
Abstract
During the last decade a tremendous technologic progress based on genome-wide profiling of genetic aberrations, structural DNA alterations, and sequence variations has allowed a better understanding of the molecular basis of pediatric and adult B/T- acute lymphoblastic leukemia (ALL), contributing to a better recognition of the biological heterogeneity of ALL and to a more precise definition of risk factors. Importantly, these advances identified novel potential targets for therapeutic intervention. This review will be focused on the cytogenetic/molecular advances in pediatric and adult ALL based on recently published articles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
ALL represents a biologically and clinically heterogeneous group of B/T-precursor-stage lymphoid cell malignancies arising from genetic insults that block lymphoid differentiation and drive aberrant cell proliferation and survival. Incidence and cure rates differ among children and adults. In children, ALL is the commonest malignancy accounting for approximately 25 % of childhood cancer and it has 5-year event-free survival rates ranging between 76 % and 86 % in patients receiving protocol-based therapy. In adults, ALL is less common and generally carries a worse prognosis with a long-term survival probability less than 35–40 % [1, 2]. Although there is remarkable progress made in the treatment of ALL in children and, with less efficacy, in adults, several ALL subtypes continue to have a poor prognosis and in a proportion of long-term surviving patients, treatment is responsible for short and long-term toxicities. Consequently, there is a need in improving the molecular dissection of subtypes, identifying genetic alterations that predict the risk of treatment failure, and developing novel and targeted therapies. Cytogenetics has long been used for diagnosis, risk stratification, and therapeutic implications, however experimental models [3, 4] have established that primary cytogenetic abnormalities alone are insufficient to induce leukemia and that cooperative mutations are required. The development of microarray technologies to profile gene expression and structural genetic alterations in a genome-wide and high-resolution fashion have revolutionized our ability to identify genetic abnormalities providing important insights into the pathways deregulated in ALL [5]. Moreover, recently the development of next-generation sequencing (NGS) technologies has provided researchers with completely new and effective tools for the discovery of novel alterations, depicting an exhaustive picture of the leukemia genome complexity (Fig. 1). This review emphasizes the most important and newest findings obtained from genomic analysis of B/T-lineage ALL.

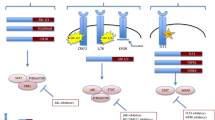
Next-generation sequencing (NGS) in B/T ALL. Whole genome/exome/transcriptome sequencing approaches provide a comprehensive view of the landscape of genetic alterations in leukemia allowing the identification of potential novel markers for diagnosis, risk-stratification, and tailored treatments. Here, studies using NGS approaches in B/T ALL are schematized
B-Progenitor Acute Lymphoblastic Leukemia
Prognostic Factors Based on Cytogenetics
Cytogenetics and fluorescence in situ hybridization (FISH) reveal recurring chromosome abnormalities, including numerical and structural changes, in approximately 80 % of ALL. Substantial differences in the frequencies of some recurring abnormalities are present between children and adults.
-
The t(8;14)[MYC-IgH], present in a high proportion of Burkitt cell leukemia/lymphoma, occurs in approximately 1 % of adults. This translocation is associated with French-American-British (FAB)-L3-type leukemia cells, high incidence of central nervous system involvement at diagnosis, and poor prognosis [6].
-
The t(4;11)[MLL-AF4] is present in up to 60 % of infants younger than 12 months, but is rarely observed in adult patients. This translocation is associated with FAB L1 or L2 morphology, immature immunophenotype, B-cell lineage, frequent co-expression of myeloid antigens, and high leukocyte counts. The prognosis is poor in both adults and children [7, 8].
-
The t(9;22)[BCR-ABL1] that produces the Philadelphia (Ph) chromosome is observed in about 2 % to 5 % of children and 30 % of adults. This translocation is associated with a dismal prognosis. Treatment with tyrosine kinase inhibitors (TKIs) has produced promising results, although with the passage of time, problems have emerged with drug resistance and disease persistence [9].
