
Abstract
Erdheim–Chester disease (ECD) is a rare (approximately 500 known cases worldwide), non-inherited, non-Langerhans form of histiocytosis of unknown origin, first described in 1930. It is characterized by xanthomatous or xanthogranulomatous infiltration of tissues by foamy histiocytes, “lipid-laden” macrophages, or histiocytes, surrounded by fibrosis. Diagnosis of ECD involves the analysis of histiocytes in tissue biopsies: these are typically foamy and CD68+ CD1a− in ECD, whereas in Langerhans cell histiocytosis (LCH) they are CD68+ CD1a+. 99Technetium bone scintigraphy revealing nearly constant tracer uptake by the long bones is highly suggestive of ECD, and a “hairy kidney” appearance on abdominal CT scan is observed in approximately half of ECD cases. Central nervous system involvement is a strong prognostic factor and an independent predictor of death in cases of ECD. Optimum initial therapy for ECD seems to be administration of interferon α (or pegylated interferon α), and prolonged treatment significantly improves survival; however, tolerance may be poor. Cases of ECD present with strong systemic immune activation, involving IFNα, IL-1/IL1-RA, IL-6, IL-12, and MCP-1, consistent with the systemic immune Th-1-oriented disturbance associated with the disease. More than half of ECD patients carry the BRAF V600E mutation, an activating mutation of the proto-oncogene BRAF. A small number of patients harboring this mutation and with severe multisystemic and refractory ECD have been treated with vemurafenib, a BRAF inhibitor, which was proved very beneficial.
Similar content being viewed by others


Introduction
Erdheim–Chester disease (ECD) was first described as the “lipoid granulomatose” by Jakob Erdheim and his pupil William Chester in 1930 [1]. It is a non-Langerhans form of histiocytosis, of unknown origin, and is rare: up to January 2013, only 500 cases had been reported [2, 3, 4••]. ECD involves xanthomatous or xanthogranulomatous infiltration of tissues by foamy histiocytes, “lipid-laden” macrophages, or histiocytes, surrounded by fibrosis [5, 6]. ECD can be distinguished from Langerhans cell histiocytosis (LCH) by the immunohistological characteristics of histiocytes, which in ECD are: positive for CD68 and negative for CD1a, and negative for the S-100 protein in 80 % of cases.
ECD is a multisystemic disease: patients may present with skeletal involvement with bone pain, exophthalmos, diabetes insipidus, xanthelasma, interstitial lung disease, bilateral adrenal enlargement, retroperitoneal fibrosis with perirenal and/or ureteral obstruction, renal impairment, testis infiltration, and involvement of the central nervous system (CNS) and/or cardiovascular system [4••, 5].
The extent and distribution of the disease determine the clinical course. Some cases present with only asymptomatic bone lesions, and others with multisystemic, life-threatening forms. Here, we review 53 cases reported in 2011 (Table 1) [4••]. Our experience has kept increasing since then; we have now seen 101 patients who have attended our center since 1991, most of whom have been followed with regular assessments. Many of the patients live in France, although 21 live elsewhere, mainly Europe but also Israel and South Africa. Twenty-two of these patients have died (22 %).
Histiocytoses are heterogeneous diseases and in some cases, different types of histiocytoses are associated (overlap forms). The associated histiocytoses are most often ECD and LCH, but Rosai–Dorfman disease (RDD), a non-Langerhans form of histiocytosis which usually has a better outcome, may also be present [7–9]. Indeed, 15 % of the 101 patients attending our center up to August 2013 presented with such associations: ECD and LCH in 11 cases, ECD and RDD in two, and LCH, ECD, and RDD in one. The frequency of these overlap forms is too high to be likely to be coincidental, and implies a common cause of the different histiocytic disorders. Thus, patients with LCH should be tested for ECD if specific symptoms are present.
Diagnostic Criteria
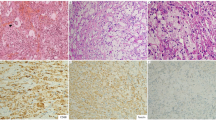
ECD is mostly diagnosed by histological analysis (Fig. 1): the typical pattern is polymorphic granuloma infiltrated with CD68-positive and CD1a-negative foamy histiocytes, and fibrosis or xanthogranulomatosis [5, 6]. These characteristic histiocytes may be found in almost any tissue in cases of ECD.

