
Opinion statement
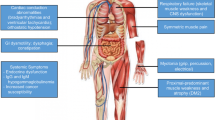
Myotonic dystrophy (DM1) is the most common form of adult muscular dystrophy. It is a multisystem disorder with a complex pathophysiology. Although inheritance is autosomal dominant, disease variability is attributed to anticipation, a maternal expansion bias, variable penetrance, somatic mosaicism, and a multitude of aberrant pre-mRNA splicing events. Patient presentations range from asymptomatic or mild late onset adult to severe congenital forms. Multiple organ systems may be affected. Patients may experience early cataracts, myotonia, muscle weakness/atrophy, fatigue, excessive daytime sleepiness, central/obstructive apnea, respiratory failure, cardiac arrhythmia, insulin resistance, dysphagia, GI dysmotility, cognitive impairment, Cluster C personality traits, and/or mood disorders. At present, there is no curative or disease-modifying treatment, although clinical treatment trials have become more promising. Management focuses on genetic counseling, preserving function and independence, preventing cardiopulmonary complications, and symptomatic treatment (e.g., pain, myotonia, hypersomnolence, etc.). Currently, there is an increasing international consensus on monitoring and treatment options for these patients which necessitates a multidisciplinary team to provide comprehensive, coordinated clinical care.
Similar content being viewed by others


References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11(10):891–905. Thorough review of the advanced state of understanding of myotonic dystrophies.
Magee AC, Hughes AE. Segregation distortion and myotonic dystrophy. J Med Genet. 1998;35(12):1045–6.
••Redman JB, Fenwick Jr RG, Fu YH, Pizzuti A, Caskey CT. Relationship between parental trinucleotide GCT repeat length and severity of myotonic dystrophy in offspring. JAMA. 1993;269(15):1960–5. Important historical reference supporting anticipation and maternal expansion bias in regards to offspring disease severity.
Pearson CE, Nichol Edmara K, Clearly JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6(10):729–42.
Morales F, Couto JM, Higham CF, Hogg G, Cuenca P, Braida C, et al. Somatic instability of the expanded CTG triple repeat in myotonic dystrophy type I is a heritable quantitative trait and modifier of disease severity. Hum Mol Genet. 2012;21(16):3558–67.
•Wong JL, Ashizawa T, Monckton DG, Caskey CT, Richards CS. Somatic heterogeneity of the CT G repeat of myotonic dystrophy is age and size dependent. Am Hum Genet. 1995;56(1):114–22. Historical paper supporting somatic instability (heterogeneity) with age (i.e. somatic mosaicism).
•Clearly JD, Tome S, Lopez Castel A, Panigrahi GB, Foiry L, Hagerman KA, et al. Tissue- and age-specific DNA replication patterns at the CTG/CAG-expanded human myotonic dystrophy type I locus. Nat Struct Mol Biol. 2010;17(9):1079–87. Historical paper supporting somatic instability (heterogeneity) between tissues and across ages in DM1 patients and transgenic mice.
•Turner C, Hilton-Jone D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. 2010;81:358–67. Important review article discussing genotype-phenotype correlations, non-genetic testing, and symptom management.
••Meola G. Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myol. 2013;32(3):154–65. This review paper precedes a more recent update [61] comparing clinical features, diagnostics, genetics, histopathology, and molecular mechanisms in DM1 and DM2.
••Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetics, pathology, and molecular pathomechanisms. Biochim Biophys Acta. 1852;2015:594–606. This review article is a supplement to a previous article [21] and provides up to date information on genotype-phenotype features, diagnosis, management, and recent advances in understanding the molecular mechanism(s), including animal models, in DM1.
Tielman AA, Knoop H, van de Logt AE, Bleijenberg G, van Engelen BG, Overeem S. Poor sleep quality and fatigue but no excessive daytime sleepiness and myotonic dystrophy type 2. J Neurol Neurosurg Psychiatry. 2010;81(9):963–7.