-
The t(1;19)[TCF3-PBX1] occurs in approximately 30 % of pre-B cell childhood ALL patients. In the past, patients with the t(1;19) typically had early treatment failure. However, this adverse prognosis can be overcome by more intensive chemotherapy such that the t(1;19) is now associated with a favorable prognosis [10, 11].
-
The t(12;21)[ETV6-RUNX1] is detectable by FISH or polymerase chain reaction analysis in about 25 % of children and 3 % of adults with B-ALL. Patients generally have a favorable prognosis [12].
-
Patients with hyperdiploidy with more than 50 chromosomes often have a good prognosis. The individual structural abnormalities do not appear to influence outcome in patients with hyperdiploidy except for the t(9;22), which is associated with a poor prognosis [13].
-
Approximately 5 % to 6 % of ALL patients, independent of age, have clonal loss of various chromosomes, resulting in a hypodiploid clone with fewer than 46 chromosomes. These patients generally have a poor prognosis, especially those with near-haploid and low-hypodiploid clones. Similarly, deletion of 9p is an unfavorable risk factor associated with a high rate of relapse in B-precursor ALL in children [14].
Prognostic Factors Based on Genomic Profiling
The advent of high-resolution genome-wide analyses of gene expression, DNA copy number alterations (CNA), and loss of heterozygosity have led to the detection of many novel genetic abnormalities refining the prognostic models for ALL (Table 1). Using single nucleotide polymorphism (SNP) arrays, many groups detected multiple submicroscopic CNA not evident on cytogenetic analysis, commonly less than one megabase (Mb) in size and targeting, in most of the cases, a single or few genes implicated in key cellular pathways, such as lymphoid development (IKZF1, PAX5, EBF1, VPREB1), cell cycle regulation and tumor suppression (CDKN2A/CDKN2B, PTEN, BTG1, RB1), lymphoid signaling (BTLA, CD200), drug responsiveness (NR3C1), and DNA mismatch repair (MTOR, HERC1, PRKCZ, and PIK3C2B) [5, 15–17, 18•, 19, 20]. Importantly, there were substantial differences in the frequency and nature of CNAs among various disease subtypes. MLL-rearranged leukemias harbor less than one CNA per case, suggesting that MLL is a potent oncogene that requires very few cooperating lesions to induce leukemia transformation. In contrast, ETV6-RUNX1 and BCR-ABL1 rearranged leukemias harbor six to eight alterations per case that may be present years after the initial chromosome rearrangements, indicating that these additional postnatal genetic alterations are required to induce the full leukemic phenotype [5].
Genetic Alterations in B-Lymphoid Development Genes
Over two-thirds of genetic abnormalities affected genes involved in lymphoid development. These alterations include deletions, focal amplifications, novel translocations, and point mutations involving transcriptional regulators of early B-cell lineage development, such as IKZF1 (Ikaros), PAX5 (Paired box 5), EBF1 (early B-cell factor), LEF1 (lymphoid enhancer factor 1), and immunoglobulin family genes, such as VPREB1 (pre-B lymphocyte 1).