Erdheim–Chester disease, surgical biopsy of the femur: spongy bone tissue in which the medullary spaces are infiltrated by foamy histiocytes within a fibrous stroma (hematein-eosin-saffron, X200)
Uptake of tracer by the long bones during 99Technetium (Tc) bone scintigraphy (Fig. 2) is observed in almost all patients (96 % of our 53 patients) [10, 11]. Abdominal CT scanning reveals “hairy kidneys” (Fig. 3) in approximately half of patients with ECD [12, 13]. Because histology biopsy is required for the diagnosis of ECD, ultrasound-guided biopsy of the perirenal infiltration (observed in approximately two thirds of patients) is an approach to be recommended [14].

Symmetric and abnormally increased metaphyso-diaphyseal labeling of the long bones, predominantly in the lower limbs, on 99Technetium bone scintigraphy. (Reprinted from Haroche et al. [60]. Copyright 2013; with permission from Elsevier)

Contrast-enhanced axial CT scan showing the origin of the superior mesenteric artery, which is stenosed. Note the “coated aorta” aspect, and the symmetrical infiltration of the perirenal fat and of the perirenal fascia taking the appearance of “hairy kidneys”
The diagnostic criteria for ECD used in our center are:
-
1.
Characteristic histological findings (Fig. 1): foamy histiocyte infiltration of polymorphic granuloma and fibrosis or xanthogranulomatosis, with CD68-positive and CD1a-negative immunostaining;
-
2.
Characteristic skeletal abnormalities: a) bilateral and symmetric cortical osteosclerosis of the diaphyseal and metaphyseal parts of the long bones on X-rays, and/or b) symmetric and abnormally intense labeling of the distal ends of the long bones of the legs, and in some cases arms, as revealed by 99Tc bone scintigraphy.
We used these ECD criteria for our literature reviews in 1996, 2004, and 2011. All cases followed at our center fulfilled the first criterion. However, four ECD patients did not fulfill the second criterion, and did not present with long-bone involvement, which is otherwise characteristic of the disease. Bone scintigraphy, X-ray, MRI, and PET–CT investigations did not reveal pathological signs in these four cases.
Epidemiology
Most of the 101 patients followed at our center are men (75 % men and 25 % women), as has been the case throughout our experience of this disease [4••]. The mean age at diagnosis has not changed substantially as the number of patients seen by our center has grown, and was 55 yr ± 14 yr (range, 16–80 yr) in the 2011 series [4••, 5]. Only eight pediatric cases have ever been reported, and none had cardiac involvement [15, 16].
In our preliminary study in 2006, the delay before diagnosis was between a few months and several years (up to 25); in recent years, this delay has been reduced, probably because awareness of the disease has improved [11].
Clinical Manifestations (Table 1)
Osseous Involvement
Although skeletal involvement is extremely frequent (96 % of the 53 patients included in the 2011 series), only 50 % of patients suffer bone pain. Nevertheless, bone pain is the most common clinical feature of ECD [11]. This pain is often mild; it can start at any time during the course of the disease, and mostly affects the legs. X-ray evidence of bilateral and symmetric cortical osteosclerosis of the diaphyseal and metaphyseal regions of the long bones is typical in cases of ECD; also, symmetric and abnormally strong labeling of the distal ends of the long bones of the legs, and sometimes the arms, is often revealed by 99Tc bone scintigraphy [5, 6]. The axial skeleton and the mandible are often involved in LCH, but not in cases of ECD. Recently, positron-emission tomography (PET) with 18F-labeled fluorodeoxyglucose (PET–CT) has been used in place of bone CT scans [17, 18]. In some cases, MRI of the long bones can be informative, because it may reveal epiphyseal involvement of the long bones and periostitis, which is not detected by X-rays [19]. MRI may also be valuable in the rare cases of ECD for which anomalies are not detected by bone CT scans.
Cardiovascular Involvement
Progress in radiological imaging has made it more likely that cardiovascular involvement will be detected. The most frequent cardiovascular sign is the circumferential sheathing of the thoracic and/or abdominal aorta (66 % of cases in our series in 2011) [4••, 5]. Serratrice et al. described cases in which the whole aorta is sheathed as having a “coated aorta” (38 % of cases in our series reported in 2006 had this feature) [11, 20]. Periarterial infiltration may extend to the main aortic branches. The clinical consequences are not classically severe, apart from reno-vascular hypertension which is the most frequent (20 % of our 101 patients); this problem can be treated by renal artery stenting [5].
