
Abstract
Blood pressure is a way of describing the end result of changes in cardiac output, intravascular volume and peripheral resistance. It has certain advantages in that it is a reproducible and easily measured parameter, but in itself, it offers only a limited understanding of the underlying haemodynamics. In pregnancy, profound haemodynamic changes occur and in hypertensive diseases of pregnancy defining a condition by blood pressure alone risks missing the underlying cause. Partly, this has been a problem of ascribing the cause of hypertensive syndromes to the placenta which has inhibited rigorous research into other possible causes of haemodynamic dysfunction. It is becoming increasingly evident that hypertension in pregnancy may be associated with primarily high cardiac output or high peripheral resistance. A knowledge of the underlying type of hypertension may allow more rational treatment of these conditions in pregnancy rather than therapeutic attempts at controlling blood pressure by any means possible as an end in itself.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypertension in Pregnancy: Definitions and History
Hypertension in pregnancy remains a ‘cinderella area’ of pathophysiology in clinical obstetrics. For many decades—and still now—the acute pathological changes that compromise the maternal cardiovascular system during pregnancy with possible devastating tissue and organ dysfunction remained firmly rooted to the nineteenth century criteria of high blood pressure and proteinuria, both ‘tertiary, downstream features of the disease’ [1]. The profusion of names for this condition, pre-eclampsia, toxaemia, gestosis and proteinuric pregnancy induced hypertension, underline the task of understanding such a condition whose diagnosis is based purely on the easily measurable manifestations of complex, possibly distinctly different causes of high blood pressure in pregnancy.
Let us consider the very name pre-eclampsia, literally a disease that occurs prior to eclampsia (‘προ-ἐκλαμψία’, ‘before-sudden occurrence’). This term was introduced by Johannes Varandaeus in the year 1620 treatise ‘Tractatus (of gynaecology) de affectibus Renum et Vesicae’. More than one century later, proteinuria was described in relation to fluid accumulation during pregnancy resulting in acute fits and possibly death if the fetus and placenta are not delivered. Pierre Rayer (1793–1867), a Frenchman, was the first to describe proteinuria in eclamptics, cited in his then classic text ‘Diseases of the Kidney’ [2]. It took a few more years for Scipione Riva-Rocci’s mercury manometer (1896) to be introduced and allowed to recognize that pre-eclampsia was a hypertensive disorder. It is surprising that in 2017, this condition is still classified according to Varandeus-Rayer and Riva Rocci. It is tempting to speculate that the consequences of defining hypertensive disorder according to such arcane criteria (ICD-10 code 014) have prevented a rational categorization of the condition based on cause, not effect.
But names and definitions are important. Indeed, some guidelines include almost everything from headache to liver damage to disseminated intravascular coagulation; intra uterine growth restriction (IUGR) plus hypertension qualifies for pre-eclampsia (PE) even without proteinuria. Broader names better fit broader criteria. We opt here for considering all these conditions within the collective term hypertensive disorders of pregnancy (HDP). The definition of HDP covers pregnancy induced hypertension, otherwise known as gestational hypertension (GH), all conditions of GH associated with maternal and foetal complications, among which can occur a typical renal glomerular lesion that causes proteinuria, and chronic hypertension (CH) (existing prior to pregnancy).
The ‘Trophoblast Invasion’ Aetiological Theory
In 1975, Brosens I and Renaer M [3] published a key paper on ‘The pathogenesis of placentae infarcts in preeclampsia’. These concepts were later developed by Khong TY, De Wolf F, Robertson WB and still among the authors, Brosens [4]. In this latter work, pathologic findings on pre-eclamptic placentae were associated with small-for-gestational-age infants. This particular link between shallow trophoblastic invasion in early gestation, placental insufficiency, led to a focused scientific insight into this possible origin of maternal endothelial dysfunction and hypertension associated with IUGR. The link between such characteristics of poor placental development in the first trimester and ‘preeclampsia’ occurring early in gestation associated with IUGR was reviewed by Burton, Woods, Jauniaux and Kingdom in 2009 [5]. Doppler interrogation of uterine arteries has for three decades been widely studied as a possible proxy of these pathological changes occurring in early placental development: foetal growth and maternal hypertension [6,7,8].