IKZF1
Ikaros is a DNA-binding zinc finger transcription factor that regulates the development and function of the immune system and acts as a master regulator of normal hematopoietic differentiation and proliferation, particularly in lymphoid lineages [21]. During the last 5 years, IKZF1 has been established as one of the most clinically relevant tumor suppressors in ALL. Deletion of a single IKZF1 allele or mutations of a single copy of IKZF1 were firstly detected in 15 % of all cases of pediatric B-cell ALL and in more than 80 % of Ph+lymphoid leukemia cases, either de novo Ph+ ALL or chronic myeloid leukemia at progression to lymphoid blast crisis [15, 22, 23]. By contrast, IKZF1 alterations were uncommon in other ALL subtypes that otherwise harbor multiple DNA copy-number alterations, such as ETV6-RUNX1 ALL, suggesting that IKZF1 alteration is a key determinant of the lineage and progression of Ph+ leukemia. The analysis of a twin pair concordant for ALL showed that in childhood Ph+ ALL, BCR-ABL1 gene fusion can be a prenatal and possibly initiating genetic event whereas deletion of IKZF1 is a secondary and probable postnatal mutation that is associated with poor prognosis [24]. The deletions either involve the entire IKZF1 locus, resulting in loss of function, or delete an internal subset of IKZF1 exons, resulting in the expression of dominant negative IKZF1 alleles. Expression of such dominant negative IKZF1 alleles in hematopoietic progenitors impairs lymphoid development [25], and loss of IKZF1 accelerates the onset of Ph+ ALL in a retroviral bone marrow transplant and transgenic models of this disease [26]. Several studies demonstrated that IKZF1 deletions are significantly associated with an increased relapse rate and adverse events and are correlated with poor outcome in patients with Ph+ ALL [27–29]. Alteration of IKZF1 is also associated with poor outcome in BCR-ABL1-negative ALL [27, 29–31], where this association is independent of commonly used risk stratification features such as age, sex, white cell count, and levels of minimal residual disease (MRD). IKZF1 deletions and nonsense mutations identified at diagnosis are preserved at relapse [27] and may be used for highly sensitive MRD tests in addition to the repertoire of MRD markers currently available for monitoring MRD in ALL [32]. Importantly, high-risk BCR-ABL1 negative ALL cases with deletion of IKZF1 and poor outcome have a gene expression profile significantly similar to that of Ph+ALL (“BCR-ABL1-like” ALL) [29, 33], suggesting that these cases may harbor alternative genetic events leading to aberrant activation of tyrosine kinase signaling pathways similar to those downstream of BCR-ABL1.
PAX5
PAX5 encodes a transcription factor known as B-cell-specific activator protein, that plays a key role in B-cell commitment by activating essential components of B-cell receptor signaling and repressing the transcription of genes necessary for T-lymphopoiesis [34]. By SNP arrays monoallelic deletion of PAX5 has been observed in about 30 % of both children and adults with B-ALL, resulting in loss of Pax5 protein expression or in the production of a Pax5 isoform lacking the DNA binding domain and/or transcriptional regulatory domain [5, 17]. Inactivating point mutations in PAX5 are also observed (7–30 %) as well as chromosomal translocations involving multiple partners such as ETV6, ENL, FOXP1, ZNF521, PML, C20ORF112, AUTS2, JAK2, POM121, HIPK1, DACH1, LOC392027, SLCO1B3, ASXL1, and KIF3B [5, 35, 36]. In these rearrangements the DNA binding paired domain of PAX5 and/or a variable amount of the C-terminal trans-activating domains are fused to functional domains of the partner genes, resulting in a loss of Pax5 function rather than in a gain of functional elements. Alterations of PAX5 have been demonstrated to not influence treatment outcome [17, 29].
Genetic Alterations in Tumor Suppressors Genes
CDKN2A/B
The CDKN2A/B locus is particularly noteworthy since it encodes for the INK4-class cyclin dependent kinase inhibitors p15INK4B, p16INK4A and for p14ARF, which inhibits MDM2’s E3 ligase activity stabilizing p53. Alterations have been demonstrated to frequently occur in all lymphoid malignancies, with homozygous deletions as the most frequent mechanism of inactivation [37]. Recently, we identified CDKN2A and CDKN2B deletion at diagnosis in 29 % and 25 % of BCR-ABL1-positive ALL patients, respectively. Deletions were predominantly monoallelic and in more than half of cases the minimal overlapping region of the lost area included a large number of genes. The detection rate of CDKN2A/ARF loss increased at relapse compared to diagnosis [18•]. The association with prognosis is still controversial. Some studies reported a correlation of CDKN2A/B deletion with poor prognosis, whereas others show no correlation [38]. Confirming previous findings from mice models [39], we demonstrated that deletions of CDKN2A/B are significantly associated with higher white blood count and poor outcome in terms of overall survival, disease-free survival, and cumulative incidence of relapse in adult BCR-ABL1-positive ALL [18•].