The most frequent heart lesion is pericardial (42 % of the 53 patients), with few cases of tamponade [5, 21]. The myocardium and endocardium may be involved. Among 37 patients who underwent systematic retrospective cardiovascular screening (MRI and/or heart CT scan) in 2009, 70 % had abnormal heart imaging results: abnormal infiltration of the right heart in 49 %, including 30 % with “pseudo-tumoral” infiltration of the right atrium, and infiltration of the auriculo-ventricular sulcus in 19 % [22].
Fifteen patients with myocardial infarctions secondary to pericoronary infiltration have been reported, leading to death in some cases [5, 23, 24]. In a series of 53 patients, 17 % had symptomatic heart valve disease (aortic and mitral regurgitations) [4••]. Valve replacement was required for three patients, including one case in our center [5, 25]. This operation is technically difficult because of the extensive infiltration of heart tissue, and therefore should only be performed at a specialist center, and only in appropriately selected cases.
Retro-Orbital Infiltration
One in four of our patients developed exophthalmos, often bilateral, caused by infiltration of the retro-orbital soft tissues [26, 27]. In a small number of cases, the infiltration is massive, and in such cases it may be refractory to conventional therapy such that surgical debulking is required.
Endocrine Involvement
Diabetes insipidus, caused by infiltration of the pituitary gland, is the most frequent endocrine manifestation of ECD (25 % of our patients). Rare cases of pituitary or hypothalamic infiltration have been reported, with other endocrine consequences, including hyperprolactinemia, gonadotropin insufficiency, and abnormally low levels of IGF-1 [28, 29].
We reported that seven of a series of 22 ECD patients had adrenal gland enlargement, with adrenal insufficiency in one case [30]. The diagnosis was radiological in all seven cases, and was confirmed by autopsy for one patient. Thus, adrenal infiltration is not rare in ECD patients and may possibly be associated with adrenal insufficiency.
Cutaneomucosal Involvement
Xanthelasma affected 28 % of our 53 patients, and usually involved the eyelids or the peri-orbital spaces. Papulonodular lesions [31] and infiltrations of the vulva and of the clitoris may be observed, but are less frequent [1].
Urological and Nephrological Infiltrations
Approximately one third of ECD cases present with pseudo “retroperitoneal fibrosis”, in some cases complicated by bilateral hydronephrosis that may require ureteral stenting [32]. Involvement of the pelvic ureters has never been described, and the inferior vena cava is rarely affected in ECD. In ECD, the “fibrosis” sheaths the aortic walls completely and circumferentially, whereas the posterior aortic wall is rarely affected in idiopathic retroperitoneal fibrosis [5].
Lung Involvement
In 2008, we performed a retrospective analysis of the lungs of 34 consecutive patients with ECD [33]. Involvement of the lung parenchyma in 53 % of the cases, and of the pleura in 41 %, was revealed by high-resolution thoracic CT scans [34•]. The lesions mainly affect the interlobular septa. Lung involvement in this series was not a significant prognostic factor for ECD, contrasting with previous findings for small numbers of patients. A MEDLINE search identified reports of lung involvement in 70 (22 %) of 319 ECD cases published before November 2008, but the descriptions were mostly incomplete.
CNS Involvement
CNS involvement is common in ECD (15–25 %) [11], and was extensively described in the largest neurological series reported [35]: this multi-center literature review in 2006 analyzed 66 ECD patients (including six personal cases) with neurological involvement. Cerebellar and pyramidal syndromes were the most frequent neurological manifestations (41 % and 45 % of cases, respectively), and the other features described included seizures, headaches, neuropsychiatric manifestations or cognitive impairment, sensory disturbances, cranial nerve paralysis, and asymptomatic lesions. Neurological involvement led to severe functional disability in almost all patients, and CNS involvement is a major prognostic factor for ECD: survival analysis suggests that it is an independent predictor of death (hazard ratio = 2.51; 95 % confidence interval, 1.28–5.52; P = 0.006) [4••].
