
Opinion statement
-
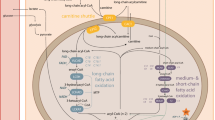
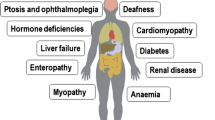
•Mitochondrial diseases are disorders of energy metabolism that include defects of pyruvate metabolism, Krebs cycle, respiratory chain (RC), and fatty acid oxidation (FAO).
-
•Treatment of pyruvate metabolism, Krebs cycle, and RC disorders is, in general, disappointing. Therapeutic approaches consist of electron acceptors, enzyme activators, vitamins, coenzymes, free-radical scavengers, dietary measures, and supportive therapy. These treatment assumptions are based on current understanding of the pathophysiology, on anecdotal clinical reports, and on a few controlled clinical trials, which have not been encouraging. Although it is difficult to perform clinical trials in these conditions due to their rarity and genotypic and phenotypic heterogeneity, there is a great need for well-performed double-blind placebocontrolled clinical trials with comparable groups of patients and with sufficient follow-up periods.
-
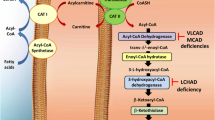
•Treatment options for FAO disorders are, in general, satisfactory and are mainly based on diet, lifestyle recommendations, and administration of L-carnitine and, in some cases, riboflavin.
-
•Special conditions that involve primary deficiencies of L-carnitine, coenzyme Q10, and cofactor- and vitamin-responsive enzyme defects must be systematically considered, because supplementation with these substances may be curative or produce dramatic improvements.
-
•While awaiting more specific therapies for mitochondrial disorders, it is useful to reach a consensus regarding the management of these patients. The expected outcome is a slowing of the disease process and stabilization of the clinical syndrome. More definitive treatments hopefully will follow in the near future.
Similar content being viewed by others
References
Powers JM, De Vivo DC: Peroxisomal and mitochondrial diseases. In: Greensfield’s Neuropathology, edn 7. London: Arnold; 2001.
DiMauro S, Bonilla E, De Vivo DC: Does the patient have a mitochondrial encephalomyopathy? J Child Neurol 1999, 14(suppl):S23-S35.
Sue C, Hirano M, DiMauro S, De Vivo DC: Neonatal presentations of mitochondrial metabolic disorders. Semin Perinatol 1999, 23:113–124.
Tein I: Neonatal metabolic myopathies. Semin Perinatol 1999, 23:125–151.
Kerrigan JF, Aleck KA, Tarby TJ, et al.: Fumaric aciduria: clinical and imaging features. Ann Neurol 2000, 47:583–588.
Morris AAM, Turnbull DM: Fatty acid oxidation defects in muscle. Curr Opinion Neurol 1998, 11:485–490.
Saudubray JM, Martin D, De Lonlay P, et al.: Recognition and management of fatty oxidation defects: a series of 107 patients. J Inher Metab Dis 1999, 22:498–502.
Walker UA, Byrne E: The therapy of respiratory chain encephalomyopathy: a critical review of the past and current perspective. Acta Neurol Scand 1995, 92:273–280.
Morris AAM, Leonard JV: The treatment of congenital lactic acidosis. J Inher Metab Dis 1996, 19:573–580.
Taylor RW, Chinnery PF, Clark KM, et al.: Treatment of mitochondrial disease. J Bioener Biomembr 1997, 29:195–205.
Esposito LA, Melov S, Panov A, et al.: Mitochondrial disease in mouse results in increased oxidative stress. Proc Natl Acad Sci USA 1999, 96:4820–4825.
Wallace DC: Mitochondrial diseases in man and mouse. Science 1999, 283:1482–1488. This is a comprehensive review of the pathophysiology of mitochondrial disorders and the animal models of mitochondrial diseases.
Peterson PL: The treatment of mitochondrial myopathies and encephalomyopathies. Biochim Biophys Acta 1995, 1271:275–280.
Shulze A, Mayatepeck E, Langhans CD, et al.: In vivo methods for therapy monitoring in lactic acidosis. J Inher Metab Dis 1998, 21:691–692.
