
Abstract
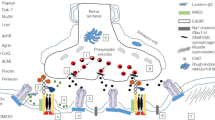
Congenital myasthenic syndromes (CMS) represent a heterogeneous group of disorders in which the safety margin of neuromuscular transmission is compromised by one or more specific mechanisms. Clinical, electrophysiologic, and morphologic studies have paved the way for detecting CMS-related mutations in proteins residing in the nerve terminal, the synaptic basal lamina, or in the postsynaptic region of the motor endplate. The disease proteins identified to date include the acetylcholine receptor, acetylcholinesterase, choline acetyltransferase, rapsyn, and Nav1.4, muscle-specific kinase, agrin, β2-laminin, downstream of tyrosine kinase 7, and glutamine-fructose-6-phosphate transaminase 1. Analysis of electrophysiologic and biochemical properties of mutant proteins expressed in heterologous systems have contributed crucially to defining the molecular consequences of the observed mutations and have resulted in improved therapy of most CMS.
Similar content being viewed by others

References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Rothbart HB. Myasthenia gravis. Familial occurrence. JAMA. 1937;108:715–7.
Selcen D, Juel VC, Hobson-Webb LD, et al. Myasthenic syndrome caused by plectinopathy. Neurology. 2011;76:327–36.
Tsujino A, Maertens C, Ohno K, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA. 2003;100:7377–82.
Engel AG, Ohno K, Wang H-L, et al. Molecular basis of congenital myasthenic syndromes: Mutations in the acetylcholine receptor. Neuroscientist. 1998;4:185–94.
Ohno K, Brengman JM, Tsujino A, Engel AG. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc Natl Acad Sci USA. 1998;95:9654–9.
•• Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–8.
Kaiser J. Affordable ‘exomes’ fill gaps in a catalog of rare diseases. Science. 2010;330:903.
Walls TJ, Engel AG, Nagel AS, et al. Congenital myasthenic syndrome associated with paucity of synaptic vesicles and reduced quantal release. Ann N Y Acad Sci. 1993;681:461–8.
Ohno K, Tsujino A, Shen XM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA. 2001;98:2017–22.
Schara U, Christen H-J, Durmus H, et al. Long-term follow-up in patients with congenital myasthenic syndrome due to CHAT mutations. Eur J Paediatr Neurol. 2010;14:326–33.
• Shen X-M, Crawford TO, Brengman J, et al. Functional consequences and structural interpretation of mutations in human choline acetyltransferase. Hum. Mutat. Published online on 23 Sep 2011. This paper identifies divergent effects of different ChAT mutations on enzyme activation and shows that the most severely affected patients carry at least one mutation near the active site of the enzyme.
Massoulié J, Pezzementi L, Bon S, et al. Molecular and cellular biology of cholinesterases. Prog Neurobiol. 1993;41:31–91.
Bon S, Coussen F, Massoulié J. Quaternary associations of acetylcholinesterase. II. The polyproline attachment domain of the collagen tail. J Biol Chem. 1997;272:3016–21.
Deprez PN, Inestrosa NC. Two heparin-binding domains are present on the collagenic tail of asymmetric acetylcholinesterase. J Biol Chem. 1995;270:11043–6.
Cartaud A, Strochlic L, Guerra M, et al. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J Cell Biol. 2004;165:505–15.
Kimbell LM, Ohno K, Engel AG, Rotundo RL. C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. J Biol Chem. 2004;279:10997–1005.
Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nature Rev Neurosci. 2003;4:339–52.
Bestue-Cardiel M, de-Cabazon-Alvarez AS, Capablo-Liesa JL, et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology. 2005;65:144–6.
Mihaylova V, Muller JS, Vilchez JJ, et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain. 2008;131:747–59.
• Liewluck T, Selcen D, Engel AG. Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and DOK-7 myasthenia. Muscle Nerve. Published online 23 SEP 2011. This paper reports significant therapeutic response in two CMS to a readily available medication.
Maselli RA, Ng JJ, Andreson JA, et al. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J Med Genet 2009, 46:203–208.
Harper CM, Engel AG. Treatment of 31 congenital myasthenic syndrome patients with 3,4-diaminopyridine. Neurology. 2000; 54(Suppl 3):A395 (Abstr.)