We reviewed brain MRI findings for 33 ECD patients followed at the Pitié-Salpêtrière Hospital up to 2009. Only three patients had normal imaging results [36], and two or more different anatomic sites were affected in most patients. Lesions are frequent in the brain, meninges, facial bones, and orbits in ECD patients. Consequently, MRI and CT should be used systematically to investigate the brain for all, even asymptomatic, ECD patients.
Other Infiltrations
The range of organs reported to be involved in ECD is broad. Autopsy has revealed the involvement of testes, thyroid, and lymph node [37]. There as also numerous case reports describing breast infiltration [38, 39].
Assessing Disease Activity
The clinical course of ECD seems to be typical of a chronic disease, but has not been described in detail. Lesions accumulate in the organs and systems affected, and rarely regress spontaneously. Serum C-reactive protein (CRP) levels are elevated in more than 80 % of cases [11], but the therapeutic consequences of this are small once the diagnosis is established. Regular clinical examination, radiological investigations (approximately every six months), and imaging to assess morphological changes are used to follow disease activity in ECD; note that no disease activity score has been established.
We have found that PET scanning is particularly informative about ECD activity [17]. Follow-up PET scans enable CNS involvement to be detected, and can reveal early responses of CNS lesions to therapy; any changes in such lesions are apparent on MRI. PET scanning can also be used to investigate the cardiovascular system, and the heart and the entire vascular tree can be studied during a single session. We therefore recommend PET investigations of ECD patients, because no other single technique provides as much information about as many of the lesions frequently associated with ECD. Our recent pilot study of use of BRAF inhibitors for ECD patients illustrates the value of PET investigations [40••].
Interferon Alpha (IFNα) Is the Best-Choice Initial Treatment for ECD
Before 2005, the standard treatments for ECD included steroids, cytotoxic agents [41], and double autologous hematopoietic stem-cell transplantation [42, 43]. The efficacy of these treatments has been difficult to establish, because some have been administered to only small numbers of patients, or in combination with other drugs; also, the follow-up periods were short. Braiteh et al. reported rapid, marked, and persistent regression of retroorbital infiltration, and progressive improvement in bone lesions, pain, and diabetes insipidus, in three ECD patients given IFNα [44]. However, for eight patients with ECD treated with low-dose IFNα (3 MU × 3 week−1), we found that the efficacy differed according to the site involved in the disease [14]. In some cases, the symptoms failed to respond to such low-dose IFNα; this is true in particular of patients with severe multisystem forms of ECD (CNS and cardiovascular involvement) [4••]. We therefore recommend higher doses, 9 MU × 3 week−1 if possible, because such doses may be more effective against meningeal infiltrations, sub and retro-sellar masses, and pericardial and pseudo-atrial infiltrations. IFNα treatment needs to be long-term, but this may lead to side effects including depression and fatigue. In cases of pseudo-degenerative forms of cerebellar involvement (similar to that observed in LCH), IFNα treatment has had disappointing results.
Nevertheless, IFNα seems to be the best choice for the initial prolonged treatment of ECD. Survival analysis of our series of 53 patients indicated that treatment with IFNα and/or PEGylated IFNα was a major independent predictor of survival (HR = 0.32; 95 % CI, 0.14–0.70; P = 0.006) [4••]. Usually, we currently start treatment with PEGylated forms of IFNα, because it is in most cases better tolerated than IFNα in the long-term.
There were reports in 2010 that imatinib mesylate was effective in cases of histiocytosis [45]; however, our preliminary experience with this treatment for six ECD patients was disappointing [46]. The treatment of two ECD patients (neither with cardiovascular or CNS involvement) with recombinant human interleukin-1 receptor (anakinra) was described at this time, and seemed to be promising [47]. We therefore treated ten ECD patients at our institution with anakinra: overall, the efficacy was poor. Cladribin may be beneficial for treating CNS localizations of the disease that are not responsive to IFNα [41], although a small number of patients at our center were administered this treatment and the outcome was not favorable. Infliximab treatment obtained benefits, after 12 to 18 months, for two ECD patients with cardiac involvement [48•]. The recent efficacy of a BRAF inhibitor (vemurafenib) for three patients is even more promising [40••].