Unfortunately, the theory [8], molecular [9] and histopathological [5] had hardly developed scientifically in 30 years and has persuaded many to consider this clinical phenotype the sole cause of pre-eclampsia by those that uncritically accepted association as equivalent to causation. Time at onset of the clinical signs and symptoms (before and after 34 weeks of gestation) became the criterion on which to define its severity. ‘Consequently, PE could be considered as a single pathophysiological entity with a wide spectrum of severity manifested in gestational age at which delivery becomes necessary for maternal and/or foetal indication’ [10]. This interpretation of pre-eclampsia is in striking contrast with epidemiological, pathological and clinical evidence and has arguably held up the proper understanding of the origin of both hypertensive disease and fetal growth restriction which are frequently closely related.
Pathophysiology and Clinical Phenotype: Towards a New Definition
Apparently anomalous characteristics of the condition are that whilst in early onset pre-eclampsia, birthweight is reduced, at term the condition is associated with normal or large weight babies [11,12,13, 14•]. Other possible explanations—for example underlying cardiovascular dysfunction or metabolic syndrome—do not contradict the relationship between early placental shallow trophoblast invasion and subsequent maternal endothelial damage. They do however suggest a different clinical phenotype of pre-eclampsia where maternal risk factors appears to play a predominant role, and both placental and newborn weight appears to be normal or even large. The existence of two main placental diseases had been recently reviewed by Redman and Staff [15]. Both placental lesions, early shallow trophoblastic invasion and late villi crowding generate a similar oxidative stress that damage the maternal endothelium that magnify underlying maternal risks; these might be so severe as to require delivery prior to 34 weeks of gestation [16].
The obvious limitation of current definitions is that irrespective of the type of hypertension, women with identical systolic pressures are considered the same as each other, when their haemodynamics (cardiac output, stroke volume, heart rate, vascular resistance and arterial function) may be significantly different. Importantly, all recent studies [17,18,19,20,21] on maternal haemodynamics suggest profound differences from normal pregnancies in cardiac output, vascular resistance and arterial function depending on the type of pregnancy hypertension and the gestation at which it occurs. The fundamental problem is that hypertension itself is characterized as the problem rather than the manifestation of an underlying cardiovascular dysfunction.
In the clinical arena, the ongoing debate on how to re-classify and interpret pre-eclampsia might be summarized by the recently published Co-Lab statement: ‘Despite intensive study, we have not been able to improve the management or early recognition of preeclampsia…. It is possible that within the syndrome, there may be different phenotypes with pathogenic pathways that differ between the subtypes. The capacity to recognize and to exploit different subtypes is of obvious importance for prediction, prevention, and treatment’ [22]. These strong statements that challenge the validity of the long trial of knowledge accumulated on pre-eclampsia were anticipated in 2010 by Steegers and co-workers on a review paper on the Lancet [23]: ‘Poor early placentation is especially associated with early onset disease (of pre-eclampsia before 34 weeks of gestation). Predisposing cardiovascular or metabolic risks for endothelial dysfunction,…, might dominate in the origins of late onset pre-eclampsia (after 34 weeks of gestation).’
Cardiovascular Function and Pre-eclampsia
Cardiac output (CO) increases during pregnancy and plateaus in the late second and early third trimester, 1.5 L/min (31%) above non-pregnant values [24•]. At 28 weeks of gestation, the percentage of total maternal CO to the uterus is approximately 12% [25•]. This increase is sustained both by the increasing heart rate (HR) and stroke volume (SV). Multiparity and multifetal pregnancies significantly influence these indices. Diastolic blood pressure shows a decrease that already starts in early first trimester and extends until the late second trimester [24•]. Total vascular resistance (TVR) decreases until the early second trimester. As a consequence, we usually observe a decrease in diastolic blood pressure. Mean arterial pressure (MAP) starts to increase towards term as compared to non-pregnant data. In this adaptation process, the venous system increases in venous capacitance, due to enhanced venous distensibility, and mostly the splanchnic system accommodates the normal plasma volume expansion [26•]. A recent meta-analysis calculated an average increase in plasma volume of 45.6% [27•].
Cardiovascular remodelling involves the heart itself. The left ventricular mass (LVM) increases to values 30–40% above non-pregnant values with a concentric hypertrophy since the first two trimesters of pregnancy [28•]. This dramatic increase outperforms that achieved only by exercise by athletes, who might gain 25% eccentric remodelling [28•]. The left atrial diameter is already significantly enlarged during the first weeks of pregnancy [29•, 30•].