Deregulated Expression of CRLF2
Four independent groups in late 2009 and early 2010 have identified that up to 50 % of BCR-ABL1-like ALL cases have dysregulated expression of CRLF2, the gene encoding the cytokine receptor-like 2 factor, also known as thymic stromal lymphopoietin receptor [40–43]. CRLF2 has a role in T-lymphoid and dendritic cell development and is involved in inflammation and allergic responses [44]. Although it also mediates B-cell precursor proliferation and survival, its role in B-lymphoid neoplasms needs to be understood [45]. CRLF2 rearrangements include a translocation of CRLF2, which is located at the pseudoautosomal region 1 (PAR1) of chromosome Xp/Yp, into the immunoglobulin heavy chain locus at chromosome 14q or a focal PAR1 deletion proximal of CRLF2 that results in a novel fusion P2RY8-CRLF2 [40–43]. Less commonly, a missense mutation in exon 6, F232C, results in constitutive CRLF2 dimerization [43]. All these events result in overexpression of full-length CRLF2 on the surface of leukemic cells harboring the genetic alterations, providing a cell-surface marker amenable to detection by flow cytometry for clinical diagnostic purposes. Overall aberrant expression of CRLF2 was found in 12.5 % to 15 % of B-ALL that lacks typical chromosomal rearrangements, but was not overexpressed in B-ALL cases that have recurring rearrangements or in other lymphoid malignancies [40–43]. CRLF2 alteration is seen at low rates (5–7 %) when all B-ALL cases are grouped together; however, it is seen in 50–60 % of Down Syndrome (DS) associated ALL, suggesting that CRLF2 overexpression is especially relevant to tumorigenesis in patients with trisomy 21 [41–43]. In high-risk B-ALL, rearrangements of CRLF2 are frequently found together with IKZF1 alterations and activating mutations in JAK1 and JAK2, most commonly at or near R683 in the pseudokinase domain of JAK2 [40–42], and are associated with very poor outcome [46, 47]. Therapeutically important, JAK2 mutant-CRLF2–mediated and CRLF2 mutant–mediated transformation are sensitive to JAK inhibition in vitro, suggesting that ALL patients with CRLF2 overexpression may benefit from future kinase inhibitor approaches [41].
Intrachromosomal Amplification of Chromosome 21
Intrachromosomal amplification of chromosome 21 (iAMP21) occurs at an incidence up to 2 % in older children with B-cell precursor ALL. iAMP21 was originally identified as a consequence of routine screening for the presence of the ETV6-RUNX1 fusion by FISH [48] and is defined by at least three copies of the RUNX1 gene, commonly concomitant with deletion of the subtelomeric regions of chromosome 21. Recently, the common region of amplification on chromosome 21 was better refined to a 5.1-Mb region that included RUNX1, miR-802, and genes mapping to the DS critical region. Recurrent abnormalities affecting IKZF1 (22 %), CDKN2A/B (17 %), PAX5 (8 %), ETV6 (19 %), and RB1 (37 %) and secondary to chromosome 21 rearrangements were identified [49]. iAMP21 has been shown to be linked to a dismal outcome when patients are treated with standard therapy, because it is associated with an increased risk of both early and late relapses [50, 51].
