
Abstract
Purpose of Review
We aim to summarize the sleep disorders reported in patients affected by primary mitochondrial dysfunctions and describe the association with their clinical and molecular characteristics.
Recent Findings
Sleep complaints are prevalent in mitochondrial disorders. Sleep-disordered breathing is the main sleep disorder reported in mitochondrial diseases. OSA and CSA are, respectively, more frequently associated with patients characterized by the prevalent involvement of the skeletal muscle and the predominant involvement of the central nervous system. Other sleep disorders, such as restless legs syndrome, have been rarely described.
Summary
Sleep disorders are frequently associated with primary mitochondrial disorders, and the clinical phenotypes affect the type of sleep disturbance associated with the mitochondrial dysfunction. A polysomnographic study should be performed in every subject with this neurogenetic disorder both at diagnosis and during follow-up for the numerous adverse clinical outcomes associated with sleep disorders and the frailty of mitochondrial patients.
Similar content being viewed by others

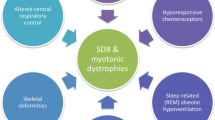

Introduction
Mitochondrial medicine, a term coined by Rolf Luft in 1994 [1], is currently a recognized field in translational medical research with important clinical implications. As one of the branches of medicine in extremely rapid evolution, the role of mitochondria dysfunction in different human conditions is deeply explored in rare and common conditions, such as primary mitochondrial diseases, neurodegenerative diseases, cardiovascular diseases, aging, and cancer [2,3,4, 5•]. In particular regard to primary genetic mitochondrial disorders, numerous medical subspecialties have helped to further understand the pleiotropic manifestations of mitochondrial diseases (MDs), involving branches like endocrinology, cardiology, gastroenterology, and ophthalmology [6,7,8]. Among the commonest forms of genetic human disorders with mutations in more than 350 genes of the mitochondrial and nuclear genomes, MDs are characterized by a primary defect in oxidative phosphorylation, the main source of cellular adenosine triphosphate (ATP). With an age of onset ranging from infancy to adulthood, the patients affected by primary mitochondrial dysfunction experience a single organ involvement or more frequently a multisystem syndrome with the most energy-dependent tissues commonly affected, such as the brain and skeletal muscle [9•, 10]. The broad clinical spectrum of MDs, including specific phenotypes characterized by the predominant involvement of the skeletal muscle defined primary mitochondrial myopathies (PMM) [11••], justifies the variety of sleep disorders documented in these genetic diseases [12•] and the emerging role of mitochondrial sleep medicine.
In this review, we aim to summarize the sleep disorders reported in patients affected by primary mitochondrial dysfunctions and describe the association with clinical and molecular characteristics of mitochondrial patients. Finally, we briefly discuss the main pathophysiological mechanisms that document the key role of the mitochondria in the sleep physiology and pathology.
Sleep Disorders in Mitochondrial Diseases: Clinical Implications
The burden of sleep disorders in MDs has been poorly documented. To date, most of the literature on this topic is mainly based on case reports or case series and few retrospective or cross-sectional studies with a significant number of patients investigated [13,14,15,16, 17••, 18, 19••, 20••, 21••]. For a detailed review of the current literature, see Table 1.
Sleep-Disordered Breathing
The most frequent sleep disorders described in the context of MDs belong to the sleep-disordered breathing (SDB) group. SDB includes a constellation of disturbances classified in four major categories: obstructive sleep apnea (OSA), central sleep apnea (CSA) syndrome, sleep-related hypoxemia disorders, and sleep-related hypoventilation disorders [22]. Each of these disorders has been associated with MDs.