BRAF Inhibition as an Alternative to IFNα for ECD Patients Carrying the BRAF V600E Mutation
The RAS-RAF-MEK-ERK pathway is a cellular signaling pathway, and is involved in diverse tumors [49]. Many human tumors carry the BRAFV600E mutation [50], an activating mutation of the proto-oncogene BRAF causing activation of the RAS-ERK pathway, independently of RAS activation. Inhibition of BRAF activation by vemurafenib improves the survival of patients with metastatic melanomas carrying the BRAFV600E mutation [51]. BRAFV600E mutations were reported in patients with LCH in 2010 [52], and we therefore screened patients with other types of histiocytosis for this mutation. We used pyrosequencing to test DNA, from paraffin-embedded samples from 127 patients with histiocytoses, for BRAFV600 mutations, and reassessed histology findings [53••]. The samples were from cases diagnosed as ECD (n = 46), LCH (n = 39), RDD (n = 23), juvenile xanthogranuloma n = 12), histiocytic sarcoma (n = 3), xanthoma disseminatum (n = 2), interdigitating dendritic cell sarcoma (n = 1), and necrobiotic xanthogranuloma (n = 1). The BRAF status was successfully identified in 93 cases: BRAFV600E mutations were detected in 13 of the 24 (54 %) ECD samples, 11 of the 29 (38 %) LCH samples, and none of the other histiocytosis samples. We have also detected BRAF mutations in nineteen of a series of 37 ECD patients [54].
Vemurafenib is an inhibitor of mutant BRAF, and has some efficacy against both BRAFV600E-associated melanoma and hairy-cell leukemia [55]. We conducted a pilot study of vemurafenib treatment for three patients who had multisystemic and refractory ECD and carried the BRAFV600E mutation. Two of the patients also had skin or lymph node LCH involvement [40••]. Vemurafenib treatment led to substantial and rapid clinical and biological improvement in all three cases, as assessed by clinical, biological (CRP values), histological (skin biopsy), and morphological (PET, CT, and MRI) follow-up. The tumor response was confirmed by PET, CT, and/or MRI after one month of treatment. For one patient, response as assessed by PET involved continued improvement between months one and four of treatment. The treatment remained effective after nine months of follow-up, although one patient continued to suffer disease activity. We therefore recommend that vemurafenib treatment should be considered for all patients with severe and refractory BRAFV600E histiocytoses, particularly if life-threatening.
Recently, Diamond et al. reported one patient with ECD NRAS mutations [56•]. Our team has also found these mutations—which have been shown to be a gain-of-function mutation in melanoma—in two BRAF wild-type ECD patients and in one patient with histiocytic sarcomas (manuscript submitted for publication). We suspect that ECD patients with NRAS mutation may benefit from targeted anti-MEK therapy, as did some patients with metastatic melanomas.
Follow-Up
We reported two series from before the “IFNα era”, and these studies provide evidence of the poor prognosis of ECD [5, 6]. In 2004, 35 (60 %) of the 58 patients for whom data were available died, and the mean survival after diagnosis was 19.2 months (range, 0 to 120 months). However, a survival analysis of our patients in 2011 indicated that overall mortality after treatment with IFNα was only 26 %, and the five-year survival was 68 % [4••].
Pathophysiology
Until recently, the pathogenesis of ECD had not been well described, largely because previous studies included only small numbers of patients. Stoppacciaro et al. reported an immunohistochemical study of three patients, revealing that a complex network of cytokines and chemokines regulates histiocyte recruitment and accumulation in the lesions [57]. Dagna et al. studied both spontaneous and stimulated cytokine production by mononuclear cells in biopsy fragments from a single patient [58]: tumor necrosis factor α was produced after stimulation, and IL-6 and IL-8 were secreted spontaneously, with IL-8 acting as a chemoattractant for polymorphonuclear cells and monocytes. Aouba et al. reported evidence from two patients indicating that targeting the IL-1 pathway might be beneficial [47]. We recently assayed serum samples from 37 ECD patients for 23 cytokines [59••], and found high IFNα, IL-1/IL1-RA, IL-6, IL-12, and MCP-1 titers, indicating strong and systemic immune activation. Thus, there is evidence that ECD is associated with systemic immune Th-1-oriented perturbation; further work on this subject may enable the development of better-targeted therapeutic agents.
The recent finding that more than half of ECD patients carry BRAF V600E mutations indicates that the pathophysiology of this disorder is even more complex than previously suspected. This finding is also evidence of clonal proliferation (associated with the BRAF V600E mutation) in addition to the non-clonal accumulation of histiocytes in affected tissues (associated with circulating chemokines and pro-inflammatory cytokines).