••Heatwole C, Bode R, Johnson N, Quinn C, Martens W, McDermott MP, et al. Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1). Neurology. 2012;79(4):348–57. Cross-sectional study assessing the prevalence and significance of 221 critical DM1 symptoms and 14 disease themes. Fatigue and limitations in mobility had the greatest effect on quality of life. Hand/arm problems, fatigue, myotonia, and impaired sleep/EDS were highly prevalent symptomatic themes in DM 1 patients.
•Thorton CA. Myotonic dystrophy. Neurol Clin. 2014;32(3):705–19. A recent review article on the epidemiology, genetics, clinical features, diagnosis, pathogenesis, and management of DM1.
•Mathiew J, Allard P, Potvin L, Prevost C, Begin P. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology. 1999;52(8):1658–62. Longitudinal study looking at causes of death in myotonic dystrophy patients.
Win AK, Perattur PG, Pulido JS, Pulido CM, Lindor NM. Increased cancer risks of myotonic dystrophy. Mayo Clin Proc. 2012;87(2):130–5.
Bianchi ML, Leoncini E, Masciullo M, Modoni A, Gadalla SM, Massa R, et al. Increase risk of tumor in DM1 is not related to exposure to common lifestyle risk factors. J Neurol. 2016;263(3):492–8.
Meola G, Sansone V, Perani. Executive dysfunction is important personality trait in myotonic dystrophy type I (DM-1) in proximal myotonic myopathy (PROMM/DM-2). Neuromuscul Disord. 2003;13(10):813–21.
Ekström AB, Hakenäs-Plate L, Samuelsson L, Tulinius M, Wentz E. Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(6):918–26.
Goossens E, Steyaert J, De Die-Smulders C, Willekens D, Fryns JP. Emotional and behavioral profile and child psychiatric diagnosis in the childhood type of myotonic dystrophy. Genet Couns. 2000;11(4):317–27.
•Douniol M, Jacquette A, Guilé JM, Tanguy ML, Angeard N, Héron D, et al. Psychiatric and cognitive phenotype in children and adolescents with myotonic dystrophy. Eur Child Adolesc Psychiatry. 2009;18(12):705–15. A literature review of cognitive and psychiatric dysfunction associated with congenital and child onset DM1.
Peric S, Stojanivic VR, Nikilic A, Kacar A, Basta I, Pavlovic S, et al. Peripheral neuropathy in patients with myotonic dystrophy type 1. Neurol Res. 2013;35(4):331–5.
••Kamsteeg EJ, Kress W, Catalli C, Hertz JM, Witsch-Baumgartner M, Buckley MF, et al. Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum Genet. 2012;20(12):1203–8. This provides clinical molecular genetic analysis and reporting guidelines in DM1, including both prenatal and presymatomatic testing.
••Tsilfidis C, MacKenzie AE, Mettler G, Barceló J, Korneluk RG. Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat Genet. 1992;1(3):192–5. Historical reference providing evidence of anticipation and maternal expansion bias in DM1.
Esposito G, Ruggiero R, Savarese M, Savarese G, Tremolaterra MR, Salvatore F, et al. Prenatal molecular diagnosis of inherited neuromuscular diseases: Duchenne/Becker muscular dystrophy, myotonic dystrophy type 1 and spinal muscular atrophy. Clin Chem Lab Med. 2013;51(12):2239–45.
Hopkins AN, Alshaeri T, Akst SA, Berger JS. Neurologic disease with pregnancy and considerations for the obstetric anesthesiologist. Semin Perinatol. 2014;38(6):359–69.
••Awater C, Zerres K, Rudnik-Schöneborn S. Pregnancy course and outcome in women with hereditary neuromuscular disorders: comparison of obstetric risks in 178 patients. Eur J Obstet Gynecol Reprod Biol. 2012;162(2):153–9. Important study for obstetrical care and genetic counseling of DM1 women considering pregnancy. The paper discusses pregnancy outcomes and complications in various neuromuscular disease including DM1 and DM2.