The physiological changes that occur during sleep make this a critical time for the process of breathing. First, the effect of gravity in the supine position determinates a reduction of the total lung capacity and a narrowing of the velopharynx [23], both contributing to the increased upper airway resistance, as well as the reduced tonic drive of pharyngeal dilator muscles [24]. Furthermore, physiological modifications observed in breathing during sleep include a reduction of respiratory rate [25], a diminished sensitivity of chemoreceptor of the respiratory center [26], and the absence of stimulus of wakefulness drive to respiration [27]. Finally, the reduction of the muscular tone involving the accessory respiratory muscles is associated with normal diaphragmatic activity in order to guarantee an adequate ventilation. This aspect is particularly important for REM sleep characterized by a complete muscular atonia and, therefore, represents the most critical sleep stage for respiration [28]. On these bases, it is expected that patients affected by neuromuscular disorders are particularly vulnerable to develop SDB [29]. OSA is the most common subtype of SDB, and it is characterized by intermittent and repetitive episodes of partial (hypopnea) or complete (apnea) obstruction of the upper airways causing falls in blood oxygen hemoglobin saturation and disruption of sleep. Daytime symptoms include excessive daytime sleepiness (EDS), fatigue, morning headache, and cognitive or mood alterations (e.g., memory loss, irritability, and depression). In our previous paper, we described a large population of adult patients affected by MDs investigated by a polysomnographic study [19••], revealing a high prevalence of OSA (35/103, 34%). Particularly, the prevalence of this specific SDB was significantly higher in the phenotypes of MDs associated with the higher grade of muscular involvement. Interestingly, the classical risk factors for OSA described in general population, such as obesity, were not significantly associated with OSA in our MD patients. Příhodová and colleagues observed a high prevalence of OSA (22%) in patients affected by Leber hereditary optic neuropathy (LHON) and dominant optic atrophy (DOA). Interestingly, the prevalence of OSA in these patients did not differ between symptomatic and asymptomatic groups [21••]. At the same time, the burden of OSA in MDs has been also revealed in pediatric populations. In particular, Jeyakumar et al. [15] observed a higher prevalence of OSA (9.8%) in pediatric patients affected by MDs when compared with that described in the general pediatric population (2%). These findings were further confirmed by Mosquera and colleagues [17••], reporting a pediatric group of patients affected by MDs by means of video-polysomnography. In their study, the authors revealed a high incidence of SDB (10/18, 56%) with a clear prevalence of OSA (6/18, 33.3%). Interestingly, the prevalence of SDB was prominent in patients with an abnormal muscular tone, while the classical risk factors of OSA, such as adenotonsillar hypertrophy, allergic rhinitis history, and post-tonsillectomy status, were not associated with SDB, suggesting that the genetic neuromuscular disease contributes to sleep respiratory disturbances. These data seem to confirm the results reported in our research paper, documenting the key role of skeletal muscle involvement in the occurrence of specific SDB [19••]. Moreover, the presence of SBD in pediatric MD population suggests that respiratory sleep disorders are an early manifestation of disease, and therefore, polysomnography should be performed as promptly as possible in these fragile patients.
Conversely, Smits et al. [20••] found a low prevalence of OSA (1/20, 5%) in a population of 20 adult patients with chronic progressive external ophthalmoplegia (PEO), with slightly increased AHI. On the other hand, in this population, CSA was the prominent SDB (4/20, 20%). A possible explanation for the discrepancy of data between the two studies is probably to be found in the different phenotypes associated with mutations in POLG gene. In our recent published article [19••], the patients associated with pathological variants in POLG presented a PMM with PEO phenotype, while Smits and colleagues investigated the presence of SDB in subjects with POLG mutations and ataxia neuropathy spectrum (ANS) phenotype. In light of these considerations, it is possible to hypothesize that patients with pathogenic variants in the same gene present OSA or CSA depending on a phenotype with predominant muscular or nervous system involvement. Another possible explanation of this conflicting findings is that, at least in part, some of the observed respiratory events could be classified as “pseudo-central” [30]. These events appear mainly during REM sleep characterized by a reduction of the oro-nasal flow due the diminished intercostal muscle activity and, in turn, to a reduction of the excursion of the rib cage. However, OSA has been described in association with other phenotypes of MDs, such as Leigh syndrome (LS) [17••, 31,32,33], Kearns–Sayre syndrome (KSS) [34], and neuropathy, ataxia, and retinitis pigmentosa (NARP) syndrome [35]. In conclusion, literature data document that OSA is a frequent sleep disorder in MDs, especially in phenotypes with predominant skeletal muscle involvement.
As aforementioned, sleep represents a state of vulnerability for patients affected by neuromuscular disorders, including MDs characterized by different degrees of involvement of the respiratory muscles. For this reason, specific subgroups of MDs have an increased risk of developing sleep-related hypoventilation/hypoxemia [34, 36]. As consequence of muscular atonia observed during REM, hypoventilation/hypoxemia usually first appears during this sleep stage [16, 19••] and, subsequently, progresses towards NREM sleep. This is mainly due to two mechanisms: the progression of the muscular weakness due to the underline disease and the decreased ventilatory drive due to a diminished sensitivity of chemoreceptor to chronic hypercarbia. In particular, the metabolic derangement, as observed in MDs, could concur to lower the chemosensitivity to hypoxia and hypercarbia. In fact, a depressed ventilatory drive response to hypoxia/hypercapnia has been described in patients affected by PEO [16, 37, 38] and mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) syndrome [39]. In particular, sleep-related hypoventilation has been associated with primary mitochondrial dysfunction with a prominent involvement of CNS [32, 34, 35, 39,40,41,42], suggesting that also a dysfunction of the brainstem respiratory center participates to the development of the sleep-related respiratory disturbance. In light of these considerations, it is not surprising that MD phenotypes with predominant CNS involvement, such as LHON “plus” [41] and LS [32], have been associated with the central hypoventilation syndrome, a peculiar life-threatening sleep disorder characterized by ineffective breathing till respiratory arrest during the sleep.