Conclusions
ECD is a rare and orphan disease. Having long been poorly recognized, numerous cases have recently been diagnosed, and more than 300 new cases have been published in the past 10 years. This is mainly the result of greater awareness among pathologists, radiologists, and clinicians of different aspects of this previously obscure disease. There has been substantial progress in recent years. In particular, the efficacy of IFNα has been revealed, systemic pro-inflammatory cytokine signatures have been described, and BRAF inhibition in severe cases of BRAF V600E-mutation-associated ECD has been found to be highly effective. More than half of ECD patients tested have been found to carry BRAF mutations. Further analysis of the disease, and especially an improved understanding of its pathogenesis, should lead to the development of better-targeted and more effective therapy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Chester W. Über lipoidgranulomatose. Virchows Arch Pathol Anat. 1930;279:561–602.
Adam Z, Koukalova R, Sprlakova A, et al. Successful treatment of Erdheim–Chester disease by 2-chlorodeoxyadenosine-based chemotherapy. Two case studies and a literature review. Vnitr Lek. 2011;57:576–89.
Haroche J, Arnaud L, Amoura Z. Erdheim–Chester disease. Curr Opin Rheumatol. 2012;24:53–9.
Arnaud L, Hervier B, Neel A, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim–Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011;117:2778–82. This is a major study, with the largest description of ECD series to date, describing the efficacy of interferon alpha, a major independent predictor of survival, and the importance of CNS involvement, a major prognostic factor and an independent predictor of death in ECD.
Haroche J, Amoura Z, Dion E, et al. Cardiovascular involvement, an overlooked feature of Erdheim–Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore). 2004;83:371–92.
Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim–Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75:157–69.
Brower AC, Worsham GF, Dudley AH. Erdheim–Chester disease: a distinct lipoidosis or part of the spectrum of histiocytosis? Radiology. 1984;151:35–8.
Tsai JW, Tsou JH, Hung LY, et al. Combined Erdheim–Chester disease and Langerhans cell histiocytosis of skin are both monoclonal: a rare case with human androgen-receptor gene analysis. J Am Acad Dermatol. 2010;63:284–91.
Wang KH, Cheng CJ, Hu CH, et al. Coexistence of localized Langerhans cell histiocytosis and cutaneous Rosai-Dorfman disease. Br J Dermatol. 2002;147:770–4.
Balink H, Hemmelder MH, de Graaf W, et al. Scintigraphic diagnosis of Erdheim–Chester disease. J Clin Oncol. 2011;29:e470–2.
Haroche J, Amoura Z, Wechsler B, et al. Erdheim–Chester disease. Press Med. 2007;36:1663–8.
Andre M, Delevaux I, de Fraissinette B, et al. Two enlarged kidneys: a manifestation of Erdheim–Chester disease. Am J Nephrol. 2001;21:315–7.
Dion E, Graef C, Haroche J, et al. Imaging of thoracoabdominal involvement in Erdheim–Chester disease. AJR Am J Roentgenol. 2004;183:1253–60.
Haroche J, Amoura Z, Trad SG, et al. Variability in the efficacy of interferon-alpha in Erdheim–Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum. 2006;54:3330–6.
Jeon IS, Lee SS, Lee MK. Chemotherapy and interferon-alpha treatment of Erdheim–Chester disease. Pediatr Blood Cancer. 2010;55:745–7.
Tran TA, Fabre M, Pariente D, et al. Erdheim–Chester disease in childhood: a challenging diagnosis and treatment. J Pediatr Hematol Oncol. 2009;31:782–6.
Arnaud L, Malek Z, Archambaud F, et al. 18F-fluorodeoxyglucose-positron emission tomography scanning is more useful in followup than in the initial assessment of patients with Erdheim–Chester disease. Arthritis Rheum. 2009;60:3128–38.
Stenova E, Steno B, Povinec P, et al. FDG-PET in the Erdheim–Chester disease: its diagnostic and follow-up role. Rheumatol Int. 2012;32:675–8.
Dion E, Graef C, Miquel A, et al. Bone involvement in Erdheim–Chester disease: imaging findings including periostitis and partial epiphyseal involvement. Radiology. 2006;238:632–9.