Prognostic Factors Based on Conventional and Next-Generation Sequencing Technologies
Whole genome/exome-sequencing approaches have been demonstrated to provide a comprehensive view of the landscape of genetic alterations in leukemia identifying crucial markers for diagnosis, risk-stratification, and potentially novel target therapies. Screening for BCR-ABL1 mutations in Philadelphia-positive ALL allows to identify patients who may benefit from second-generation TKIs or from novel compounds targeting the T315I mutation [52]. An extensive Sanger resequencing of 120 candidate cancer genes in diagnostic leukemia samples from 187 children and adolescents with high-risk B-ALL treated with augmented post-induction chemotherapy on the Children’s Oncology Group P9906 protocol and integrated analysis with genome-wide CNAs and gene expression profiles revealed recurrent somatic alterations in key signaling pathways, including B-cell development/differentiation (68 %), TP53/RB tumor suppressor pathway (54 %), Ras signaling (50 %), and Janus kinases (11 %). The frequency of mutations within the four major pathways varied markedly across genetic subtypes with a striking association of B-cell development/differentiation and JAK2 mutations with leukemias expressing a BCR-ABL1-like gene expression profile and of Ras signaling pathway mutations with cases defined by a distinct gene expression profile coupled with focal ERG deletion [53]. For most of these genes the prognostic impact remains to be determined, but they highlighted important new therapeutic targets for selected patient subsets. The landscape of genetic alterations in B-ALL was extended by the identification of loss-of-function mutations (either deletions or sequence mutations) of CREBBP, encoding the transcriptional coactivator and histone acetyltransferase CREB-binding protein or CBP, in 19 % of relapsed samples from children with B-cell progenitor ALL [54•]. Functionally, the mutations impair histone acetylation and transcriptional regulation of CREBBP targets, including glucocorticoid responsive genes, likely influencing treatment responsiveness in ALL. CREBBP mutations are preserved at relapse when present at diagnosis, acquired at relapse but occurring in minor subclones at diagnosis, or reduplicated to homozygosity at relapse when heterozygous at diagnosis [54•]. Although the prognostic impact remains to be determined, the observation that the CREBBP mutations impair regulation of glucocorticoid-responsive genes, and that the mutations are selected for at relapse, suggests that these alterations may influence the likelihood of relapse. Therefore, novel therapeutic approaches, such as histone deacetylase inhibitors (HDAC) [55], directed at modulating protein acetylation may be useful in high-risk ALL. Recently, new molecular prognostic markers have been identified. The analysis of 265 first-relapse patients enrolled in the German Acute Lymphoblastic Leukemia Relapse Berlin-Frankfurt-Münster 2002 (ALL-REZ BFM 2002) trial for sequence and copy number alterations of the TP53 gene, by using direct sequencing and multiplex ligation-dependent probe amplification, identified copy number and sequence alterations of TP53 in 12.4 % of B-cell precursor ALL samples [56]. In more than half of cases (54 %) TP53 alterations were gained at relapse. Mutations were strikingly associated with non-response to chemotherapy and poor event-free survival and overall survival rates, allowing the identification of patients at high risk of treatment failure. To overcome the limits of conventional Sanger sequencing whole exome and transcriptome NGS approaches have been subsequently pursued in two pediatric ETV6-RUNX1 (TEL-AML1) positive ALL [57] and in an adult patient with BCR-ABL1 positive ALL [58], respectively. Both studies identified novel somatic mutations, some of them occurred in genes previously described to be mutated in other types of cancer, but none was found to be recurrent in an extended series of ALL samples. Although the true pathogenetic significance of the mutations must await future functional evaluations, these studies provided a first estimate of the mutational burden of ETV6-RUNX1 and BCR-ABL1-positive ALLs.
T-Progenitor Acute Lymphoblastic Leukemia
Prognostic Factors Based on Cytogenetics
T-ALL is a malignant disorder of T-cell lymphoid progenitor cells that affects 15 % of children and 25 % of adults with ALL. Structural chromosomal aberrations are identified in approximately 50 % of cases and frequently involve the juxtaposition of strong promoter and enhancer elements from T-cell receptor (TCR) genes with transcription factor genes as consequence of an illegitimate event during V(D)J recombination in normal T-cell development. This leads to the aberrant expression of the fusion partners resulting in thymocytes differentiation block at various stages of maturation.