Therefore, it is possible to hypothesize that hypoventilation observed during sleep in MDs recognizes a combined etiology involving CNS dysfunction and muscular weakness [32, 43]. Moreover, sleep-related hypoventilation/hypoxemia have been described also in mitochondrial pediatric population [17••, 33], underlining that these disorders could manifest even in the early stages of the disease.
As previously mentioned, CSA has also been described in association with MDs [16, 20••, 33, 35, 42, 44]. This specific subgroup of SDB is characterized by air-flow cessation in consequence of diminished or absent respiratory effort, due to the lack of drive to breath. CSA can be associated with wakefulness hypercapnia (hypercapnic CSA) or normocapnia (non-hypercapnic CSA) [45]. Hypercapnic CSA is the result of an impaired central drive due to a dysfunction of brainstem respiratory centers (e.g., CNS diseases, specific medications) or an impaired respiratory efference (e.g., muscular disorders, motor neuron diseases). Conversely, non-hypercapnic CSA is idiopathic or frequently related to congestive heart failure. In the latter case, a peculiar pattern of crescendo/decrescendo ventilatory pattern is observed, mostly during NREM sleep, and defined as Cheyne–Stokes breathing (CSB). MDs encompass a large variety of phenotypes with different grades of muscular, neurological, and cardiac involvement. On these bases, it is reasonable to presume that they are widely associated to CSA, and therefore, the clinical suspicion of disordered breathing of central origin should be raised in those patients with genetic mitochondrial dysfunction who manifest cardiomyopathy, as observed in a LHON patient with a severe cardiac involvement [21••] or the involvement of the CNS. In fact, most of the cases described in literature of CSA in MDs are related to clinical phenotypes such as LS [33, 46], NARP [35], and KSS [34]. CSA and a diminished ventilatory response to inhaled CO2 have been described by Manni and colleagues [16] in a cohort of patients affected by PEO, without molecular characterization but defined as “ophthalmoplegia plus” for the involvement of the central and/or peripheral nervous system. Similarly, Smits et al. [20••] revealed high prevalence of CSA (4/20; 20%) in PEO patients associated with POLG mutations and ANS phenotype. Although the absence of genetic data in the article of Manni et al. does not allow us to draw definitive conclusions, it is possible to speculate that a combined mechanism of impaired CNS control of respiration and respiratory muscle weakness concur to determinate sleep-disordered breathing of central origin.
Subjective Sleep Disturbances
Subjective nocturnal sleep dysfunction, evaluated by the Pittsburgh sleep quality index (PSQI), has been reported in about 75% of patients with MDs [20••] and in 70% of patients with mitochondrial optic neuropathies [21••]. Similarly, EDS, evaluated by means of Epworth sleepiness scale (ESS), has been described in adult patients affected by primary mitochondrial disorders with a prevalence ranging from 27 [14] to 33% [18] and up to 66% in pediatric population [17••]. In both studies, the prevalence of EDS appears significantly higher than in general population. EDS appears to be prevalent (4/36, 11.1%) also in mitochondrial optic neuropathies, regardless of the presence of ocular symptoms [21••]. A single study by Guilleminault and colleagues [47] objectively evaluated EDS by means of multiple sleep latency test (MSLT) in patients with neuromuscular disorders, including two patients with MDs. Both subjects presented subjective EDS (ESS > 10) objectively confirmed by a mean sleep latency of about 8 min at MSLT. Interestingly, after correcting the underlying sleep respiratory disorder, these patients presented a normalization of both subjective and objective EDS. Other experiences indicate that a treatment of an underlying sleep disorder could ameliorate daytime symptoms correlated to a chronic sleep deprivation promoted by sleep disruption [31, 35, 43]. Conversely, subjective sleep complaints were not present in a study group of patients with concomitant sleep apnea and REM-related hypoventilation [16]. Moreover, in large cross-sectional studies the prevalence of perceived sleep dysfunction [20••] and EDS [17••, 20••] is higher than the prevalence of a concomitant sleep disorder or abnormal findings on polysomnography. Similarly, in LHON and DOA [21••], no significant correlation was observed between polysomnographic parameters and poor subjective sleep quality. Therefore, it is difficult to establish whereby EDS and subjective sleep dysfunction are a reflection of underlying sleep disorder or a direct manifestation of MDs.