Sadeh M, Shen X-M, Engel AG. Beneficial effect of albuterol in congenital myasthenic syndrome with epsilon subunit mutations. Muscle Nerve In press. 2011.
Ohno K, Hutchinson DO, Milone M, et al. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the ε subunit. Proc Natl Acad Sci USA. 1995;92:758–62.
Harper CM, Engel AG. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Ann Neurol. 1998;43:480–4.
Harper CM, Fukudome T, Engel AG. Treatment of slow channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:170–3.
Shen X-M, Fukuda T, Ohno K, et al. Congenital myasthenia-related AChR δ subunit mutation interferes with intersubunit communication essential for channel gating. J Clin Invest. 2008;118:1867–76.
Sine SM, Engel AG. Recent advances in Cys-loop receptor structure and function. NAT. 2006;440:448–55.
Engel AG, Brengman J, Edvardson S, Shen X-M. Highly fatal low-affinity fast-channel congenital myasthenic syndrome caused by a novel AChR epsilon subunit mutation at the agonist binding site. Neurology. 2011;76 Suppl 4:A644.
Froehner SC, Luetje CW, Scotland PB, Patrick J. The postsynaptic 43 K protein clusters muscle nicotinic acetylcholine receptors in Xenopus oocytes. Neuron. 1990;5:403–10.
Cartaud A, Coutant S. Petrucci TC, Cartaud J: Evidence for in situ and in vitro association between β-dystroglycan and the subsynaptic 43K rapsyn protein. Consequence for acetylcholine receptor clustering at the synapse. J Biol Chem. 1998;273:11321–6.
Okada K, Inoue A, Okada M, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–5.
Zhang B, Luo S, Wang Q, et al. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–97.
Burke G, Cossins J, Maxwell S, et al. Rapsyn mutations in hereditary myasthenia. Distinct early- and late-onset phenotypes. Neurology. 2003;61:826–8.
Ohno K, Engel AG, Shen X-M, et al. Rapsyn mutations in humans cause endplate acetylcholine receptor deficiency and myasthenic syndrome. Am J Hum Genet. 2002;70:875–85.
Milone M, Shen XM, Selcen D, et al. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology. 2009;73:228–35.
Müller JS, Mildner G, Müller-Felber W, et al. Rapsyn N88K is a frequent cause of CMS in European patients. Neurology. 2003;60:1805–11.
Ohno K, Sadeh M, Blatt I, et al. E-box mutations in RAPSN promoter region in eight cases with congenital myasthenic syndrome. Hum Mol Genet. 2003;12:739–48.
Banwell BL, Ohno K, Sieb JP, Engel AG. Novel truncating RAPSN mutation causing congenital myasthenic syndrome responsive to 3,4-diaminopyridine. Neuromuscul Disord. 2004;14:202–7.
Milone M, Shen XM, Selcen D, et al. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology 2009;73:228-235
Kim N, Stiegler AL, Cameron TO, et al. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–42.
Hallock PT, Xu C-F, Neubert TA, Curran T. Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes Dev. 2010;24:2451–61.
Linnoila J, Wang Y, Yao Y, Wang Z-S. A mammalian homolog of Drosophila tumorous imaginal disks, Tid1, mediates agrin signaling at the neuromuscular junction. Neuron. 2010;60:625–41.
Huze C, Bauche S, Richard P, et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. 2009;85:155–67.
Chevessier F, Faraut B, Ravel-Chapuis A, et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum Mol Genet. 2004;13:3229–40.
• Maselli R, Arredondo J, Cagney O, et al.: Mutations in MUSK causing congenital myasthenic syndrome impair MuSK-Dok-7 interaction. Hum Mol Genet 2010, 19:2370–2379. This paper provides important insight into the mechanism of interaction between MuSK and Dok-7.
Mihaylova V, Salih MA, Mukhtar MM, et al. Refinement of the clinical phenotype in Musk-related congenital myasthenic syndromes. Neurology. 2009;73:1926–8.
•• Selcen D, Milone M, Shen X-M, et al.: Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann Neurol 2008, 64:71–87. This paper shows that Dok-7 is important not only for the development but also for the maintenace of the neuromuscular junction throughout life and that some mutations can be detected only by cloning patient complementary DNA.