TCR Gene Rearrangements
The most common rearrangements include TCR alpha/delta chain at 14q11.2, TCR beta chain at 7q34, and TCR gamma chain at 7p14. With few exceptions, the involved gene on the partner chromosome encodes a cell cycle inhibitor or a transcription factor whose expression is deregulated or activated as a result of the rearrangement. TAL1 (1p32) is ectopically expressed in T-ALL as consequence of t(1;14)(p32;q11) [59] (3 % in childhood T-ALL) and more frequently as a consequence of the intrachromosomal deletion resulting in SIL-TAL1 fusion gene, while LYL1 (19p13), TAL2 (9q32), and BHLH1 (21q22) are up-regulated in the rare translocations t(7;19)(q34;p13), t(7;9)(q34;q32), and t(14;21)(q11;q22), respectively. Their aberrant expression may contribute to leukemia through the formation of heterodimers with class I basic helix-loop-helix members that regulate T-cell specific genes, with consequent differentiation and proliferation impairment. LIM domain only genes LMO1 (11p15) and LMO2 (11p13) are involved in t(11;14)(p15;q11) and t(11;14)(p13;q11) with TCR alpha/delta loci and in translocations with TCR beta. LMO abnormal expression associates with deregulation of LYL1 and TAL1, even in absence of specific translocations [60]. The homeobox (HOX) genes are a highly conserved family of transcription factors that play an important role in morphogenesis during embryonic development and in normal hemopoiesis [61]. The inv(7)(p15q34) and t(7;7)(p15;q34) bring the TCR beta regulatory elements in the vicinity of the HOXA genes cluster disrupting the normal regulatory elements of the cluster with subsequent up-regulation, especially of HOXA9, HOXA10, and HOXA11 [62, 63]. TLX1 (HOX11) is expressed at high level in more than 30 % of adult T-ALL as consequence of t(10;14)(q24;q11) and t(7;10)(q34;q24), while TLX3 (HOX11L2) is involved in t(5;14)(q35;q32) with the fusion partner BCL11B in about 20 % of children and 4 % of adults [64]. Additional genes rarely involved in TCR loci rearrangements are LCK [65], CCND2 [66], and IRS4 [67].
Non-TCR Loci
A variety of cytogenetic abnormalities can occur in T-cell ALL that do not involve the TCR loci. These include del(6q), t(11q23) (MLL gene), t(14q32), trisomy 8, and t(10;11). A favorable prognostic correlation has been assessed for t(10;14), which is extremely rare in adult patients, and more frequent in the pediatric subset. Trisomy of chromosome 8 and monosomy of chromosome 7 usually carry a bad prognosis [68]. The cryptic deletion del(9)(q34.11q34.13) results in the SET-NUP214 fusion product, which transcriptionally activates specific members of the HOXA cluster maybe contributing to T-ALL pathogenesis by the inhibition of T-cell maturation [69]. The ABL1 cytoplasmic tyrosine kinase plays a role in T-cell signaling, leading to the induction of IL-2 production and proliferation following TCR activation [70]. ABL1 fusion genes can be identified in about 8 % of T-ALL. EML1-ABL1 fusion due to a cryptic t(9;14)(q34;q32), BCR-ABL1 t(9;22)(q34;q11), and ETV6-ABL1 t(9;12)(q34;p13) are seldom reported chimeric genes in T-ALL, although if frequent in other hematologic malignancies. In contrast, the most frequent and strictly associated alteration with T-ALL is the NUP214-ABL1 fusion identified in 6 % of cases, in both children and adults, also if it is not clear its prognostic relevance [71]. JAK2 resulted constitutively activated in the rare t(9;12)(p24;p13) encoding ETV6-JAK2 fusion oncoprotein, while JAK1 is widely mutated in adult T-ALL (18 %) and less in children (3 %), and associates with a poor response to therapy [72]. Finally, a recent study identified a novel fusion partner of ETV6 in one T-ALL patient, harboring ETV6-ARNT t(1;12)(q21;p13) [73].