Abe K, Matsuo Y, Kadekawa J, et al.: Effects of coenzyme Q10 in patients with mitochondrial myopathy encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): evaluation by noninvasive oximetry. J Neurol Sci 1999, 162:65–68.
Bresolin N, Doriguzzi C, Ponzetto C, et al.: Ubidecarenone in the treatment of mitochondrial myopathies: a multi-center double-blind trial. J Neurol Sci 1990, 100:70–78.
Bernsen PLJA, Gabreels FJM, Ruitenbeek W, Hamburger HL: Treatment of complex I deficiency with riboflavin. J Neurol Sci 1993, 118:181–187.
Campos Y, Huertas R, Lorenzo G, et al.: Plasma Lcarnitine insufficiency and effectiveness of l-Lcarnitine therapy in patients with mitochondrial myopathy. Muscle Nerve 1993, 16:150–153.
De Stefano N, Matthews PM, Ford B, et al.: Shortterm dichloroacetate treatment improves indices of cerebral metabolism in patients with mitochondrial disorders. Neurology 1995, 45:1193–1198.
Chen RS, Huang CC, Chu NS: Coenzyme Q10 treatment in mitochondrial encephalomyopathies. Eur Neurol 1997, 37:212–218.
Tanaka J, Nagai T, Arai H, et al.: Treatment of mitochondrial encephalomyopathy with combination of cytochrome C and vitamins B1 and B2. Brain Dev 1997, 19:262–267.
Artuch R, Vilaseca MA, Pineda M: Biochemical monitoring of the treatment in pediatric patients with mitochondrial disease. J Inher Metab Dis 1998, 21:837–845.
Chan A, Reichman H, Kogel A, et al.: Metabolic changes with mitochondrial myopathies and effects of coenzyme Q10 therapy. J Neurol 1998, 245:681–685.
Harding CO, Gillingham MB, Van Calcar S, et al.: Docosahexaneoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inher Metab Dis 1999, 22:276–280. Interesting report analyzing the physiopathologic implications of DHA deficiency in the pigmentary retinopathy of patients with LCHAD deficiency. In addition, the authors present preliminary data of an open trial with DHA supplementation in 8 children with LCHAD deficiency.
Kerr DS, Wexler ID, Zinn AB: Disorders of pyruvate metabolism and the tricarboxylic acid cycle. In Inborn Metabolic Diseases: Diagnosis and treatment, edn 3. Edited by Fernandez J, Saudubray JM, van den Berghe G. Heidelberg, Germany: Springer; 2000.
Wexler ID, Hemalatha SG, McConnell J, et al.: Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets: studies in patients with identical mutations. Neurology 1997, 49:1655–1661.
Stacpoole PW, Barnes CL, Hurbanis MD, et al.: Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child 1997, 77:535–541.
Wijburg FA, Barth PG, Bindoff LA, et al.: Leigh syndrome associated with a deficiency of the pyruvate dehydrogenase complex: results of treatment with a ketogenic diet. Neuropediatrics 1992, 23:147–152.
Craigen WJ: Leigh Disease with deficiency of lipoamide dehydrogenase: treatment failure with dichloroacetate. Pediatr Neurol 1996, 14:69–71.
Ahmand A, Kahler SG, Kishnani PS, et al.: Treatment of pyruvate carboxylase deficiency with high doses of citrate and aspartate. Am J Med Genet 1999, 87:331–338. This is the report of the treatment regimen in a patient with PC deficiency. The authors present a thorough analysis of the clinical and biochemical effects of citrate and aspartate supplementation and they review the treatment options of PC deficiencies reported in the literature.
Munich A: Defects of the respiratory chain, In Inborn Metabolic Diseases: Diagnosis and treatment, edn 3. Edited by Fernandez J, Saudubray JM, van den Berghe G. Heidelberg, Germany: Springer; 2000.
Stanley CA: Disorders of fatty acid oxidation. In Inborn Metabolic Diseases: Diagnosis and treatment, edn 3. Edited by Fernandez J, Saudubray JM, van den Berghe G. Heidelberg, Germany: Springer; 2000.