Lashley D, Palace J, Jayawant S, et al. Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology. 2010;74:1517–23.
Slater CR, Fawcett PRW, Walls TJ, et al. Pre- and postsynaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain. 2006;127:2061–76.
•• Senderek J, Muller JS, Dusl M, et al.: Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet 2011, 88:162–172. This paper describes a novel myasthenic syndrome with a limb-girdle phenotype associated with tubular aggregates in muscle caused by a defect in protein glycosylation.
Banwell BL, Russel J, Fukudome T, et al. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J Neuropathol Exp Neurol. 1999;58:832–46.
Forrest K, Mellerio JE, Robb S, et al. Congenital muscular dystrophy, myasthenic symptoms and epidermolysis bullosa simplex (EBS) associated with mutations in the PLEC1 gene encoding plectin. Neuromuscul Disord. 2010;20:709–11.
Maselli R, Arredondo J, Cagney O, et al. Congenital myasthenic syndrome associated with epidermolysis bullosa caused by homozygous mutaions in PLEC1 and CHRNE. Clin. Genet. 2010 In press.
Romero NB. Centronucelar myopathies: a widening concept. Neuromuscul Disord. 2010;20:223–8.
Claeys KG, Maisonobe T, Bohm J, et al. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology. 2010;74:519–21.
Baradello A, Vita G, Girlanda P, et al. Adult-onset centronuclear myopathy: evidence against a neurogenic pathology. Acta Neurol Scand. 1989;80:162–6.
• Liewluck T, Shen X-M, Milone M, Engel AG: Endplate structure and parameters of neuromuscular transmission in sporadic centronuclear myopathy associated with myasthenia. Neuromuscul Disord 2011, 21:387–395. This paper identifies both pre- and postsynaptic changes accounting for the defect in neuromuscular transmission.
Robb SA, Sewry CA, Dowling JJ, et al. Impaired neuromuscular transmission and response to aceylcholinesterase inhibitors in centronuclear myopathy. Neuromuscul Disord. 2011;21:379–86.
Oskoui M, Jacobson L, Chung WK, et al. Fetal acetylcholine receptor inactivation syndrome and maternal myasthenia gravis. Neurology. 2008;71:2010–2.
Reimann J, Jacobson L, Vincent A, Kornblum C. Endplate destruction due to maternal antibodies in arthrogryposis multiplex congenita. Neurology. 2009;73:1806–8.
Hesselmans LFGM, Jennekens FGI, Vand Den Oord CJM, et al. Development of innervation of skeletal muscle fibers in man: Relation to acetylcholine receptors. Anat Rec. 1993;236:553–62.
Hoffmann K, Muller JS, Stricker S, et al. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am J Hum Genet. 2006;79:303–12.
Morgan NV, Brueton LA, Cox P, et al. Mutations in the embryonal subunit of the acetylcholine receptor (CHNRG) cause lethal and Escobar variants of the multiple pterygium syndrome. Am J Hum Genet. 2006;79:390–5.
• Michalk A, Stricker S, Becker J, et al.: Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am J Hum Genet 2008, 82:464–476. This paper identifies heretofore unsuspected causes of the fetal dyskinesia and deformation syndrome causing myasthenia in utero.
Vogt J, Harrison BJ, Spearman H, et al. Mutation Analysis of CHRNA1, CHRNB1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am J Hum Genet. 2008;82:222–7.
Vogt J, Morgan NV, Marton T, et al. Germline mutation in DOK7 associated with fetal akinesia deformation sequence. J Med Genet. 2009;46:338–40.
Acknowledgment
Work in the author’s laboratory was supported by a National Institutes of Health Research Grant NS6277 and by the Muscular Dystrophy Association.
Disclosure
Conflicts of interest: A.G. Engel: has received honorarium from the American Academy of Neurology for serving on the editorial board of the journal Neurology; and has received royalties from McGraw-Hill for editing the textbook entitled “Myology.”
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Engel, A.G. Congenital Myasthenic Syndromes in 2012. Curr Neurol Neurosci Rep 12, 92–101 (2012). https://doi.org/10.1007/s11910-011-0234-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-011-0234-7