Other Sleep Disorders
Other sleep disorders, in addition to those belonging to the SDB category, have been rarely described in association with MDs. Sleep-onset and maintenance insomnia have been reported in a high prevalence of patients (15/36, 41.7%) with mitochondrial optic neuropathies [21••]. Smits et al. [20••] reported a high prevalence (7/20, 35%) of restless legs syndrome (RLS) in PEO patients: two carrying POLG mutations, whereas the other five are associated with single mtDNA deletion or other mutations. Surprisingly, the presence of RLS was not associated to increased sleep latency neither to worst subjective sleep quality. Regarding polysomnographic findings, they found a high prevalence of periodic limb movements (PLM) in their population (mean PLM index 25.5 events/h), and in nine cases (45%), the PLM index was higher than 15 events/h which is considered the pathological cut-off [48]. PLM were more common in those patients who complained poor subjective nocturnal sleep quality, suggesting a high prevalence of periodic limb movement disorder (PLMD) in their cohort of patients affected by genetic mitochondrial dysfunction. RLS and nocturnal leg cramps seems to be prevalent (4/36, 11.1%) in patients affected by LHON and DOA, while PLM detected on PSG were present in few patients of the same cohort (2/36, 5.6%) [21••]. A case of RLS in a patient affected by PEO with a POLG pathological variants has been reported by Aitken et al. [49]. In this case, the patient presented an asymmetric uptake of tracer in the putamen at DaTscan, suggesting a dysfunction of the dopaminergic system, as usually observed in Parkinson disease and idiopathic RLS [50]. Interestingly, Haschka et al. [51] reported an association between RLS and the mitochondrial iron deficiency in peripheral monocytes, suggesting that mitochondrial dysfunction can concur to aggravate RLS symptoms. Increased PLM index (pediatric cut-off > 5 events/h) has been described also by Mosquera et al. [17••] in two pediatric patients, without subjective sleep movement complaints. Finally, a peculiar sleep-related movement disorder defined as excessive fragmentary hypnic myoclonus has been reported by Pincherle and colleges [52]. In this case report, the authors describe a patient with brainstem lesions on MRI, who underwent to V-PSG, presenting sub-continuous and arrhythmic myoclonic jerks occurring during both NREM and REM sleep and associated to sleep-onset insomnia, reduced sleep efficiency, and increased wake after sleep onset (WASO). Finally, a delayed sleep–wake phase disorder, a circadian rhythm sleep disorder (CRSD), has been described in a family of diabetes mellitus associated with m.3243A>G mutation [53]. Interestingly, the circadian rhythm disorder dramatically improved after the administration of coenzyme Q10, suggesting that circadian rhythm disorder can be a direct manifestation of MD, as indicated by recent evidence of a crosstalk between the mitochondria and the circadian clock [54]. Conversely, no CRSD have been observed in a large cohort of patients with mitochondrial optic neuropathies [21••], reinforcing the hypothesis that the retinohypothalamic tract, essential for light-dependent regulation of the circadian rhythm, is sufficiently preserved in these pathologies.
Approaches to Treatment
SDB are a frequent comorbidity of MDs, and an early recognition is crucial in order to prevent further clinical deterioration of these fragile patients. Data from the literature support that mechanical ventilation is effective to improve nocturnal breathing, to restore a normal sleep architecture [35] and to ameliorate daytime symptoms [31, 47]. Particularly, in cases associated with severe central nocturnal hypoventilation [32, 41, 44, 46], treatment is lifesaving, because this condition can lead to sudden death during sleep. An early recognition and treatment could reduce the risk of further respiratory deterioration and the need for invasive mechanical ventilation or tracheotomy [32, 34, 35, 41, 46]. Moreover, a pharmacological treatment has been proposed [39, 40] to treat hypoventilation in these patients with drugs that stimulate hypoxic ventilatory response (e.g., aminophylline, theophylline, almitrine) with inconsistent results. The treatment of the underlying metabolic disorder associated with MD could also concur to ameliorate the SDB [31, 42, 43]. For pediatric population, adenotonsillectomy has been proposed for the treatment of OSA, in the absence of sufficient data to support this approach [15]. Therefore, a combined treatment with ventilatory support and pharmacological therapy appears desirable to treat SDB in MDs.
