
Abstract
Human monocytotropic ehrlichiosis is an emerging tick-borne infection caused by the obligate intracellular pathogen, Ehrlichia chaffeensis. The non-specific symptoms can range from a self-limiting fever to a fatal septic-like syndrome and may be misdiagnosed. The limited treatment choices including doxycycline are effective only in the initiation phase of the infection. It seems that novel therapeutic targets and new vaccine strategies could be effective to control this pathogen. This study is comprised of two major phases. First, the common proteins retrieved through subtractive analysis and potential drug targets were evaluated by subcellular localization, homology prediction, metabolic pathways, druggability, essentiality, protein–protein interaction networks, and protein data bank availability. In the second phase, surface-exposed proteins were assessed based on antigenicity, allergenicity, physiochemical properties, B cell and T cell epitopes, conserved domains, and protein–protein interaction networks. A multi-epitope vaccine was designed and characterized using molecular dockings and immune simulation analysis. Six proteins including WP_011452818.1, WP_011452723.1, WP_006010413.1, WP_006010278.1, WP_011452938.1, and WP_006010644.1 were detected. They belong to unique metabolic pathways of E. chaffeensis that are considered as new essential drug targets. Based on the reverse vaccinology, WP_011452702.1, WP_044193405.1, WP_044170604.1, and WP_006010191.1 proteins were potential vaccine candidates. Finally, four B cell epitopes, including SINNQDRNC, FESVSSYNI, SGKKEISVQSN, and QSSAKRKST, were used to generate the multi-epitope vaccine based on LCL platform. The vaccine showed strong interactions with toll-like receptors and acceptable immune-reactivity by immune simulation analysis. The findings of this study may represent a turning point in developing an effective drug and vaccine against E. chaffeensis. However, further experimental analyses have remained.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human monocytotropic ehrlichiosis (HME) is an emerging life-threatening zoonotic infection that is characterized by a flu-like illness [1]. HME may lead to a severe disease similar to the sepsis-like syndrome in the elderly and people with underlying conditions such as transplant patients [2]. The etiologic agent of HME is Ehrlichia chaffeensis, one of the most prevalent zoonotic pathogens in North America. Approximately 50–70% of the cases require hospitalization and fatality rates are estimated to be about 3% [3].
E. chaffeensis, a Gram-negative obligatory intracellular bacterium, belonging to the Rickettsiales order, was first isolated in 1990 and transmitted primarily through the Lone Star tick, Amblyomma americanum [4]. The infection often is misdiagnosed due to non-specific clinical symptoms and a lack of specific diagnostic tests, especially in the early stages of HME. An adverse outcome is correlated with delayed diagnosis and therapy. The only treatment choice is broad-spectrum antibiotics, doxycycline, or tetracycline, which are effective only in the initiation phase of the infection [5]. It seems that more advanced researches are necessary to overcome the challenges related to this pathogen. On the other hand, due to the rapid spread of bacterial antibiotic resistance, new drug target discovery is necessary to control infections [6].
With the development of technologies in the comparative and subtractive genomics of different strains, this great opportunity has been provided for researchers to use in silico approaches for the prediction of new potential drug and vaccine targets with multiple screenings [7]. Recently, bioinformatics analyses have shown practical and useful results in the field of new target discovery in several life-threatening microorganisms such as SARS-CoV-2 [8, 9], Mycobacterium tuberculosis [10, 11], and Helicobacter pylori [12]. These strategies that reduce the time and cost associated with the experimental errors serve to list the new potential non-homologous drug and vaccine targets that may be utilized for experimental and in vivo validation [13].
There are limited studies on the protein variability, essential proteins, and virulence factors of E. chaffeensis in different cycles of infection [14]. Although virulence factors such as lipopolysaccharide, peptidoglycan, pili, and capsular polysaccharide components are not known for this bacterium, it seems that surface proteins have essential roles in virulence and host–pathogen interactions [15]. No vaccines exist for HME and only limited studies have reported possible vaccine candidates against E. chaffeensis based on subunit and live attenuated vaccines [16, 17]. Today, new studies do not often support live attenuated vaccines because of the associated challenges (e.g., reversion to wild type or causing illness in immunosuppressed individuals), and researchers are looking for safer vaccines. It seems that no comprehensive and systematic investigation has been conducted on the discovery of new drug targets against E. chaffeensis. Thus, in this study, we mainly focus on the in silico discovery of new putative drug targets against E. chaffeensis using subtractive genomics. We also applied reverse vaccinology approaches to identify potential vaccine candidates and finally designed a multi-epitope vaccine against E. chaffeensis.
Materials and Methods
Data Collection of Proteomes
The eight E. chaffeensis strains with available complete genome sequences in the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) were extracted and subjected to the bacterial pan-genome analysis (BPGA) software, a quick genome analysis pipeline [18], to identify the core proteome (identity cutoff = 0.5).
Prediction of Subcellular Localization
All non-redundant core proteins were imported to PSORTb v.3.0.2 online server (www.psort.org/psortb/) for the determination of subcellular localization [19].
Identification of Novel Drug Targets Against E. chaffeensis.
Similarity of Proteins With the Human Proteome
To prevent tolerance or auto-immune responses, the sequence similarity of proteins with the human proteome (Homo sapiens taxid: 9606) was evaluated by PSI-BLAST provided by the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) (identity ≥ 15%, max score > 100, E-value < \({10}^{-3}\)) [20]. Identification of sequence similarity via PSI-BLAST is more sensitive than usual BLASTp when they are distantly related to the query sequence.