It is plausible that a case of pre-eclampsia that occurs earlier in gestation and is associated with fetal growth restriction is related to low cardiac output and high peripheral vascular resistance [31] with a much similar profile as observed in women with fetal growth restriction without HDP [32]. In cases of later and term gestation pre-eclampsia, babies tend to be larger and there is a predominantly high cardiac output, low peripheral vascular resistance and raised intravascular volume state [33]. Certainly, the clinical phenotype of a very ‘dry’, intravascularly depleted woman at 26 weeks with a growth restricted baby and conversely of a well-perfused oedematous woman with a bounding pulse and large baby at 38 weeks rings true: both have hypertension, but the mechanisms may be diametrically opposite.
Implications for Pharmacotherapy
It is plausible to consider that if these diametrically opposed cardiovascular states both cause hypertension, then the pharmacological treatment for one condition will not necessarily cure the other. Beta blockers, combined alpha and beta blockers, calcium antagonists, adrenergic false transmitters, presynaptic alpha receptor modulators, nitrates and diuretics have all been used to treat hypertensive pregnancy. International guidelines (NICE, ACOG) have in recent years supported a simplification of the therapeutic options which in the UK favour labetalol as a first line agent, with alpha methyl dopa as second line, a place for calcium antagonists and almost no place for diuretics. An inherent difficulty is that though these agents have quite different modes of action, they are used interchangeably. Further, national guidance is based on little data: studies of hypertensive agents of the scale performed in the general adult population do not exist hence empirical advice is given on the basis of what seems to be safe clinical practice and unlikely to harm the foetus. The major problem is that hypertension per se is considered to be the goal of therapy, irrespective of the underlying cause. The recent CHIPS study [34] investigated whether ‘tight’ blood pressure control conferred a benefit in outcome in women with hypertension in pregnancy, concluding that it did not. Would it not have been instructive in determining benefit to understand the underlying differences in womens’ cardiovascular status and in this light the response to treatment?
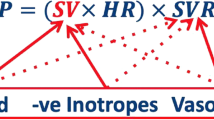
Contrast this approach with rapidly evolving specialist adult hypertensive practice. Knowledge of whether the condition is ‘high output’ or ‘high resistance’ allows selection of the anti-hypertensive agent that will work in the most physiological way—not by targeting blood pressure—but more the underlying condition [35, 36]. Non-invasive devices, now readily available and validated in pregnancy, give accurate cardiac output and other data and allow profiling of arterial function in hypertension [37]. In a low cardiac output, high vascular resistance scenario, a negatively inotropic alpha/beta blocker such as labetalol may make the haemodynamics worse whilst achieving the therapeutic goal of lowering blood pressure; a calcium channel blocker may be far better. In the well-perfused term, woman with hypertension and a high cardiac output, labetalol and a diuretic may be therapeutically better suited than a vasodilator—where there is already vasodilatation. It may also be that similar to anti-hypertensive therapy in the general population, prescription of the ‘wrong’ drug for the haemodynamic abnormality present may, due to physiological responses to treatment, make women more likely to be resistant to subsequent therapies. This is not to mention the fetal-placental unit, where a reduction in perfusion might have serious consequences.
Conclusions
The current therapeutic approach, a ‘one size fits all’, is certainly too simplistic: why not tailor therapeutic approaches to haemodynamic profile rather than blood pressure alone? It is now timely to define hypertensive conditions in pregnancy based on the cardiovascular profile, treat women and perform studies based on rational assessment of pharmacological action—not purely of anti-hypertensive efficacy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Chappell LC, Duckworth S, Seed PT, Griffin M, Myers J, Mackillop L, et al. Diagnostic accuracy of placental growth factor in women with suspected preeclampsia: a prospective multicenter study. Circulation. 2013;128(19):2121–31.
Pierre-François Rayer (1793-1867) Physician to the kings of France. JAMA 1965;191(10):856. doi: 10.1001/jama.1965.03080100074023.
Brosens I, Renear M. In the pathogenesis of placentae infarcts in preeclampsia. Br J Obstet Gynaecol. 1975;79:794–9.
Khong TY, De Wolf F, Brosens I. Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational-age infants. Br J Obstet Gynaecol. 1986;93:1049–59.
Burton GJ, Woods AW, Jauniaux E, Kingdom JCP. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. 2010;30(6):473–82.
Ferrazzi E, Bulfamante G, Mezzopane R, Barbera A, Ghidini A, Pardi G. Uterine Doppler velocimetry and placental hypoxic-ischemic lesion in pregnancies with fetal intrauterine growth restriction. Placenta. 1999;20(5–6):389–94.