••Campbell N, Brandom B, Day JW, Moxley R. Practical suggestions for the anesthetic management of a myotonic dystrophy patient. Myotonic dystrophy foundation. MDFToolkit. 2013;23:794–803. This paper is endorsed by the Myotonic Dystrophy Foundation and provides recommendations for anesthesia management for DM1 patients undergoing surgery.
•Veyckemans F, Scholtes JL. Myotonic dystrophies type 1 and 2: anesthetic care. Paediatr Anaesth. 2013;23(9):794–803. Review article discussing genetics, clinical presentation, potential peri-operative complications and anesthesia care in myotonic disorders.
••Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, et al. Expanded CUG repeats trigger aberrant splicing of CIC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10(1):35–44. Historical reference of DM1 spliceopathy leading to a chloride channelopathy contributing to membrane hyperexcitability and myotonia.
Kimura T, Takahashi MP, Okuda Y, Kaido M, Fujimura H, Yanagihara T, et al. Expression of ion channel mRNAs in skeletal muscles from patients with myotonic muscular dystrophy. Neurosci Lett. 2000;295(3):93–6.
Favero M, Jiang DJ, Chiamulera C, Cangiano A, Fumagalli GF. Expression of small-conductance calcium-activated potassium channels (SK3) in skeletal muscle: regulation by muscle activity. J Physiol. 2008;586(19):4763–74.
Kimura T, Takahashi MP, Fujimura H, Sakoda S. Expression and distribution of a small-conductance calcium-activated potassium channel (SK3) protein in skeletal muscles from myotonic muscular dystrophy patients and congenital myotonic mice. Neurosci Lett. 2003;347(3):191–5.
•Renaud JF, Desnuelle C, Schmid-Antomarchi H, Huhues M, Serratrice G, Lazdunski M. Expression of apamin receptor in muscles of patients with myotonic muscular dystrophy. Nature. 1986;319(6055):678–80. Historical article showing the presence of SK3 channels, normally present in developing or denervated myofibers, in DM1 and suggesting a role for SK3 in myotonia.
•Behrens MI, Vergara C. Increase of apamin receptors in skeletal muscle induced by colchicine: possible role in myotonia. Am J Physiol. 1992;263:C794–802. Historical paper supporting a role for SK3 channels in myotonia.
Behrens MI, Jalil P, Serani A, Vergara F, Alvarez O. Possible role of apamin-sensitive K+ channels in myotonic dystrophy. Muscle Nerve. 1994;17(11):1264–70.
••Logigian EL, Martens WB, Moxley IV RT, McDermott MP, Dilek N, Wiegner AW, et al. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. 2010;74(18):1441–8. Clinical trial supporting the use of mexilitine for myotonia.
••Mathieu J, Boivin H, Meunier D, Gaudreault M, Bégin P. Assessment of a disease-specific muscular impairment rating scale in myotonic dystrophy. Neurology. 2001;56(3):336–40. This paper validates and documents the intra/interrater reliability of the Muscular Impairment Rating Scale (MIRS) in assessing DM1 patients.
Voet NB, van der Kooi EL, Riphagen II, Lindeman E, van Engelen BG, Geurts AC. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst Rev. 2013; CD003907.
Mohanty S, Mohanty P, Tamaki M, Natale V, Gianni C, Trivedi C, et al. Differential association of exercise intensity with risk of atrial fibrillation in men and women: evidence from a meta-analysis. J Cardiovasc Electrophysiol. 2016. doi:10.1111/jce.13023.
Turagam MK, Velagapudi P, Alpert MA. Does exercise cause atrial fibrillation? Int J Cardiol. 2015;181:245–6.
Ofman P, Khawaja O, Rahilly-Tierney CR, Peralta A, Hoffmeister P, Reynolds MR, et al. Regular physical activity and risk of atrial fibrillation: a systematic review and meta-analysis. Circ Arrhythm Electrophysiol. 2013;6(2):252–6.