Host and Pathogen Metabolic Pathway Analysis
Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg), a database of metabolic pathways, linking the genomic information to the functional information [21], was used to exclude proteins from common metabolic pathways between E. chaffeensis and human. The host and pathogen metabolic pathways were manually compared to identify the unique metabolic pathways of E. chaffeensis.
Druggability Analysis
Druggability analysis of the non-homologous cytoplasmic proteins evaluated against all present and FDA-approved drug targets in the DrugBank database (https://go.drugbank.com/) [22]. A BLASTp was performed to align the sequence of selected proteins against the above-mentioned drug targets. Proteins with similarity to the drug targets in the DrugBank database were considered as druggable targets. On the other hand, the non-hit proteins at the threshold value were considered novel drug targets.
Prediction of Non-homologous Essential Proteins and Shortlisted Proteins
The essentiality of the novel drug targets was determined using the Database of Essential Genes (DEG) (http://origin.tubic.org/deg/public/index.php) [23] with identity = 0.5. The DEG contains experimentally essential gene products that are involved in the key cellular functions and are necessary to support cellular life. Finally, the relevant Protein Data Bank (PDB) of novel drug targets was identified using BLASTp against RCSB PDB database (https://www.rcsb.org/). Proteins having a similarity with PDB files (coverage ≥ 80 and identity ≥ 50) were shortlisted and considered as promising novel drug targets. Finally, the protein–protein interaction of novel drug targets was evaluated using the STRING (https://string-db.org/) web tool [24].
Prediction of Vaccine Targets by Reverse Vaccinology Approaches
Identification of Antigenic and Non-allergen Proteins
The surface-exposed proteins are considered ideal subunit vaccine targets due to their stronger interactions with host immune cells. Furthermore, these proteins were selected based on subcellular localization from PSORTb v.3.0.2 online server. The outer membrane and extracellular proteins were then assessed by the TMHMM Server v. 2.0 web tool (http://www.cbs.dtu.dk/services/TMHMM/) to identify their transmembrane helices [25]. In the next step, the antigenic properties of the selected proteins were determined by VaxiJen online server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) with a threshold of ≥ 0.4 [26]. VaxiJen is the first software that predicts antigenic properties via the machine learning method. In addition, the allergenicity of antigenic proteins was determined using the AlgPred 2.0 web tool (https://webs.iiitd.edu.in/raghava/algpred2/batch.html) with a threshold of ≥ 0.5 [27].
Linear B Cell Epitopes and MHC-II Binding Site Determination
The selected proteins from the above analyses were evaluated to identify B cell epitopes and MHC-II binding sites. In this study, the BepiPred v2.0 tool was used to predict linear B cell epitopes of proteins (http://www.cbs.dtu.dk/services/BepiPred/) with the threshold value of ≥ 0.6 [28]. B cell epitopes’ ratio to the total number of amino acids was calculated for each protein. Human MHC-II binding sites were predicted by TepiTool, the prediction tool of Immune Epitope Database (http://tools.iedb.org/tepitool/) with a threshold of the top 10% of peptides [29]. The T cell ratio to the total number of amino acids was calculated for each protein.
Physiochemical Characteristics of Selected Proteins
The physicochemical properties including molecular weight, theoretical pI, the estimated half-life, and aliphatic and instability indices of selected proteins were evaluated using the Expasy ProtParam server (https://web.expasy.org/protparam/) [30]. The functional class of the proteins and adhesion probability were predicted through VICMpred (https://webs.iiitd.edu.in/raghava/vicmpred/) and Vaxign (http://www.violinet.org/vaxign2), respectively [31].
Tertiary Structure Prediction and Determination of the Conformational B Cell Epitopes
The tertiary structure (3D) of putative immunogenic proteins was predicted by the Robetta tool (https://robetta.bakerlab.org/) [32]. In the next step, the conformational B cell epitopes of selected proteins were characterized using the ElliPro server (http://tools.iedb.org/ellipro/) [33] with a threshold ≥ 0.8. The surface-exposed conformational B cell epitopes were visualized and shown in different colors by Jmol software [34].
Sequence Conservation of B Cell Epitopes
The linear and conformational B cell epitopes of candidate proteins were assessed to determine the conservancy among E. chaffeensis strains using the Epitope Conservancy Database (IEDB) (http://tools.iedb.org/conservancy/) [35].
Conserved Domain Search and Protein–Protein Interaction Networks
The Conserved Domain Database, CDD (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml), analysis was applied to find the conserved domains of the selected proteins [36]. The interactions between surface-exposed unknown-function proteins and other proteins of E. chaffeensis were evaluated using the STRING (https://string-db.org/) web tool [24].
Shortlisted Putative Vaccine Candidates
Considering different indicators such as antigenicity, allergenicity, B cell and T cell epitopes, physicochemical characteristics, and epitope conservation, we have proposed four appropriate targets as promising immunogenic proteins.
Construction of the Multi-epitope Vaccine
In the next step, we used four linear B cell epitopes and the TbpB C-lobe mutant from Neisseria meningitidis M982, as a scaffold for a better presentation of surface epitopes to the immune system, to design an effective multi-epitope vaccine [37]. The epitopes were selected considering four features including antigenicity, allergenicity, conservancy, and being exposed on the surface of the proteins. The 3D structure of the multi-epitope TbpB was determined using the Robetta server. ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) was used to discover the potential errors in the 3D model and validate the quality of the 3D structures. Moreover, the Ramachandran plot was created using the Zlab Ramachandran Plot server (https://zlab.umassmed.edu/bu/rama/index.pl). This plot demonstrates the energetically permitted and refused dihedral angles constituting an amino acid psi (ψ) and phi (φ), based on the Van der Waal radius of the side chains.