Serratrice J, Granel B, De Roux C, et al. "Coated aorta": a new sign of Erdheim–Chester disease. J Rheumatol. 2000;27:1550–3.
Gupta A, Kelly B, McGuigan JE. Erdheim–Chester disease with prominent pericardial involvement: clinical, radiologic, and histologic findings. Am J Med Sci. 2002;324:96–100.
Haroche J, Cluzel P, Toledano D, et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim–Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119:e597–8.
Fink MG, Levinson DJ, Brown NL, et al. Erdheim–Chester disease. Case report with autopsy findings. Arch Pathol Lab Med. 1991;115:619–23.
Loeffler AG, Memoli VA. Myocardial involvement in Erdheim–Chester disease. Arch Pathol Lab Med. 2004;128:682–5.
Kenn W, Stabler A, Zachoval R, et al. Erdheim–Chester disease: a case report and literature overview. Eur Radiol. 1999;9:153–8.
Alper MG, Zimmerman LE, Piana FG. Orbital manifestations of Erdheim–Chester disease. Trans Am Ophthalmol Soc. 1983;81:64–85.
Sheidow TG, Nicolle DA, Heathcote JG. Erdheim–Chester disease: two cases of orbital involvement. Eye (Lond). 2000;14:606–12.
Khamseh ME, Mollanai S, Hashemi F, et al. Erdheim–Chester syndrome, presenting as hypogonadotropic hypogonadism and diabetes insipidus. J Endocrinol Invest. 2002;25:727–9.
Tritos NA, Weinrib S, Kaye TB. Endocrine manifestations of Erdheim–Chester disease (a distinct form of histiocytosis). J Intern Med. 1998;244:529–35.
Haroche J, Amoura Z, Touraine P, et al. Bilateral adrenal infiltration in Erdheim–Chester disease. Report of seven cases and literature review. J Clin Endocrinol Metab. 2007;92:2007–12.
Opie KM, Kaye J, Vinciullo C. Erdheim–Chester disease. Australas J Dermatol. 2003;44:194–8.
Droupy S, Attias D, Eschwege P, et al. Bilateral hydronephrosis in a patient with Erdheim–Chester disease. J Urol. 1999;162:2084–5.
Arnaud L, Pierre I, Beigelman-Aubry C, et al. Pulmonary involvement in Erdheim–Chester disease: a single-center study of thirty-four patients and a review of the literature. Arthritis Rheum. 2010;62:3504–12.
Brun AL, Touitou-Gottenberg D, Haroche J, et al. Erdheim–Chester disease: CT findings of thoracic involvement. Eur Radiol. 2010;20:2579–87. A major radiologic thoracic review of 40 ECD patients describing infiltration into the mediastinal spaces including the pericardium, coronary sulci, and right atrium; association with pleural and interstitial lung diseases is frequently observed.
Lachenal F, Cotton F, Desmurs-Clavel H, et al. Neurological manifestations and neuroradiological presentation of Erdheim–Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253:1267–77.
Drier A, Haroche J, Savatovsky J, et al. Cerebral, facial, and orbital involvement in Erdheim–Chester disease: CT and MR imaging findings. Radiology. 2010;255:586–94.
Sheu SY, Wenzel RR, Kersting C, et al. Erdheim–Chester disease: case report with multisystemic manifestations including testes, thyroid, and lymph nodes, and a review of literature. J Clin Pathol. 2004;57:1225–8.
Provenzano E, Barter SJ, Wright PA, et al. Erdheim-chester disease presenting as bilateral clinically malignant breast masses. Am J Surg Pathol. 2010;34:584–8.
Johnson TR, Lenhard MS, Weidinger M, et al. An unusual breast tumor. Radiologe. 2009;49:942–5.
Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim–Chester disease and Langerhans cell histiocytosis harbouring the BRAF V600E mutation. Blood. 2013;121:1495–500. A major pilot study of three ECD patients with a proof of concept demonstration of the importance of BRAF inhibitors in BRAF mutated histiocytoses.
Myra C, Sloper L, Tighe PJ, et al. Treatment of Erdheim–Chester disease with cladribine: a rational approach. Br J Ophthalmol. 2004;88:844–7.
Boissel N, Wechsler B, Leblond V. Treatment of refractory Erdheim–Chester disease with double autologous hematopoietic stem-cell transplantation. Ann Intern Med. 2001;135:844–5.