••Puymirat J, Bouchard JP, Mathieu J. Efficacy and tolerability of a 20 mg dose of methylphenidate for the treatment of daytime sleepiness in adult patients with myotonic dystrophy type 1: a 2 center, randominzed, double-blind, placebo-controlled, 3-week crossover trial. Clin Ther. 2012;34(5):1103–11. Clinical trial on the efficacy and tolerability of methylphenidate for EDS in DM1.
Lazarus A, Varin J, Ounnoughene Z, Radvanyi H, Junuen C, Coste J, et al. Relationships among electyrophysiological findings and clinical status, heart function, and extent of DNA mutation in Myotonic Dystrophy. Circulation. 1999;99(8):1041–6.
••Lau JK, Sy RW, Corbett A, Kritharides L. Myotonic dystrophy and the heart: a systematic review of evaluation and management. Int J Cardiol. 2015;184:600–8. This review discusses up to date guidelines regarding cardiac monitoring and management in DM1 patients. It also discusses the pathophysiology of cardiac problems in DM1.
•Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358(25):2688–97. This paper shows that severe ECG abnormalities and a clinical diagnosis of atrial tachyarrythmia are independent risk factors for sudden death in DM1 patients.
Verhart D, Richards K, Rafaael-Fortney JA, Raman SV. Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotype considerations. Circ Cardiovasc Imaging. 2011;4(1):67–76.
••Epstein AE, DiMarco JP, Ellenbogen KA, Estes 3rd NA, Freedman RA, Gettes LS, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2013;61(3):e6–75. This paper provides the American Heart Association guidelines for pacer and/or defibrillator placement in DM1 patients.
•Savkur RS, Philips AV, Copper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29(1):40–7. Historical reference of DM1 spliceopathy supporting the theory of “RNA toxic or deleterious gain of function” independent of DMPK.
Vujnic M, Peric S, Popovic S, Raseta N, Ralic V, Dobricic V, et al. Metabolic syndrome in patients with myotonic dystrophy type 1. Muscle Nerve. 2015;52(2):273–7.
••Sansone V, Gandossini S, Cotelli M, Calabria M, Zanetti O, Meola G. Cognitive impairment in adult myotonic dystrophies: a longitudinal study. Neurol Sci. 2007;28(1):9–15. Important longitudinal study revealing the progressive nature of frontal cognitive impairment (attentional) in adult DM1 and DM2.
Meola G, Sansone V, Perani D, et al. Reduce cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology. 1999;53:1042–50.
•Conforti R, de Cristofaro M, Cristofano A, Brogna B, Sardaro A, Tedeschi G, et al. Brain MRI abnormalities in the adult form of myotonic dystrophy type 1: a longitudinal case series study. Neuroradiol J. 2016;29(1):36–45. Longitudinal study using MRI showing a variable rate of progressive white matter and brain atrophy in DM1 patients regardless of age, CTG repeat number, or muscular involvement.
•Wozniak JR, Mueller BA, Lim KO, Hemmy LS, Day JW. Tractography reveals diffuse white matter abnormalities in myotonic dystrophy type 1. J Neurol Sci. 2014;341(1–2):73–8. This study uses Diffusion Tensor Imaging (DTI) tractography to characterize white matter disturbances in adult DM1 patients. DTI metrics were correlated with CTG repeat length, cognitive function, the Muscular Impairment Rating Scale (MIRS), and the Epworth Sleepiness Scale.
Zanigni S, Evangelisti S, Giannoccaro MP, Oppi F, Poda R, Giorgio A, et al. Relationship of white and gray matter abnormalities to clinical and genetic features in myotonic dystrophy type 1. Neuroimage Clin. 2016;11:678–85.
Gallais B, Montreuil M, Gargiulo M, Eymard B, Gagnon C, Laberge L. Prevalence and correlates of apathy and myotonic dystrophy type 1. BMC Neurol. 2015;15:148–16.