Molecular Dockings and Immune-Simulation of the Multi-epitope Vaccine
The interaction between the multi-epitope construct and TLR1, 2, 4, and 6 was evaluated using the pyDockWEB server (https://life.bsc.es/pid/pydockweb) [38]. The immunoreactivity of the multi-epitope vaccine was tested in silico immune simulation which was predicted by C-ImmSim web server (https://150.146.2.1/C-IMMSIM/index.php) [39]. The levels of B cell population, T cell population, and cytokines were predicted on the 7th day after immunization.
Results
The procedure of drug discovery and reverse vaccinology and the total number of proteins described in each step of our work is summarized in Fig. 1.

This flowchart shows the workflow for the identification of novel drug and vaccine targets against E. chaffeensis using bioinformatics databases. All tools, web servers, and thresholds are mentioned. Eventually, six drug targets and four putative vaccine candidates were identified against E. chaffeensis, and a multi-epitope vaccine was generated based on LCL platform
Data Collection of Proteomes
The proteomes of eight strains of E. chaffeensis were retrieved and BPGA analysis resulted in the identification of 841 proteins in the core proteome of E. chaffeensis.
Prediction of Subcellular Localization
Out of 841 proteins, 456 cytoplasmic proteins and 385 non-cytoplasmic proteins were identified, consisting of 27 surface-exposed proteins. Generally, cytoplasmic proteins usually are considered potential therapeutic targets while surface-exposed proteins are promising vaccine candidates.
Identification of Novel Drug Targets Against E. chaffeensis.
Similarity of Proteins With the Human Proteome
Out of 456 proteins associated with the cytoplasmic proteins, 173 proteins with no similarity to the host proteome were identified by BLASTp analysis and characterized for drug target evaluation. The rest of the 283 proteins were homologous to human’s proteome and consequently discarded.
Host and Pathogen Metabolic Pathway Analysis
According to the results of the metabolic pathway analysis, there is no common metabolic pathway between humans and E. chaffeensis. Proteins of unique pathogen-specific metabolic pathways are very important in determining drug targets and might serve as potential drug targets.
Druggability Analysis
Druggability analysis results revealed that out of 173 non-homologous proteins, a total of 41 druggable proteins were identified, while 132 proteins with no similarity to known drug targets were considered novel drug targets.
Prediction of Non-homologous Essential Proteins and Shortlisted Proteins
Out of non-homologous and novel drug targets that were subjected to BLAST against the DEG (Database of Essential Gene), 27 proteins were essential for the survival of E. chaffeensis. With PDB analysis, 18 proteins with relevant PDB files are represented and summarized in Supplementary Table 1. Finally, six proteins including WP_011452818.1, WP_011452723.1, WP_006010413.1, WP_006010278.1, WP_011452938.1, and WP_006010644.1 (with coverage ≥ 80 and identity ≥ 50) were introduced as shortlisting drug targets to be evaluated for the designing of promising novel drugs.
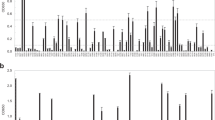
The STRING analysis of the novel drug targets revealed that ThiC (WP_011452818.1) has a neighborhood relationship with several proteins involved in thiamine, proline, and biotin synthesis including thiD, thiG, thiE, putA, bioB, thiL, and thiF. LysS (WP_011452723.1) belongs to the class-I aminoacyl-tRNA synthetase family and has a co-occurrence relationship with gltX-1, ECH_0784, and ECH_0820 which are involved in tRNA synthesis. GlyQ (WP_006010413.1), Glycine-tRNA ligase alpha subunit, has a neighborhood relationship with GlyS (Glycine-tRNA ligase beta subunit) and DnaJ that participates in hyperosmotic and heat shock response. PyrH (WP_006010278.1) catalyzes the reversible phosphorylation of UMP to UDP, and has a co-occurrence relationship with Gmk (guanylate kinase), and neighborhood relationships with several translation-related proteins such as frr, tsf, and rpsB genes. HslV (WP_011452938.1) is a protease subunit of a proteasome-like degradation complex that has co-occurrence relationships with Hslu, Dnak, Grpe, Htpg, Dnaj, Clpb, Clpp, Groes, and Grol, which are all chaperones and chaperonins with essential roles in response to hyperosmotic and heat shock. SecA (WP_006010644.1) has co-occurrence relations with SecY, Ffh, YidC, and LepB all are essential for Sec translocase complex (see Fig. 2A).

Protein–protein interaction evaluation by STRING database. A Interaction of novel putative drug targets (red balls) with other proteins of E. chaffeensis. ThiC (WP_011452818.1) has a neighborhood relationship with Thid, Thig, Thie, Puta, Biob, Thil, and Thif proteins. LysS (WP_011452723.1) has a co-occurrence relationship with GltX-1, ECH_0784, and ECH_0820 involved in tRNA synthesis. GlyQ (WP_006010413.1) has a neighborhood relationship with GlyS (Glycine-tRNA ligase beta subunit) and DnaJ. PyrH (WP_006010278.1) catalyzes the reversible phosphorylation of UMP to UDP, and has a co-occurrence and neighborhood relationship with Gmk (guanylate kinase) and translation-related proteins (Frr, Tsf, and RpsB), respectively. HslV (WP_011452938.1) has co-occurrence relationships with Hslu, Dnak, Grpe, Htpg, Dnaj, Clpb, Clpp, Groes, and Grol. SecA (WP_006010644.1) has co-occurrence relations with Secy, Ffh, Yidc, and Lepb. B Protein–protein interactions of novel hypothetical putative vaccine candidates (red balls) with other proteins of E. chaffeensis. WP_044170604.1 and WP_044147713.1 have a neighborhood relationship with lpdA-2 as a dihydrolipoyl dehydrogenase, and ECH_0866 (uncharacterized protein) from E. chaffeensis, respectively. WP_044193405.1 has a neighborhood relationship with sucC and fabD genes. Both of these molecules are involved in metabolic processes
Prediction of Vaccine Targets by Reverse Vaccinology Approaches
Identification of Antigenic and Non-allergen Proteins
A total of 27 surface-exposed proteins (by PSORTb online server) were imported to VaxiJen, and the server evaluated the potential antigenicity of 24 proteins. Consequently, out of 24 antigenic proteins, 14 non-allergens were investigated and only seven proteins had no similarity to the human proteome (see Fig. 1).