Gillingham M, Van Calcar S, Ney D, et al.: Dietary management of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD): a case report and survey. J Inher Metab Dis 1999, 22:123–131.
Tein I, Vajsar J, MacMillan L, Sherwood G: Long-chain l-3-hydroxyacyl-coenzyme A dehydrogenase deficiency neuropathy: response to cod oil. Neurology 1999, 52:640–643.
Duran M, Wadman K: Thiamine-responsive inborn errors of metabolism. J Inher Metab Dis 1986, 8(suppl 1):S70-S75.
Pastoris O, Savasta S, Foppa P, et al.: Pyruvate dehydrogenase deficiency in a child responsive to thiamine treatment. Acta Paediatr 1996, 85:625–628.
Naito E, Ito I, Yokota I, et al.: Thiamine-responsive lactic acidaemia: role of pyruvate dehydrogenase complex. Eur J Pediatr 1998, 157:648–652. In this report, the authors evaluate the biochemical basis of thiamine-responsive mitochondrial disorders.
Naito E, Ito M, Yokota I, et al.: Concomitant administration of sodium dichloroacetate and thiamine in West syndrome caused by thiamineresponsive pyruvate dehydrogenase complex deficiency. J Neurol Sci 1999, 171:56–59.
Sato Y, Nakagawa M, Higuchi I, et al.: Mitochondrial myopathy and familial thiamine deficiency. Muscle Nerve 2000, 23:1069–1075.
Vergani L, Barile M, Angelini C, et al.: Riboflavin therapy: biochemical heterogeneity in two adult lipid storage myopathies. Brain 1999, 122:2401–2411.
Uziel G, Garavaglia B, Ciceri E, Loroni I: Riboflavinresponsive glutaric aciduria type II presenting as a leukodystrophy. Pediatr Neurol 1995, 13:333–335.
Gregersen N, Christensen MF, Christensen E, Kolvraa S: Riboflavin responsive multiple acyl-CoA dehydrogenase deficiency. Acta Paediatr Scand 1986, 75:676–681.
Ogle RF, Christodoulou J, Fagan E, et al.: Mitochondrial myopathy with tRNA Leu (UUR) mutation and complex I deficiency responsive to riboflavin. J Pediatr 1997, 130:138–145.
Higgins JJ, Glasgow AM, Lusk M, Kerr DS: MRI, clinical, and biochemical features of partial pyruvate carboxylase deficiency. J Chil Neurol 1994, 9:436–439.
Byrd DJ, Krohn HP, Winkler L, et al.: Neonatal pyruvate dehydrogenase deficiency with lipoate responsive lactic acidaemia and hyperammonaemia. Eur J Pediatr 1989, 148:543–547.
Elpeleg ON, Ruitenbeek W, Jackobs C, et al.: Congenital lactacidemia caused by lipoamide dehydrogenase deficiency with favorable outcome. J Pediatr 1995, 126:72–74.
Takanashi J, Sugita K, Tanabe Y, Maemoto T: Dichloroacetate treatment in Leigh syndrome caused by mitochondrial DNA mutation. J Neurol Sci 1997, 145:83–86.
Saitoh S, Momoi MY, Yamagat T, et al.: Effects of dichloroacetate in three patients with MELAS. Neurology 1998, 50:531–534.
Morten KJ, Caky M, Matthews PM: Stabilization of the pyruvate dehydrogenase E1a subunit by dichloroacetate. Neurology 1998, 51:1331–1335. In this report, the authors evaluate the molecular basis of the effects of DCA in PDH-E1a deficiency.
Morten KJ, Beattie P, Brown GK, Matthews PM: Dichloroacetate stabilizes the mutant E1a subunit in pyruvate dehydrogenase deficiency. Neurology 1999, 53:612–616. In this report, the authors evaluate the molecular basis of the effects of DCA in PDH-E1a deficiency.