•Heatwole CR, Eichinger KJ, Friedman DI, Hilbert JE, Jackson CE, Logigian EL, et al. Open-label trial of recombinant human insulin-like growth factor 1/recombinant human insulin-like growth factor binding protein 3 in myotonic dystrophy type 1. Arch Neurol. 2011;68(1):37–44. Clinical study with promising results, leading to a randomized trial for this treatment.
Tsuji K, Furutama D, Tagami M, Ohsawa N. Specific binding and effects of dehydroepiandrosterone sulfate (DHEA-S) on skeletal muscle cells: possible implication for DHEA-S replacement therapy in patients with myotonic dystrophy. Life Sci. 1999;65(1):17–26.
Sugino M, Ohsawa N, Ito T, Ishida S, Yamasaki H, Kimura F, et al. A pilot study of dehydroepiandrosterone sulfate in myotonic dystrophy. Neurology. 1998;51(2):586–9.
Thornton C, Mankodi A, Barbieri C, et al. Pilot trial of oral dehydroepiandrosterone sulfate (DHEAS) in myotonic dystrophy (DM). Neurology. 2001;56 suppl 3:A80–1.
Pénisson-Besnier I, Devillers M, Porcher R, Orlikowski D, Doppler V, Desnuelle C, et al. Dehydroepiandrosterone for myotonic dystrophy type 1. Neurology. 2008;71(6):407–12.
•Nakamori M, Taylor K, Mochizuki H, Sobczak K, Takahashi MP. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann Clin Transl Neurol. 2015;3(1):42–54. Important reference on erythromycin’s ability to rescue aberrant RNA splicing (reducing RNA toxicity) in DM1 fibroblasts and transgenic mice. This study identifies erythromycin as a potential therapy for DM1.
Chau A, Kalsotra A. Developmental insights into the pathology of and therapeutic strategies for DM1: back to the basics. Dev Dyn. 2015;244(3):377–90.
•Fardaei M, Larkin K, Brook JD. In vivo colocalization of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 2001;29(13):2766–71. Historical paper showing MBNL co-localization with DMPK expanded repeats which becomes sequestered within the nucleus. This early work lends to the theory of “RNA toxic gain or loss of function.
Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, et al. Three proteins, MBLN, MBLL, and MBXL, co-localized in vivo with nuclear foci of expanded repeat transcripts in DM1 in DM2 cells. Hum Mol Genet. 2002;11(7):805–14.
Krahe R, Ashizawa T, Abbruzzese C, Roeder E, Carango P, Giacanelli M, et al. Effect of myotonic dystrophy trinucleotide repeat expansion on DMPK transcription and processing. Genomics. 1995;28(1):1–14.
Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol. 1995;128(6):995–1002.
••Phillips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by CUG binding protein in myotonic dystrophy. Science. 1998;280(5364):737–41. Historical reference supporting the inappropriate binding of proteins to CUG repeats resulting in a “RNA toxic gain of function.”.
•Buj-Bello A, Furling D, Tronchere H, Laporte J, Lerouge T, Butler-Browne GS, et al. Muscle-specific alterative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet. 2002;11(19):2297–307. This article and the next two explore changes in other genes that affect muscle, besides the DMPK gene.
Bondy-Chorney E, Crawford Parks TE, Ravel-Chapuis A, Klinck R, Rocheleau L, Pelchat M, et al. Staufen1 regulates multiple alternative splicing events either positively or negatively in DM1 indicating its role as a disease modifier. PLoS Genet. 2016;12(1):e10058278. eCollection.
Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17(6):720–5.
••Pandey SK, Wheeler TM, Justice SL, Kim A, Younis HS, Gattis D, et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J Pharmacol Exp Ther. 2015;355(2):329–40. Important description of translational work in animal models that has led to the first targeted clinical trial for DM1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neuromuscular Disorders
Rights and permissions
About this article

Cite this article
Smith, C.A., Gutmann, L. Myotonic Dystrophy Type 1 Management and Therapeutics. Curr Treat Options Neurol 18, 52 (2016). https://doi.org/10.1007/s11940-016-0434-1
Published:
DOI: https://doi.org/10.1007/s11940-016-0434-1