Linear B Cell Epitopes and MHC-II Binding Sites
The number of linear and conformational B cell epitopes, B cell epitope ratio, MHC class II binding sites, and the T cell ratio of the 7 proteins were determined and included in Supplementary Table 2.
Physiochemical Characteristics of Immunogenic Proteins
VICMpred database classifies the immunogenic proteins into four different functional classes. Three proteins were virulence factors, followed by three metabolism molecules, and one protein involved in the cellular process. The estimated half-life of all proteins was over 10 h in Escherichia coli, in vivo. All physicochemical features of proteins were reported in Supplementary Table 2.
Tertiary Structure Prediction and Conformational B Cell Epitopes
The 3D structure of all seven selected proteins was predicted using Robetta. The conformational B cell epitopes of seven proteins are presented in Supplementary Table 3 and Fig. 3. All immunogenic proteins were assigned for five outer membrane proteins (WP_044170828.1, WP_006010497.1, WP_044170604.1, WP_011452702.1, and WP_044193405.1) and two extracellular proteins (WP_006010191.1 and WP_044147713.1). The number of conformational epitopes was as follows: WP_044170604.1, 11 epitopes; WP_011452702.1, 8 epitopes; WP_044147713.1, 5 epitopes; WP_044170828.1 4 epitopes; WP_044193405.1, 3 epitopes; WP_006010191.1, 3 epitopes; and WP_006010497.1, 2 epitopes. See Supplementary Table 2 for additional information.

Identification of conformational B cell epitopes on the tertiary structure of selected proteins via the Ellipro and Robetta database. Each color demonstrated one conformational epitope. This figure showed 7 vaccine candidates, including five outer membrane proteins (WP_044170828.1, WP_006010497.1, WP_044170604.1, WP_011452702.1, and WP_044193405.1) and two extracellular proteins (WP_006010191.1 and WP_044147713.1)
Sequence Conservation of B Cell Epitopes
The IEDB conservancy analysis of linear and conformational B cell epitopes among eight E. chaffeensis strains showed that WP_006010191.1, WP_044147713.1, and WP_044170604.1 have 100% conserved linear B cell epitopes. In addition, WP_044193405.1, WP_011452702.1, and WP_044170828.1 have highly conserved linear B cell epitopes. Moreover, the conformational epitopes of WP_006010191.1, WP_044193405.1, WP_011452702.1, WP_044147713.1, WP_044170604.1, and WP_006010497.1 were completely conserved among E. chaffeensis strains. The conservation analysis of linear and conformational B cell epitopes is demonstrated in Supplementary Table 3. The conformational B cell epitopes have been shown on the 3D structure of proteins in Fig. 3.
Conserved Domains and Protein–Protein Interaction Networks
Based on CDD analysis, the WP_011452702.1 and WP_044170828.1 have an outer membrane channel family domain that belongs to the porin superfamily. These outer membrane channels share a beta-barrel structure with different strands and cracks. The WP_006010497.1 has an OmpH conserved domain that belongs to the outer membrane protein (OmpH-like) superfamily. Skp (OmpH) is a molecular chaperone that interacts with unfolded proteins in the periplasm, by the Sec transposition machinery.
WP_006010191.1 has a peptidase_M23 conserved domain. Members of this family are zinc-dependent metallopeptidases. This family belongs to Gly-Gly endopeptidases. WP_044147713.1, WP_044170604.1, and WP_044193405.1 as hypothetical proteins had no information in CDD databases and were evaluated by STRING databases. The STRING analysis showed that WP_044170604.1 (ECH_0991) and WP_044147713.1 (ECH_0865) have a neighborhood relationship with LpdA-2 as a dihydrolipoyl dehydrogenase, and ECH_0866 (uncharacterized protein), respectively. In addition, WP_044193405.1 as a conserved protein (ECH_0526) has a neighborhood relationship with sucC and fabD genes. Both of these molecules are involved in metabolic processes. The SucC enzyme, a succinyl-CoA synthetase, is involved in the citric acid cycle (TCA) which couples the hydrolysis of succinyl-CoA to the ATP or GTP synthesis. FabD is a malonyl CoA-acyl carrier protein transacylase. See Fig. 2B.
Shortlist of Selected Proteins
Finally, through multiple analyses, four surface proteins were selected as promising immunogenic targets against E. chaffeensis. These putative proteins include a porin (WP_011452702.1), two hypothetical proteins (WP_044193405.1 and WP_044170604.1), and a M23 family metallopeptidase (WP_006010191.1).