Taivassalo T, Matthews PM, DeStefano N, et al.: Combined aerobic training and dichloroacetate improve exercise capacity and indices of aerobic metabolism in muscle cytochrome oxidase deficiency. Neurology 1996, 47:529–534.
Kimura S, Ohtuki N, Nezu A, et al.: Clinical and radiologic improvements in mitochondrial encephalomyelopathy following sodium dichloroacetate therapy. Brain Dev 1997, 19:535–540.
Seneca S, De Meirleir L, De Schepper J, et al.: Pearson marrow pancreas syndrome: a molecular study and clinical management. Clin Genet 1997, 51:338–342.
Ernster L, Dallner G: Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta 1995, 1271:195–204.
Matthews RT, Yamg L, Browne S, et al.: Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci U S A 1998, 95:8892–8897.
Matsuoka T, Maeda H, Goto Y, Nonaka I: Muscle coenzyme Q10 in mitochondrial encephalomyopathies. Neuromuscul Disord 1991, 1:443–447.
Ogasahara S, Engel AG, Frens D, Mack D: Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci U S A 1989, 86:2379–2382.
Sobreira C, Hirano M, Shanske S, et al.: Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology 1997, 48:1238–1243.
Boitier E, Degoul F, Desguerre I, et al.: A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J Neurol Sci 1998, 156:41–46.
Rotig A, Appelkvist EL, Geromel V, et al.: Quinoneresponsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000, 356:391–396. This is the report of 2 siblings with primary CoQ10 deficiency with a dramatic improvement of multisystemic symptoms with CoQ10 supplementation.
Pons R, De Vivo DC: Primary and secondary L-carnitine deficiency syndromes. J Child Neurol 1995, 10(suppl):2S8–2S24.
Tein I, Donner EJ, Hale DE, Murphy EG: Clinical and neurophysiologic response of myopathy and neuropathy in long-chain l-3-hydroxyacyl-CoA dehydrogenase deficiency to oral prednisone. Pediatr Neurol 1995, 12:68–76.
Tarnapolsky MA, Roy BD, MacDonald JR: A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve 1997, 20:1502–1509.
Borchert A, Wilichowski E, Hanefeld F: Supplementation with creatine monohydrate in children with mitochondrial encephalomyopathies. Muscle Nerve 1999, 22:1299–1300.
Tarnapolsky MA, Garise G: Direct measurement of high-energy phosphate compounds in patients with neuromuscular disease. Muscle Nerve 1999, 22:1228–1233.
Carroll PV, Umpleby AM, Albany E, et al.: Growth hormone therapy may benefit protein metabolism in mitochondrial encephalomyopathy. Clin Endocrinology 1997, 47:113–117.
Tein I, DiMauro S, Xie Z-W, De Vivo DC: Valproic acid impairs L-carnitine uptake in cultured human skin fibroblasts. An in vitro model for the pathogenesis of valproic acid associated L-carnitine deficiency. Pediatr Res 1993, 34:281–287.
North K, Korson MS, Krawiecki N, et al.: Oxidative phosphorylation defect associated with primary adrenal insufficiency. J Pediatr 1996, 128:688–692.
Allen RJ, DiMauro S, Coulter DL, et al.: Kerans Sayre Syndrome with reduced plasma cerebrospinal fluid folate. Ann Neurol 1983, 13:679–682.
Thompson VA, Wahr JA: Anesthetic considerations in patients presenting with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome. Anesth Analg 1997, 85:1404–1406.
Taivassalo T, De Stefano N, Argov Z, et al.: Effects of aerobic training in patients with mitochondrial myopathies. Neurology 1998, 50:1055–1060.
Andrews RM, Griffiths PG, Chinnery PF, Turnbull DM: Evaluation of bupivacaine-induced muscle regeneration in the treatment of ptosis in patients with chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome. Eye 1999, 13:769–772.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Pons, R., De Vivo, D.C. Mitochondrial disease. Curr Treat Options Neurol 3, 271–288 (2001). https://doi.org/10.1007/s11940-001-0008-7
Issue Date:
DOI: https://doi.org/10.1007/s11940-001-0008-7