Sleep Structure
Few data are available regarding sleep structure in patients with MDs. Sporadic and not recent articles reported the alteration of sleep macrostructure in these patients, and it seems to depend on the degree of the involvement of CNS [42, 55, 56] or on the presence and the severity of an overlapping sleep disorder [16, 44, 52]. In our previous study [19••], we observed an increase of wake after sleep onset (WASO) in the study group, whereas a reduction of slow-wave sleep (SWS) was present only in patients who suffered from SDB. These data indicated that the SDB worsen sleep quality and sleep structure in primary mitochondrial disorders. Similarly, Smits et al. [20••] observed an unstructured sleep as a consequence of SDB in a PEO population, reporting a diminished total sleep, sleep efficiency, and REM sleep in patients with pathological AHI index. The treatment of underlying sleep disorder in some cases was associated with the amelioration of sleep architecture [31, 35]. In LS [33] a decreased SWS and absence of REM stage sleep have been reported in patients who presented lower medullary lesions, regardless the presence of associated sleep disorder. In LHON and DOA, no significant alterations of sleep architecture have been observed, despite the presence of subjective sleep dysfunction, excessive daytime sleepiness, or concomitant sleep disorders [21••]. Conversely, in patients affected by mitochondrial optic neuropathies, the authors revealed the presence of REM sleep without atonia (RSWA), speculating that it is a marker of brainstem dysfunction.
To date, data regarding sleep architecture and its association with clinical phenotype and concomitant sleep disorders are inconsistent; therefore, further studies are needed to clarify this point.
Pathophysiological Mechanisms
The mitochondrial function has a recognized pivotal role in skeletal muscle physiology for the involvement of an extraordinary number of critical processes in this high-energy-dependent tissue, such as the generation of the ATP and the regulation of energy-sensitive signaling pathways, calcium homeostasis, modulation and production of reactive oxygen species, apoptosis, and more generally cell metabolism [57]. Consequently, mitochondrial dysfunction is associated with a plethora of pathological conditions affecting the skeletal muscle, including mitochondrial myopathy [11••]. With this background, it is not surprising that patients affected by primary mitochondrial disorders characterized by the involvement of skeletal muscle (e.g., PEO and MERRF) [58, 59] presented specific subgroups of SDB, such as OSA and REM-related oxygen desaturations [19••]. Similarly, the central role of the mitochondria in numerous mechanisms of the CNS [60] may partly explain the coexistence in these patients of sleep disorders such as CSA [20••]. In addition to the data reported so far, there are no experimental models that document the direct role of mitochondria dysfunction in their genesis of sleep disorders or that explain the association with MDs. On the contrary, in recent years, a large number of research articles have documented the consequences of sleep disorders in mitochondrial functions. Rodrigues and colleagues demonstrated in a Drosophila melanogaster sleep-disordered model that dysregulation of homeostatic sleep regulation resulted in mitochondrial bioenergetics function, as a consequence of mitochondrial OXPHOS system dysfunction and/or of an inhibition of the mitochondrial complexes, and generation of reactive oxygen species [61]. These data are reinforced by the evidence of alterations in the antioxidant defense biomarkers in a mouse model of chronic sleep deprivation [62] and by the results reported by Trivedi and colleagues that showed oxidative stress induction and ATP depletion in patients undergoing sleep deprivation [63]. Furthermore, the imbalance between reactive oxygen species production and antioxidant defense mechanisms, resulting in an oxidative stress condition, documented in patients affected by OSA is reported in a multitude of studies, suggesting also that it may contribute to increased cardiovascular risk and neurocognitive impairment [64]. Wrede et al. have provided a further contribution in understanding the consequences of sleep disorders in the mitochondrial function. They documented that the reduction of sleep duration and sleep efficiency were associated with decreased mitochondrial DNA copy number, an indirect manifestation of mitochondrial dysfunction [65].
Conclusions
In MDs, the involvement of organs and tissues with the highest energy demands, such as the brain and skeletal muscle, is reflected in the heterogeneity of sleep disorders associated with these metabolic diseases. There are currently few published retrospective or cross-sectional studies on this topic. Most of the knowledge regarding the association of sleep disorders and MDs is the result of case reports or case series, in which patients are poorly characterized clinically and molecularly. Nevertheless, taken together, the literature data suggest some interesting conclusions: sleep disorders are frequently associated with primary mitochondrial disorders; the clinical phenotypes affect the type of sleep disturbance associated with the mitochondrial dysfunction, as shown by the high prevalence of OSA in patients with prevalent involvement of skeletal muscle and CSA in patients in whom the central nervous system is predominantly affected; for the numerous adverse clinical outcomes associated with sleep disturbances and the frailty of mitochondrial patients, a polysomnographic study should be performed in every subject with this genetic disorder both at diagnosis and during follow-up.
Prospective and multicenter studies are needed to document the incidence of sleep disorders in primary mitochondrial disorders and their role in disease prognosis.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Luft R. The development of mitochondrial medicine. Proc Natl Acad Sci U S A. 1994;91(19):8731–8.
Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–7.
Picca A, Mankowski RT, Burman JL, Donisi L, Kim JS, Marzetti E, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol. 2018;15(9):543–54.
Schapira AH. Mitochondrial diseases. Lancet. 2012;379(9828):1825–34.
• Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol. 2018;19(2):77–92 A comprehensive review article on mitochondrial diseases with particular regard to current concepts of molecular and biochemical mechanisms.
