/2/Nrf2 signaling. International Journal of Clinical and Experimental Medicine, 8(9), 14793.
Xin, Y., Bai, Y., Jiang, X., Zhou, S., Wang, Y., Wintergerst, K. A., & Cai, L. (2018). Sulforaphane prevents angiotensin II-induced cardiomyopathy by activation of Nrf2 via stimulating the Akt/GSK-3ss/Fyn pathway. Redox Biology, 15, 405–417.
Li, J., Ichikawa, T., Villacorta, L., Janicki, J. S., Brower, G. L., Yamamoto, M., & Cui, T. (2009). Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arteriosclerosis, Thrombosis, and Vascular Biology, 29(11), 1843–1850.
Gounder, S. S., Kannan, S., Devadoss, D., Miller, C. J., Whitehead, K. S., Odelberg, S. J., & Rajasekaran, N. S. (2012). Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training.
Lopes, R. A., Neves, K. B., Tostes, R. C., Montezano, A. C., & Touyz, R. M. (2015). Downregulation of nuclear factor erythroid 2–related factor and associated antioxidant genes contributes to redox-sensitive vascular dysfunction in hypertension. Hypertension, 66(6), 1240–1250.
Ooi, B. K., Chan, K. G., Goh, B. H., & Yap, W. H. (2018). The role of natural products in targeting cardiovascular diseases via Nrf2 pathway: Novel molecular mechanisms and therapeutic approaches. Frontiers in Pharmacology, 9, 1308.
Abukhalil, M. H., Hussein, O. E., Aladaileh, S. H., Althunibat, O. Y., Al-Amarat, W., Saghir, S. A., & Mahmoud, A. M. (2021). Visnagin prevents isoproterenol-induced myocardial injury by attenuating oxidative stress and inflammation and upregulating Nrf2 signaling in rats. Journal of Biochemical and Molecular Toxicology, 35(11), e22906.
Gaona-Gaona, L., Molina-Jijón, E., Tapia, E., Zazueta, C., Hernández-Pando, R., Calderón-Oliver, M., & Pedraza-Chaverri, J. (2011). Protective effect of sulforaphane pretreatment against cisplatin-induced liver and mitochondrial oxidant damage in rats. Toxicology, 286(1–3), 20–27.
Greco, T., Shafer, J., & Fiskum, G. (2011). Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radical Biology and Medicine, 51(12), 2164–2171.
Geisel, J., Brück, J., Glocova, I., Dengler, K., Sinnberg, T., Rothfuss, O., & Ghoreschi, K. (2014). Sulforaphane protects from T cell–mediated autoimmune disease by inhibition of IL-23 and IL-12 in dendritic cells. The Journal of Immunology, 192(8), 3530–3539.
Moustafa, P. E., Abdelkader, N. F., El Awdan, S. A., El-Shabrawy, O. A., & Zaki, H. F. (2018). Extracellular matrix remodeling and modulation of inflammation and oxidative stress by sulforaphane in experimental diabetic peripheral neuropathy. Inflammation, 41(4), 1460–1476.
Qi, T., Xu, F., Yan, X., Li, S., & Li, H. (2016). Sulforaphane exerts anti-inflammatory effects against lipopolysaccharide-induced acute lung injury in mice through the Nrf2/ARE pathway. International Journal of Molecular Medicine, 37(1), 182–188.
Mazarakis, N., Anderson, J., Toh, Z. Q., Higgins, R. A., Do, L. A. H., Luwor, R. B., & Licciardi, P. V. (2021). Examination of novel immunomodulatory effects of L-sulforaphane. Nutrients, 13(2), 602.
Hernandez-Rabaza, V., Cabrera-Pastor, A., Taoro-Gonzalez, L., Gonzalez-Usano, A., Agusti, A., Balzano, T., & Felipo, V. (2016). Neuroinflammation increases GABAergic tone and impairs cognitive and motor function in hyperammonemia by increasing GAT-3 membrane expression. Reversal by sulforaphane by promoting M2 polarization of microglia. Journal of Neuroinflammation, 13(1), 1–13.
Zhou, A., Hong, Y., & Lv, Y. (2019). Sulforaphane Attenuates Endometriosis in Rat Models Through Inhibiting PI3K/Akt Signaling Pathway. Dose-response : A publication of International Hormesis Society, 17(2), 1559325819855538. https://doi.org/10.1177/1559325819855538
Checker, R., Gambhir, L., Thoh, M., Sharma, D., & Sandur, S. K. (2015). Sulforaphane, a naturally occurring isothiocyanate, exhibits anti-inflammatory effects by targeting GSK3β/Nrf-2 and NF-κB pathways in T cells. Journal of Functional Foods, 19, 426–438.
Briones-Herrera, A., Ramírez-Camacho, I., Zazueta, C., Tapia, E., & Pedraza-Chaverri, J. (2020). Altered proximal tubule fatty acid utilization, mitophagy, fission and supercomplexes arrangement in experimental Fanconi syndrome are ameliorated by sulforaphane-induced mitochondrial biogenesis. Free Radical Biology and Medicine, 153, 54–70.
Pan, H., He, M., Liu, R., Brecha, N. C., Yu, A. C., & Pu, M. (2014). Sulforaphane protects rodent retinas against ischemia-reperfusion injury through the activation of the Nrf2/HO-1 antioxidant pathway. PLoS ONE, 9(12), 14186. https://doi.org/10.1371/journal.pone.0114186
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). The author declares that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
The author designed and conducted the study, performed the experiments, collected data, and wrote and approved the manuscript. He would like to thank his Mother for supporting him during his research career.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The author has no relevant financial or non-financial interests to disclose.
Ethical Approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of National Research Centre (19-313).
Additional information
Handling Editor: Phillip Kopf .
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ibrahim Fouad, G. Sulforaphane, an Nrf-2 Agonist, Modulates Oxidative Stress and Inflammation in a Rat Model of Cuprizone-Induced Cardiotoxicity and Hepatotoxicity. Cardiovasc Toxicol 23, 46–60 (2023). https://doi.org/10.1007/s12012-022-09776-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-022-09776-0