Construction of the Multi-epitope Vaccine
Four suitable linear B cell epitopes from shortlisted proteins including SINNQDRNC (from WP_006010191.1), FESVSSYNI (WP_044193405.1), SGKKEISVQSN (WP_011452702.1), and QSSAKRKST (WP_044170604.1) were preferred for designing a multi-epitope recombinant protein. The selected epitopes showed reasonable characteristics such as antigenicity, non-allergenicity, conservancy, and being exposed on the surface of the proteins. Several previous studies have confirmed that TbpB can function as a powerful platform to represent the surface epitopes of immunogenic proteins to induce immunity against bacterial infection [37]. The final chimeric multi-epitope TbpB sequence has been shown in Supplementary Table 3. The 3D structure of the multi-epitope vaccine was predicted using Robetta. The ProSA-web analysis showed a z-score of − 5.33. ProSA-web plot showed that the z-score of the protein was in the range of native conformations, determined by NMR spectroscopy (dark blue) and X-ray crystallography (light blue) based on the protein length. At least 90% of the residues of an ideal and reliable 3D structure are located in the favored zone of the Ramachandran plot. The plot of this multi-epitope vaccine showed that 91.53%, 6.45%, and 2.016% of the amino acids were located in the highly preferred and questionable zones, respectively. See Fig. 4A.

A 1 3D structure of chimeric TbpB. The colored regions represent the linear B cell epitopes of E. chaffeensis embedded in TbpB. 2 ProSA-web analysis, showing a z-score of − 5.33, is in the range of other native proteins. 3 The Ramachandran plot represents that 91.53%, 6.45%, and 2.016% of the amino acids of the chimeric TbpB were located in the preferred (green crosses), allowed (brown triangles), and outlier (red circles) zones, respectively. Black, dark gray, gray, and light gray zones show the preferred conformation degree. B Assessment of immunoreactivity of the multi-epitope construct through in C-ImmSim server. 1 B cell population counts based on the isotype (IgM, IgG1, and IgG2). 2 T cell populations demonstrated an increased Th1 cell population. 3 The cytokine and interleukin concentration showed higher levels of IL-2 and IFN-γ. C Molecular docking of the multi-epitope construct using the pyDockWEB server. The total interaction scores of TLR 1, 2, 4, and 6 were − 26.956, − 22.950, − 36.800, and − 27.634, respectively. The strongest interaction was observed between the multi-epitope vaccine and TLR4
Dockings and Immune Simulations of the Multi-epitope Vaccine
The immune simulation demonstrated acceptable immunoreactivity of the multi-epitope vaccine, inducing high levels of cytokine (IL-2 and IFN-γ), B cell, and T cell populations (Fig. 4B). The multi-epitope vaccine showed strong interactions with human TLRs and the total molecular docking score varied between − 36.8 and − 22.9 (Fig. 4C).
Discussion
Despite the importance and current challenges associated with E. chaffeensis, limited studies have been performed on designing drug targets or vaccines against this pathogen using conventional and bioinformatics approaches. In 2015, Nair et al. studied two clones of attenuated E. chaffeensis mutants that protected against wild-type infection in both reservoir and accidental hosts [40]. In other studies, isoforms of the outer membrane protein P28 [41] and the outer membrane protein entry-triggering protein EtpE [16] have been identified as vaccine candidates. In Budacherti et al. study, EtpE from the specific strain of E. chaffeensis seems to be involved in entering the host cell and demonstrated partial protection in dogs. There were some challenges with this experimental study; this candidate was selected from one strain of E. chaffeensis (Arkansas strain) and the mice infected with the E. chaffeensis Wakulla strain could not induce an immune response completely. In the past, limited information was available about the biology of the proteins in microorganisms. However, in this post-genomic era advancements in integrated “omics” data such as genomics and proteomics allow extensive computational investigations to find new non-homologous drug and vaccine targets with the lowest cost and high efficiency [42]. Subtractive proteomics, an in silico strategy, uses comprehensive comparative screening and proposes bacterial proteins with the potential to become drug targets or suitable vaccine candidates without laboratory experimentation [43]. One of the advantages of our study was the extraction of the core proteome from all available E. chaffeensis strains in the GenBank database and finding common immunogenic proteins among them, which can increase the range of immunity against different strains.
Analysis of the core proteome of various microorganisms has surprisingly revealed new drug target proteins that could not be identified by conventional methods [44,45,46]. This can solve many problems and impossibilities in drug and vaccine development where conventional approaches have failed. Unlike many pathogens, there is no specific study on drug targets against E. chaffeensis. However, Abid Ali et al. investigated novel drug targets and vaccine candidates by subtractive proteomics in ticks and tick-borne pathogens (TBPs). They introduced 11 potential drug targets and one potential vaccine candidate against TBPs [47].
In this study, our major focus was the identification of new potential vaccine and drug targets with proteome-based approaches to develop therapeutic goals and introduce an epitope-based vaccine against E. chaffeensis through a reverse vaccinology strategy. Based on the results, we found all unique metabolic pathways in E. chaffeensis that are not present in human. The presence of these pathogen-specific pathways provided an opportunity to identify antimicrobial agents that specifically target the pathogen, and thus, they are safe and have no side effects for the host [48]. Targeting essential proteins in unique metabolic pathways, required for bacterial survival and cell cycle function, provides an advantage to design new therapeutic agents that specifically target the pathogen [49]. In this study, after multi-stage analysis, we shortlisted six essential non-homologous proteins with unique metabolic pathways as novel promising drug targets. These proteins are involved in different essential cellular life processes. WP_011452818.1, thiamine biosynthesis ThiC, is involved in the synthesis of thiamine and biotin. Thiamine is a crucial co-factor for life and plays a central role in the metabolism of all organisms such as bacteria [50]. The importance of thiamine biosynthetic pathways is demonstrated well in ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.) [51]. Another drug target protein, WP_006010413.1, is a glycine-tRNA ligase subunit alpha that is an important part of the enzyme complex that catalyzes the attachment of glycine to tRNA(Gly) [52].