Bates MG, Bourke JP, Giordano C, d'Amati G, Turnbull DM, Taylor RW. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J. 2012;33(24):3023–33.
Chow J, Rahman J, Achermann JC, Dattani MT, Rahman S. Mitochondrial disease and endocrine dysfunction. Nat Rev Endocrinol. 2017;13(2):92–104.
Sadun AA, La Morgia C, Carelli V. Mitochondrial optic neuropathies: our travels from bench to bedside and back again. Clin Exp Ophthalmol. 2013;41(7):702–12.
• Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080 A detailed review on the clinical, genetic and therapeutic aspects of mitochondrial diseases.
Rahman J, Rahman S. Mitochondrial medicine in the omics era. Lancet. 2018;391(10139):2560–74.
•• Mancuso M, McFarland R, Klopstock T, Hirano M, Consortium on Trial Readiness in Mitochondrial M. International Workshop:: Outcome measures and clinical trial readiness in primary mitochondrial myopathies in children and adults. Consensus recommendations. 16-18 November 2016, Rome, Italy. Neuromuscul Disord. 2017;27(12):1126–37 A consensus paper that defines a specific subgroup of mitochondrial diseases defined “primary mitochondrial myopathies,” with important implications in the clinical and therapeutic field.
• Ramezani RJ, Stacpoole PW. Sleep disorders associated with primary mitochondrial diseases. J Clin Sleep Med. 2014;10(11):1233–9 A review article on sleep disorders in mitochondrial diseases.
Florian A, Ludwig A, Stubbe-Drager B, Boentert M, Young P, Waltenberger J, et al. Characteristic cardiac phenotypes are detected by cardiovascular magnetic resonance in patients with different clinical phenotypes and genotypes of mitochondrial myopathy. J Cardiovasc Magn Reson. 2015;17(1):40.
Gorman GS, Elson JL, Newman J, Payne B, McFarland R, Newton JL, et al. Perceived fatigue is highly prevalent and debilitating in patients with mitochondrial disease. Neuromuscul Disord. 2015;25(7):563–6.
Jeyakumar A, Williamson ME, Brickman TM, Krakovitz P, Parikh S. Otolaryngologic manifestations of mitochondrial cytopathies. Am J Otolaryngol. 2009;30(3):162–5.
Manni R, Piccolo G, Banfi P, Cerveri I, Bruschi C, Zoia C, et al. Respiratory patterns during sleep in mitochondrial myopathies with ophthalmoplegia. Eur Neurol. 1991;31(1):12–7.
•• Mosquera RA, Koenig MK, Adejumo RB, Chevallier J, Hashmi SS, Mitchell SE, et al. Sleep disordered breathing in children with mitochondrial disease. Pulm Med. 2014;2014:467576 A retrospective chart review of a significant number of pediatric mitochondrial patients.
Parikh S, Galioto R, Lapin B, Haas R, Hirano M, Koenig MK, et al. Fatigue in primary genetic mitochondrial disease: no rest for the weary. Neuromuscul Disord. 2019;29(11):895–902.
•• Primiano G, Brunetti V, Vollono C, Losurdo A, Moroni R, Della Marca G, et al. Sleep-disordered breathing in adult patients with mitochondrial diseases: a cohort study. Neurology. 2021;96(2):e241–e9 The largest cross-sectional study of the prevalence of sleep-disordered breathing in adult patients with clinically and molecularly characterized mitochondrial diseases.
•• Smits BW, Westeneng HJ, van Hal MA, van Engelen BG, Overeem S. Sleep disturbances in chronic progressive external ophthalmoplegia. Eur J Neurol. 2012;19(1):176–8 A study of the prevalence of sleep disturbances in a significant number of PEO patients.
•• Prihodova I, Nepozitek J, Kelifova S, Dostalova S, Kemlink D, Sonka K, et al. Subjective and polysomnographic evaluation of sleep in mitochondrial optic neuropathies. J Sleep Res. 2021;30(2):e13051 A study of the prevalence of sleep disorders in mitochondrial optic neuropathies.
Sateia MJ. International classification of sleep disorders-third edition: highlights and modifications. Chest. 2014;146(5):1387–94.
Joosten SA, O'Driscoll DM, Berger PJ, Hamilton GS. Supine position related obstructive sleep apnea in adults: pathogenesis and treatment. Sleep Med Rev. 2014;18(1):7–17.
Henke KG, Dempsey JA, Kowitz JM, Skatrud JB. Effects of sleep-induced increases in upper airway resistance on ventilation. J Appl Physiol. 1990;69(2):617–24.