Aardema MW, Oosterhof H, Timmer A, van Rooy I, Aarnoudse JG. Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta. 2001;22(5):405–11.
Papageorghiou AT, Yu CKH, Nicolaides KH. The role of uterine artery Doppler in predicting adverse pregnancy outcome. Best Pract Res Clin Obstet Gynaecol. 2004;18(3):383–96.
Levine RJ, Maynard SE, Qian C, Lim K-H, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia. New England Journal of Medicine. Mass Medical Soc; 2004;350(7):672–683.
Wright D, Akolekar R, Syngelaki A, Poon LCY, Nicolaides KH. A competing risks model in early screening for preeclampsia. Fetal Diagn Ther. 2012;32(3):171–8.
Conde-Agudelo A, Belizan JM. Risk factors for pre-eclampsia in a large cohort of Latin American and Caribbean women. BJOG: Int J O&G. 2000;107(1):75–83.
Vadillo-Ortega F, Perichart-Perera O, Espino S, Avila-Vergara MA, Ibarra I, Ahued R, et al. Effect of supplementation during pregnancy with L-arginine and antioxidant vitamins in medical food on pre-eclampsia in high risk population: randomised controlled trial. BMJ [Internet]. 2011;19;342(may19 1):d2901–1. doi:10.1136/bmj.d2901.
Ye C, Ruan Y, Zou L, Li G, Li C, Chen Y, et al. The 2011 survey on hypertensive disorders of pregnancy (HDP) in China: prevalence, risk factors, complications, pregnancy and perinatal outcomes. Obukhov AG, editor. PLoS One 2014;9(6):e100180–e100189.
• Terefe W, Getachew Y, Hiruye A. Patterns of hypertensive disorders of pregnancy and associated factors at debre berhan referral hospital, north shoa, amhara region. Ethiop Med J. 2015; Vol. Suppl 2: p. 5765. These three references (12,13,14) show how in areas other then developed western countries Hypertensive Disorders of Pregnancy might be severe, associated with proteinuria, normally grown fetuses, and becoming clinically manifest late in gestation.
Redman CW, Sargent IL, Staff AC. IFPA senior award lecture: making sense of pre-eclampsia. Placenta Elsevier Ltd. 2014;35(S):S20–5.
Oliveira N, Magder LS, Blitzer MG, Baschat AA. First-trimester prediction of pre-eclampsia: external validity of algorithms in a prospectively enrolled cohort. 2014;44(3):279–85.
Easterling TR, Benedetti TJ, Schmucker BC, Millard SP. Maternal hemodynamics in normal and preeclamptic pregnancies: a longitudinal study. Obstet Gynecol. 1990;76:1061–9.
Tomsin K, Mesens T, Molenberghs G, Peeters L, Gyselaers W. Characteristics of heart, arteries, and veins in low and high cardiac output preeclampsia. Eur J Obstet Gynecol Reprod Biol. 2013;169:218–22.
Bamfo JE, Kametas NA, Chambers JB, Nicolaides KH. Maternal cardiac function in normotensive and pre-eclamptic intrauterine growth restriction. Ultrasound Obstet Gynecol. 2008;32:682–6.
Valensise H, Vasapollo B, Gagliardi G, Novelli GP. Early and late preeclampsia: two different maternal hemodynamic states in the latent phase of the disease. Hypertension. 2008;52:873–80.
Bamfo JE, Kametas NA, Chambers JB, Nicolaides KH. Maternal cardiac function in fetal growth-restricted and non-growth-restricted small-for-gestational age pregnancies. Ultrasound Obstet Gynecol. 2007;29:51–7.
Myatt L, Redman CW, Staff AC, Hansson S. Strategy for standardization of preeclampsia research study design. Hypertension. 2014;6332(6):1293–301.
Steegers EA, Dadelszen von P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376(9741):631–44.
• Meah VL, Cockcroft JR, Backx K, Shave R, Stöhr EJ. Cardiac output and related haemodynamics during pregnancy: a series of meta-analyses. Heart. 2016;102(7):518–26.A seminal meta-analysis of published works on cardiovascular changes in pregnancy.