In 2020, Luo et al. uncovered linear and conformal B cell epitopes of immunodominant proteins in E. chaffeensis and Ehrlichia canis based on a bioinformatics approach [53]. In another study, Chapes et al. designed peptides based on T cell epitopes in the host response of mice to E. chaffeensis. They showed that the most stimulating epitope was conserved between the outer membrane proteins p28-OMP-14 and p28-OMP-19 [54]. Pathogens induce the arming of the immune system and generate an efficient immune response. Interestingly, synthetic peptides or epitopes can elicit immune responses due to their versatility and high specificity [55]. Moreover, the pathogenesis of pathogenic organisms is not dependent on a specific virulence factor, and usually, vaccination with a single antigen cannot sufficiently stimulate a protective immune response [56]. Therefore, the prediction of appropriate antigenic epitopes of target proteins may be a ray of hope in the development of new vaccines and could improve the efficacy of further studies. Through various analyses, four proteins were introduced as promising putative targets for further in vitro and in vivo vaccine development. Finally, we developed a multi-epitope vaccine from selected short-listed proteins.
Among the selected vaccine candidates, WP_044170604.1, a hypothetical protein, had the highest score in terms of the number of linear and conformational B cell epitopes and MHC II binding sites. Therefore, this protein could be a potential candidate for stimulating an immune response. All shortlisted proteins, except for WP_044170604.1 (instability index: 40.72), had an instability index < 40, indicating the desired candidate stability. Adhesin probability is considered a criterion for vaccine candidates because it plays an important role in virulence and host–pathogen interaction [57]. In our study, WP_011452702.1.1 and WP_044193405.1 had the highest adhesion probability and B cell rate among the shortlisted proteins.
WP_011452702.1 and WP_044170828.1 belonged to the porin superfamily. It has been demonstrated that classical porin proteins contain 16 beta-stranded barrels and function as passive diffusion channels [58]. Since the essential role of porins has been confirmed, they are ideal vaccine candidates for Gram-negative bacteria, including intracellular bacteria [59]. In 2008, Kumagai et al. demonstrated the porin activity of the outer membrane proteins P28/OMP-19 and OMP-1F/OMP-18 of E. chaffeensis and presented them as suitable vaccine candidates [60]. Another putative protein, WP_006010497.1, is an OmpH-like protein characterized as a molecular chaperone. This protein, with a molecular weight of 18 kDa, is encoded by a gene homologue to the ompH genes of some Gram-negative and -positive bacteria. Dumetz et al. and Luo et al. introduced OmpH as a candidate vaccine against Flavobacterium psychrophilum [61] and Pasteurella multocida [62], respectively.
The enzymatic protein WP_006010191.1 from the M23 metallopeptidase family (pfam01551) is another potential vaccine candidate against E. chaffeensis. In general, the zinc-dependent M23 metallopeptidase is classified in the M23A and M23B families and is found in some Gram-negative and -positive bacteria. LasA from P. aeruginosa, which belongs to the M23A family proteins, and LytM/Lysostaphin from S. aureus, which belongs to the M23B family, are the best-studied bacteriocins as M23 metallopeptidase. The M23B family protein has glycyl-glycine endopeptidase activity and acts as an autolysin for peptidoglycan [63]. However, this family may also include some bacterial lipoproteins that lack proteolytic activity. In 2006, Nathan et al. demonstrated that peptidase M23B from Burkholderia pseudomallei is probably not a peptidase enzyme but rather an immunogenic lipoprotein that could be considered a vaccine candidate [64]. Finally, the results of this study can be helpful development of new effective drug and vaccines against E. chaffeensis for a better prevention and treatment management of this tick-borne disease.
Conclusion
In the current study, subtractive genomic strategy and reverse vaccinology approach were carried out to predict potential drug targets and vaccine candidates. We finally shortlisted six promising drug target proteins (WP_011452818.1, WP_011452723.1, WP_006010413.1, WP_006010278.1, WP_011452938.1, and WP_006010644.1) and four promising vaccine candidates (WP_011452702.1, WP_044193405.1, WP_006010191.1, and WP_044170604.1). The predicted targets were evaluated by different analyses and immune databases, which facilitate the development of new preventive and therapeutic approaches against E. chaffeensis at a lower cost and time. However, further in vitro and in vivo analyses remain to confirm the safety and efficacy of these proteins.
Data Availability
The datasets analyzed during the current study are available in the NCBI repository (https://www.ncbi.nlm.nih.gov/genome/browse/).
Abbreviations
- HME:
-
Human monocytotropic ehrlichiosis
- TBP:
-
Tick-borne pathogen
- IEDB:
-
Immune Epitope Database
- OMP:
-
Outer membrane protein
- DEG:
-
Database of Essential Genes
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PDB:
-
Protein Data Bank
- CDD:
-
Conserved Domain Database
- MHC:
-
Major Histocompatibility Complex
- TLR:
-
Toll-like receptors
- IL:
-
Interleukin
References
Thirumalapura, N.R. and Walker, D.H. (2015). Ehrlichia. In Molecular medical microbiology (pp. 2011–2032). Academic Press.
Pandey, R., et al. (2013). Ehrlichiosis presenting with toxic shock-like syndrome and secondary hemophagocytic lymphohistiocytosis. The Journal of the Arkansas Medical Society, 109(13), 280–282.
Heitman, K. N., et al. (2016). Increasing incidence of ehrlichiosis in the United States: A summary of national surveillance of Ehrlichia chaffeensis and Ehrlichia ewingii infections in the United States, 2008–2012. The American Journal of Tropical Medicine and Hygiene, 94(1), 52.