Gaspar N, Boudou P, Haroche J, et al. High-dose chemotherapy followed by autologous hematopoietic stem cell transplantation for adult histiocytic disorders with central nervous system involvement. Haematologica. 2006;91:1121–5.
Braiteh F, Boxrud C, Esmaeli B, et al. Successful treatment of Erdheim–Chester disease, a non-Langerhans-cell histiocytosis, with interferon-alpha. Blood. 2005;106:2992–4.
Janku F, Amin HM, Yang D, et al. Response of histiocytoses to imatinib mesylate: fire to ashes. J Clin Oncol. 2010;28:e633–6.
Haroche J, Amoura Z, Charlotte F, et al. Imatinib mesylate for platelet-derived growth factor receptor-beta-positive Erdheim–Chester histiocytosis. Blood. 2008;111:5413–5.
Aouba A, Georgin-Lavialle S, Pagnoux C, et al. Rationale and efficacy of interleukin-1 targeting in Erdheim–Chester disease. Blood. 2010;116:4070–6.
Dagna L, Corti A, Langheim S, et al. Tumor necrosis factor α as a master regulator of inflammation in Erdheim–Chester disease: rationale for the treatment of patients with infliximab. J Clin Oncol. 2012;30:e286–90. A study focusing on the crucial importance of TNF alpha in ECD; two patients were treated with infliximab, with interesting results.
Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23:6771–90.
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16.
Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919–23.
Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120:2700–3. An important study of a large number of histiocytic disorders with half of ECD and LCH patients tested mutated for BRAF V600E; no mutations found in all the other non-Langerhans histiocytosis tested (for example Rosai–Dorfman disease).
Emile JF, Charlotte F, Amoura Z, et al. BRAF mutations in Erdheim–Chester disease. J Clin Oncol. 2013;31:398.
Dietrich S, Glimm H, Andrulis M, et al. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366:2038–40.
Diamond EL, Abdel-Wahab O, Pentsova E, et al. Detection of an NRAS mutation in Erdheim–Chester disease. Blood. 2013;122:1089–91. An interesting study with detection of an NRAS mutation in an ECD case, focusing on the importance of the RAS-RAF-MEK-ERK pathway in this condition.
Stoppacciaro A, Ferrarini M, Salmaggi C, et al. Immunohistochemical evidence of a cytokine and chemokine network in three patients with Erdheim–Chester disease: implications for pathogenesis. Arthritis Rheum. 2006;54:4018–22.
Dagna L, Girlanda S, Langheim S, et al. Erdheim–Chester disease: report on a case and new insights on its immunopathogenesis. Rheumatology (Oxford). 2010;49:1203–6.
Arnaud L, Gorochov G, Charlotte F, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim–Chester disease: a single-center series of 37 patients. Blood. 2011;117:2783–90. This important cytokine analysis from serum samples of 37 ECD patients reveals an intense systemic immune activation mainly involving IFNα, IL-1/IL1-RA, IL-6, IL-12, and MCP-1, emphasising the systemic immune Th-1-oriented perturbation associated with this condition.
Haroche J, Arnaud L, Cohen-Aubart F, Hervier B, Charlotte F, Emile JF, et al. Erdheim–Chester disease. Rheum Dis Clin N Am. 2013;39(2):299–311.
Compliance with Ethics Guidelines
Conflict of Interest
Julien Haroche has received honoraria from GlaxoSmithKline. Jean-François Emile has received honoraria from GlaxoSmithKline and Roche. Laurent Arnaud, Fleur Cohen-Aubart, Baptiste Hervier, Frédéric Charlotte, and Zahir Amoura declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with animal subjects performed by any of the authors. With regard to the authors’ research cited in this paper, all procedures were followed in accordance with the ethical standards of the responsible committee on human experimentation and with the Helsinki Declaration of 1975, as revised in 2000 and 2008.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Orphan Diseases
Rights and permissions
About this article
Cite this article
Haroche, J., Arnaud, L., Cohen-Aubart, F. et al. Erdheim–Chester Disease. Curr Rheumatol Rep 16, 412 (2014). https://doi.org/10.1007/s11926-014-0412-0
Published:
DOI: https://doi.org/10.1007/s11926-014-0412-0