Tabachnik E, Muller NL, Bryan AC, Levison H. Changes in ventilation and chest wall mechanics during sleep in normal adolescents. J Appl Physiol Respir Environ Exerc Physiol. 1981;51(3):557–64.
Gutierrez G, Williams J, Alrehaili GA, McLean A, Pirouz R, Amdur R, et al. Respiratory rate variability in sleeping adults without obstructive sleep apnea. Physiol Rep. 2016;4(17):e12949.
Dubois M, Chenivesse C, Raux M, Morales-Robles A, Nierat MC, Garcia G, et al. Neurophysiological evidence for a cortical contribution to the wakefulness-related drive to breathe explaining hypocapnia-resistant ventilation in humans. J Neurosci. 2016;36(41):10673–82.
Douglas NJ, White DP, Pickett CK, Weil JV, Zwillich CW. Respiration during sleep in normal man. Thorax. 1982;37(11):840–4.
Aboussouan LS. Sleep-disordered breathing in neuromuscular disease. Am J Respir Crit Care Med. 2015;191(9):979–89.
Quera-Salva MA, Guilleminault C, Chevret S, Troche G, Fromageot C, Crowe McCann C, et al. Breathing disorders during sleep in myasthenia gravis. Ann Neurol. 1992;31(1):86–92.
Mermigkis C, Bouloukaki I, Mastorodemos V, Plaitakis A, Alogdianakis V, Siafakas N, et al. Medical treatment with thiamine, coenzyme Q, vitamins E and C, and carnitine improved obstructive sleep apnea in an adult case of Leigh disease. Sleep Breath. 2013;17(4):1129–35.
Tan AL, Goy R. Anaesthetic management of a patient with Leigh’s syndrome with central hypoventilation and obstructive sleep apnoea. Singap Med J. 2013;54(12):e250–3.
Yasaki E, Saito Y, Nakano K, Katsumori H, Hayashi K, Nishikawa T, et al. Characteristics of breathing abnormality in Leigh and its overlap syndromes. Neuropediatrics. 2001;32(6):299–306.
Sanaker PS, Husebye ES, Fondenes O, Bindoff LA. Clinical evolution of Kearns-Sayre syndrome with polyendocrinopathy and respiratory failure. Acta Neurol Scand Suppl. 2007;187:64–7.
Sembrano E, Barthlen GM, Wallace S, Lamm C. Polysomnographic findings in a patient with the mitochondrial encephalomyopathy NARP. Neurology. 1997;49(6):1714–7.
Thorpy MJ. Classification of sleep disorders. Neurotherapeutics. 2012;9(4):687–701.
Barohn RJ, Clanton T, Sahenk Z, Mendell JR. Recurrent respiratory insufficiency and depressed ventilatory drive complicating mitochondrial myopathies. Neurology. 1990;40(1):103–6.
Carroll JE, Zwillich C, Weil JV, Brooke MH. Depressed ventilatory response in oculocraniosomatic neuromuscular disease. Neurology. 1976;26(2):140–6.
Osanai S, Takahashi T, Enomoto H, Satoh N, Yahara O, Akiba Y, et al. Hypoxic ventilatory depression in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. Respirology. 2001;6(2):163–6.
Johnston K, Newth CJ, Sheu KF, Patel MS, Heldt GP, Schmidt KA, et al. Central hypoventilation syndrome in pyruvate dehydrogenase complex deficiency. Pediatrics. 1984;74(6):1034–40.
Sadler M, Wiles CM, Stoodley N, Linnane SJ, Smith AP. Ondine’s curse in a woman with Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. 2002;73(3):347–8.
Tatsumi C, Takahashi M, Yorifuji S, Nishikawa Y, Kitaguchi M, Hashimoto S, et al. Mitochondrial encephalomyopathy with sleep apnea. Eur Neurol. 1988;28(2):64–9.
Hernandez-Voth A, Sayas Catalan J, Corral Blanco M, Castano Mendez A, Martin MA, De Fuenmayor Fernandez de la Hoz C, et al. Deoxynucleoside therapy for respiratory involvement in adult patients with thymidine kinase 2-deficient myopathy. BMJ Open Respir Res. 2020;7(1):e000774.
Vetrugno R, Valentino ML, La Morgia C, Liguori R, Stecchi S, Mascalchi M, et al. Sleep-related periodic respiration with central sleep apnea in Leber hereditary optic neuropathy (LHON). Sleep Med. 2010;11(4):426–7.
Eckert DJ, Jordan AS, Merchia P, Malhotra A. Central sleep apnea: pathophysiology and treatment. Chest. 2007;131(2):595–607.
Shuk-kuen Chau C, Kwok KL, Ng DK, Lam CW, Tong SF, Chan YW, et al. Maternally inherited Leigh syndrome: an unusual cause of infantile apnea. Sleep Breath. 2010;14(2):161–5.