Varela-Stokes, A. (2007). Transmission of Ehrlichia chaffeensis from lone star ticks (Amblyomma americanum) to white-tailed deer (Odocoileus virginianus). Journal of wildlife diseases, 43(3), 376–381.
Walker, D.H. (2018) Chlamydial, mycoplasmal, rickettsial, and ehrlichial diseases. In Pulmonary Pathology: A Volume in the Series: Foundations in Diagnostic Pathology, pp. 315–326, Elsevier Inc.
Zhang, Z., & Ren, Q. (2015). Why are essential genes essential?—The essentiality of Saccharomyces genes. Microbial Cell, 2(8), 280.
Zhang, X. et al. (2021) In silico methods for identification of potential therapeutic targets. Interdisciplinary Sciences: Computational Life Sciences, 1–26.
Mody, V., et al. (2021). Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Communications biology, 4(1), 1–10.
Kumar, V. et al. (2021) Reverse vaccinology approach towards the in-silico multiepitope vaccine development against SARS-CoV-2. F1000Research, 10.
Bibi, S., et al. (2021). In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Scientific reports, 11(1), 1–16.
Hosen, M., et al. (2014). Application of a subtractive genomics approach for in silico identification and characterization of novel drug targets in Mycobacterium tuberculosis F11. Interdisciplinary Sciences: Computational Life Sciences, 6(1), 48–56.
Ghosh, P., et al. (2021). A novel multi-epitopic peptide vaccine candidate against Helicobacter pylori: In-silico identification, design, cloning and validation through molecular dynamics. International journal of peptide research and therapeutics, 27(2), 1149–1166.
Khan, K., et al. (2022). An integrated in silico based subtractive genomics and reverse vaccinology approach for the identification of novel vaccine candidate and chimeric vaccine against XDR Salmonella typhi H58. Genomics, 114(2), 110301.
McBride, J. W., & Walker, D. H. (2010). Progress and obstacles in vaccine development for the ehrlichioses. Expert review of vaccines, 9(9), 1071–1082.
Rikihisa, Y. (2010). Anaplasma phagocytophilum and Ehrlichia chaffeensis: Subversive manipulators of host cells. Nature Reviews Microbiology, 8(5), 328–339.
Budachetri, K., et al. (2020). An entry-triggering protein of Ehrlichia is a new vaccine candidate against tick-borne human monocytic ehrlichiosis. MBio, 11(4), e00895-e920.
McGill, J. L., et al. (2016). Vaccination with an attenuated mutant of Ehrlichia chaffeensis induces pathogen-specific CD4+ T cell immunity and protection from tick-transmitted wild-type challenge in the canine host. PLoS ONE, 11(2), e0148229.
Chaudhari, N. M., et al. (2016). BPGA—An ultra-fast pan-genome analysis pipeline. Scientific reports, 6(1), 1–10.
Yu, N. Y., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics, 26(13), 1608–1615.
Bhagwat, M., & Aravind, L. (2007). Psi-blast tutorial. In Comparative genomics (pp. 177–186). Humana Press.
Kanehisa, M., & Goto, S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic acids research, 28(1), 27–30.
Wishart D.S. et al. (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic acids research, 34 (suppl_1), D668–D672.
Luo, H., et al. (2021). DEG 15, an update of the Database of Essential Genes that includes built-in analysis tools. Nucleic Acids Research, 49(D1), D677–D686.
Szklarczyk, D., et al. (2019). STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic acids research, 47(D1), D607–D613.
Emanuelsson, O., et al. (2007). Locating proteins in the cell using TargetP. SignalP and related tools. Nature protocols, 2(4), 953–971.
Doytchinova, I. A., & Flower, D. R. (2007). VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics, 8(1), 1–7.
Sharma, N., et al. (2021). AlgPred 2.0: An improved method for predicting allergenic proteins and mapping of IgE epitopes. Briefings in Bioinformatics, 22(4), bbaa294.
Jespersen, M. C., et al. (2017). BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic acids research, 45(W1), W24–W29.
Astle W.J. et al. (2016) The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 167 (5), 1415–1429. e19.
Duvaud, S., et al. (2021). Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Research, 49(W1), W216–W227.
He, Y. et al. (2010) Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. Journal of Biomedicine and Biotechnology 2010.
Kondabala, R. and Kumar, V. (2019) Computational intelligence tools for protein modeling. In Harmony Search and Nature Inspired Optimization Algorithms, pp. 949–956.
Ponomarenko, J., et al. (2008). ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics, 9(1), 1–8.
Yazdani, Z., et al. (2020). Design an efficient multi-epitope peptide vaccine candidate against SARS-CoV-2: An in silico analysis. Infection and drug resistance, 13, 3007.
Fleri, W., et al. (2017). The Immune Epitope Database and analysis resource in epitope discovery and synthetic vaccine design. Frontiers in immunology, 8, 278.
Marchler-Bauer, A., et al. (2015). CDD: NCBI’s Conserved Domain Database. Nucleic acids research, 43(D1), D222–D226.
Renauld-Mongénie, G., et al. (2004). Transferrin-binding protein B of Neisseria meningitidis: Sequence-based identification of the transferrin-binding site confirmed by site-directed mutagenesis. Journal of Bacteriology, 186(3), 850–857.
Jiménez-García, B., et al. (2013). pyDockWEB: A web server for rigid-body protein–protein docking using electrostatics and desolvation scoring. Bioinformatics, 29(13), 1698–1699.