• Flo K, Wilsgaard T, Vårtun A, Acharya G. A longitudinal study of the relationship between maternal cardiac output measured by impedance cardiography and uterine artery blood flow in the second half of pregnancy. BJOG: Int J O&G. 2010;117(7):837–44. This work provides the required physiological background to the daily users of Doppler velocimetry of uterine arteries. The link between maternal cardiac function and the placenta.
• Gyselaers W, Mullens W, Tomsin K, Mesens T, Peeters L. Role of dysfunctional maternal venous hemodynamics in the pathophysiology of pre-eclampsia: a review. 2011;38(2):123–9. The venous system is a forgotten player of maternal physiology in pregnancy. This provide the background needed to understand the dysfunctional arterial performance in pre-eclampsia and the venous system.
• de Haas S, Ghossein-Doha C, van Kuijk SMJ, van Drongelen J, Spaanderman MEA. Physiological adaptation of maternal plasma volume during pregnancy: a systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2017;49(2):177–87. The venous capacitance role is an essential part of the dramatic cardiovascular changes in pregnancy. De Haas and co workers review the best evidences of this critical adaptation.
• Arbab-Zadeh A, Perhonen M, Howden E, Peshock RM, Zhang R, Adams-Huet B, et al. Cardiac remodeling in response to 1 year of intensive endurance training. Circulation. 2014;130(24):2152–61. This work provides describes the cardiac adaptation resulting from intensive training in sport: this is quoted as a "benchmark" for the dramatic cardiac changes in pregnancy.
• Duvekot JJ, Pieters F, Peeters L. Early pregnancy changes in hemodynamics and volume homeostasis are consecutive adjustments triggered by a primary fall in systemic vascular tone. Am J Obstet Gynecol. 1993;169, Mar:1382–92. An old seminal paper on vascular adaptation in pregnancy.
• Melchiorre K, Sharma R, Khalil A, Thilaganathan B. Maternal cardiovascular function in normal pregnancy evidence of maladaptation to chronic volume overload. Hypertension. 2016;67(4):754–62. Cardiac ventricular function and increased plasma volume in pregnancy conjure as to determine in "a significant proportion of cases at term" cardiovascular maladaptation to the volume-overloaded state in some apparently normal pregnancies.
Valensise H, Vasapollo B, Gagliardi G, Novelli GP. Early and late preeclampsia: two different maternal hemodynamic states in the latent phase of the disease. Hypertension. 2008;52:873–80.
Melchiorre K, Sutherland GR, Liberati M, Thilaganathan B. Maternal cardiovascular impairment in pregnancies complicated by severe fetal growth restriction. Hypertension. 2012;60:437–43.
Melchiorre K, Sutherland G, Sharma R, Nanni M, Thilaganathan B. Mid-gestational maternal cardiovascular profile in preterm and term pre-eclampsia: a prospective study. BJOG: Int J O&G. 2012;120(4):496–504.
Magee LA, CHIPS Study Group, von Dadelszen P, Singer J, Lee T, Rey E, et al. Control of Hypertension In Pregnancy Study randomised controlled trial-are the results dependent on the choice of labetalol or methyldopa? BJOG. 2015; doi:10.1111/1471-0528.13568.
Smith RD, Levy P, Ferrario CM. Value of noninvasive hemodynamics to achieve blood pressure control in hypertensivevalue of noninvasive hemodynamics to achieve blood pressure control in hypertensive subjects. Hypertension. 2006;47:771–7.
Aoka Y, Hagiwara N, Kasanuki H. Heterogeneity of hemodynamic parameters in untreated primary hypertension, and individualization of antihypertensive therapy based on noninvasive hemodynamic measurements. Clin Exp Hypertens. 2013;35(1):61–6.
Serg M, Graggaber J, Kampus P, Zagura M, Kals J, MakiPeta K, et al. Value of haemodynamic profiling to the response of antihypertensive therapy. Artery Res. 2014;8:189–96.
Acknowledgments
CCL is supported by the UK National Institute for Health Research Biomedical Research Centre based at Imperial College Healthcare National Health Service Trust and Imperial College London.
Author information
Authors and Affiliations
Contributions
CCL and EF both contributed equally to drafting, reviewing and finalizing the document.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Blood Pressure Monitoring and Management
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lees, C., Ferrazzi, E. Relevance of Haemodynamics in Treating Pre-eclampsia. Curr Hypertens Rep 19, 76 (2017). https://doi.org/10.1007/s11906-017-0766-6
Published:
DOI: https://doi.org/10.1007/s11906-017-0766-6