Guilleminault C, Philip P, Robinson A. Sleep and neuromuscular disease: bilevel positive airway pressure by nasal mask as a treatment for sleep disordered breathing in patients with neuromuscular disease. J Neurol Neurosurg Psychiatry. 1998;65(2):225–32.
Stefani A, Hogl B. Diagnostic criteria, differential diagnosis, and treatment of minor motor activity and less well-known movement disorders of sleep. Curr Treat Options Neurol. 2019;21(1):1.
Aitken H, Gorman G, McFarland R, Roberts M, Taylor RW, Turnbull DM. Clinical reasoning: blurred vision and dancing feet: restless legs syndrome presenting in mitochondrial disease. Neurology. 2009;72(18):e86–90.
Earley CJ, Kuwabara H, Wong DF, Gamaldo C, Salas R, Brasic J, et al. The dopamine transporter is decreased in the striatum of subjects with restless legs syndrome. Sleep. 2011;34(3):341–7.
Haschka D, Volani C, Stefani A, Tymoszuk P, Mitterling T, Holzknecht E, et al. Association of mitochondrial iron deficiency and dysfunction with idiopathic restless legs syndrome. Mov Disord. 2019;34(1):114–23.
Pincherle A, Mantoani L, Villani F, Confalonieri P, Erbetta A. Excessive fragmentary hypnic myoclonus in a patient affected by a mitochondrial encephalomyopathy. Sleep Med. 2006;7(8):663.
Suzuki Y, Taniyama M, Hata T, Miyaoka H, Atsumi Y, Matsuoka K. Sleep-wake dysrhythm in mitochondrial diabetes mellitus. Diabetes Res Clin Pract. 1997;35(1):61–2.
Sardon Puig L, Valera-Alberni M, Canto C, Pillon NJ. Circadian rhythms and mitochondria: connecting the dots. Front Genet. 2018;9:452.
Araki S, Hayashi M, Yasaka A, Maruki K. Electrophysiological brainstem dysfunction in a child with Leigh disease. Pediatr Neurol. 1997;16(4):329–33.
Kotagal S, Archer CR, Walsh JK, Gomez C. Hypersomnia, bithalamic lesions, and altered sleep architecture in Kearns-Sayre syndrome. Neurology. 1985;35(4):574–7.
Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–33.
Catteruccia M, Sauchelli D, Della Marca G, Primiano G, Cuccagna C, Bernardo D, et al. “Myo-cardiomyopathy” is commonly associated with the A8344G “MERRF” mutation. J Neurol. 2015;262(3):701–10.
Orsucci D, Angelini C, Bertini E, Carelli V, Comi GP, Federico A, et al. Revisiting mitochondrial ocular myopathies: a study from the Italian Network. J Neurol. 2017;264(8):1777–84.
Davis RE, Williams M. Mitochondrial function and dysfunction: an update. J Pharmacol Exp Ther. 2012;342(3):598–607.
Rodrigues NR, Macedo GE, Martins IK, Gomes KK, de Carvalho NR, Posser T, et al. Short-term sleep deprivation with exposure to nocturnal light alters mitochondrial bioenergetics in Drosophila. Free Radic Biol Med. 2018;120:395–406.
Lu C, Lv J, Dong L, Jiang N, Wang Y, Fan B, et al. The protective effect of 20(S)-protopanaxadiol (PPD) against chronic sleep deprivation (CSD)-induced memory impairments in mice. Brain Res Bull. 2018;137:249–56.
Trivedi MS, Holger D, Bui AT, Craddock TJA, Tartar JL. Short-term sleep deprivation leads to decreased systemic redox metabolites and altered epigenetic status. PLoS One. 2017;12(7):e0181978.
Zhou L, Chen P, Peng Y, Ouyang R. Role of oxidative stress in the neurocognitive dysfunction of obstructive sleep apnea syndrome. Oxidative Med Cell Longev. 2016;2016:9626831.
Wrede JE, Mengel-From J, Buchwald D, Vitiello MV, Bamshad M, Noonan C, et al. Mitochondrial DNA copy number in sleep duration discordant monozygotic twins. Sleep. 2015;38(10):1655–8.
Acknowledgements
This work is generated within the European Reference Network for Neuromuscular Diseases.
Funding
Open access funding provided by Università Cattolica del Sacro Cuore within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Sleep
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brunetti, V., Della Marca, G., Servidei, S. et al. Sleep Disorders in Mitochondrial Diseases. Curr Neurol Neurosci Rep 21, 30 (2021). https://doi.org/10.1007/s11910-021-01121-2
Accepted:
Published:
DOI: https://doi.org/10.1007/s11910-021-01121-2