Rapin, N., et al. (2011). Immune system simulation online. Bioinformatics, 27(14), 2013–2014.
Nair, A. D., et al. (2015). Attenuated mutants of Ehrlichia chaffeensis induce protection against wild-type infection challenge in the reservoir host and in an incidental host. Infection and immunity, 83(7), 2827–2835.
Crocquet-Valdes, P. A., et al. (2011). Immunization with Ehrlichia P28 outer membrane proteins confers protection in a mouse model of ehrlichiosis. Clinical and Vaccine Immunology, 18(12), 2018–2025.
Yan, F., & Gao, F. (2020). A systematic strategy for the investigation of vaccines and drugs targeting bacteria. Computational and Structural Biotechnology Journal, 18, 1525–1538.
Maurya S. et al. (2020) Subtractive proteomics for identification of drug targets in bacterial pathogens: A review. International Journal of Engineering Research & Technology 9.
Solanki, V., & Tiwari, V. (2018). Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Scientific reports, 8(1), 1–19.
Shahid, F., et al. (2020). In silico subtractive proteomics approach for identification of potential drug targets in Staphylococcus saprophyticus. International Journal of Environmental Research and Public Health, 17(10), 3644.
Gupta, E., et al. (2020). Identification of drug and vaccine target in Mycobacterium leprae: A reverse vaccinology approach. International Journal of Peptide Research and Therapeutics, 26(3), 1313–1326.
Ali, A., et al. (2020). Modeling novel putative drugs and vaccine candidates against tick-borne pathogens: A subtractive proteomics approach. Veterinary Sciences, 7(3), 129.
Mondal, S. I., et al. (2015). Identification of potential drug targets by subtractive genome analysis of Escherichia coli O157:H7: An in silico approach. Advances and applications in bioinformatics and chemistry: AABC, 8, 49.
Sosa, E. J., et al. (2018). Target-Pathogen: A structural bioinformatic approach to prioritize drug targets in pathogens. Nucleic acids research, 46(D1), D413–D418.
Palmieri, F. et al. (2022) Mitochondrial transport and metabolism of the vitamin B‐derived cofactors thiamine pyrophosphate, coenzyme A, FAD and NAD+, and related diseases: A review. IUBMB life.
Barra, A. L. C., et al. (2020). Essential metabolic routes as a way to ESKAPE from antibiotic resistance. Frontiers in Public Health, 8, 26.
Tang, S.-N., & Huang, J.-F. (2005). Evolution of different oligomeric glycyl-tRNA synthetases. FEBS letters, 579(6), 1441–1445.
Luo, T., et al. (2020). Ehrlichia chaffeensis and E. canis hypothetical protein immunoanalysis reveals small secreted immunodominant proteins and conformation-dependent antibody epitopes. NPJ vaccines, 5(1), 1–12.
Chapes, S.K. et al. (2016) Identification of T-Cell Epitopes in the Murine Host Response to Ehrlichia chaffeensis. In Rickettsiales (pp. 197–214). Springer, Cham.
Parvizpour, S., et al. (2020). Epitope-based vaccine design: A comprehensive overview of bioinformatics approaches. Drug Discovery Today, 25(6), 1034–1042.
Skwarczynski, M., & Toth, I. (2016). Peptide-based synthetic vaccines. Chemical science, 7(2), 842–854.
Casadevall, A., & Pirofski, L. A. (2001). Host-pathogen interactions: The attributes of virulence. The Journal of infectious diseases, 184(3), 337–344.
Marchler-Bauer, A., et al. (2017). CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic acids research, 45(D1), D200–D203.
Pal, S., et al. (2005). Vaccination with the Chlamydia trachomatis major outer membrane protein can elicit an immune response as protective as that resulting from inoculation with live bacteria. Infection and immunity, 73(12), 8153–8160.
Kumagai, Y., et al. (2008). Expression and porin activity of P28 and OMP-1F during intracellular Ehrlichia chaffeensis development. Journal of bacteriology, 190(10), 3597–3605.
Dumetz, F., et al. (2006). A protective immune response is generated in rainbow trout by an OmpH-like surface antigen (P18) of Flavobacterium psychrophilum. Applied and environmental microbiology, 72(7), 4845–4852.
Luo, Y., et al. (1999). Sequence analysis of Pasteurella multocida major outer membrane protein (OmpH) and application of synthetic peptides in vaccination of chickens against homologous strain challenge. Vaccine, 17(7–8), 821–831.
Grabowska, M., et al. (2015). High resolution structure of an M23 peptidase with a substrate analogue. Scientific reports, 5(1), 1–8.
Nathan, S., et al. (2006). Cloning and expression of a Burkholderia pseudomallei putative peptidase M23B. Malay. J. Biochem. Mol. Biol, 14, 33–37.
Acknowledgements
The authors of this article will thank the personnel of the Pasteur Institute of Iran.
Author information
Authors and Affiliations
Contributions
S. S. and S. S. H. collected the data; S. S., S. S. H., N. N. G., A. M., F. H. J., M. H., and N. B. wrote the manuscript and analyzed the data; F. B. participated in all steps, supervised the project and wrote and revised the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This article does not contain any studies on human or animals.
Consent for Publication
This article does not contain any individual person’s data in any form.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sabzi, S., Shahbazi, S., Noori Goodarzi, N. et al. Genome-Wide Subtraction Analysis and Reverse Vaccinology to Detect Novel Drug Targets and Potential Vaccine Candidates Against Ehrlichia chaffeensis. Appl Biochem Biotechnol 195, 107–124 (2023). https://doi.org/10.1007/s12010-022-04116-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-022-04116-y