Prognostic Factors Based on Genomic Profiling
On the basis of gene expression profiling, T-ALL cases can be classified into major subgroups that are indicative of leukemic arrest at specific stages of normal thymocyte development (HOX11 + early cortical thymocytes; LYL1 + early pro-T thymocytes; TAL1 + late cortical thymocytes) and have clinical relevance, because they are associated with a favorable or worse prognosis [60]. Using SNP array platforms, many novel genomic alterations have recently been identified, including focal deletions of RB1 [5], duplications of the proto-oncogene MYB [5, 74], deletion of 9p21.2 in more than 70 % of patients [75], deletion and mutation of PTEN [5, 76], and deletion or mutation of the U3 ubiquitin ligase FBXW7 [77, 78]. Thus far, mutations of NOTCH1 and FBXW7 have generally been associated with a favorable prognosis [79]. NOCTH1 role in leukemogenesis was initially identified in the rare chromosomal translocation t(7;9)(q34;q34.3) that fuses the intracellular form of NOTCH1 to the TCR beta and leads to the expression of TAN1, a truncated and constitutively activated form of NOTCH1; subsequently, activating or loss of function mutations has been identified in more than 50 % of T-ALL cases, representing the most common alteration in T-ALL. The high frequency of NOTCH1 mutations in T-ALL has sparked an interest in the development of anti-NOTCH1 targeted therapies for the treatment of T-ALL. Other mutated genes in T-ALL are WT1, NRAS, the negative regulator of the RAS pathway, NF1 that is inactivated because of deletions or mutations [80], and rarely FLT3 [81, 82] and PTPN2 [83], that are affected by activating mutations and focal deletions, respectively. All these genes play crucial roles as regulators and alterations in their function may affect critically different signal transduction pathways. Of note, identification of 6q15-16.1 deletion containing CASP8AP2 gene represents a novel prognostic factor that defines a high-risk group (Table 2) [84].
Prognostic Factors Based on Next-Generation Sequencing Technology
Using exon capture of chromosome X a recent study by Van Vlierberghe and colleagues [85] identified inactivating mutations of the X-linked plant homeodomain finger 6 (PHF6) in 16 % of pediatric and 38 % of adult T-ALL cases. PHF6 mutations were almost exclusively found in male and were associated with leukemias driven by aberrant expression of TLX1 and TLX3. Although the prognostic significance remains to be determined, PHF6 has emerged as a new X-linked tumor suppressor in T-ALL. Recently, the application of whole-genome sequencing (WGS) to the characterization of “early T-cell precursor” (ETP) ALL, that comprises up to 15 % of T-ALL and is associated with a high risk of treatment failure, has provided potential new molecular targets for therapy [86••]. Zhang and colleagues [86••] performed WGS of 12 ETP ALL cases identifying activating mutations in genes regulating cytokine receptor and RAS signaling, such as NRAS, KRAS, FLT3, IL7R, JAK3, JAK1, SH2B3, and BRAF (67 %), inactivating lesions disrupting hematopoietic development, such as GATA3, ETV6, RUNX1, IKZF1, and EP300 (58 %), and histone-modifying genes, such as EZH2, EED, SUZ12, SETD2, and EP300 (48 %). Moreover, they identified new targets of recurrent mutation in DNM2, ECT2L, and RELN genes. Importantly, the gene expression profile of ETP ALL resulted similar to that of normal and myeloid leukemia hematopoietic stem cells, suggesting the possibility that myeloid-directed therapies might improve the poor outcome of ETP ALL.
Conclusions
Thanks to the use of SNP microarray-based technology, extensive candidate gene sequencing, and whole genome/exome/transcriptome NGS approaches, novel and recurring lesions have been identified in B and T-ALL. The identification of these alterations allowed a better classification of leukemia patients in subgroups according to specific genetic lesions (eg, BCR-ABL1 like subgroup), provided new markers for risk-assessment and/or for monitoring minimal residual disease (eg, IKZF1, CRLF2, TP53), and highlighted novel therapeutic approaches (eg, inhibitors of JAK2, HDAC, and NOTCH1). However, much remains to be investigated. From the biological prospective, whole exome/transcriptome NGS approaches will provide a comprehensive view of the complexity of genetic alterations in ALL, identifying genes and pathways relevant for the establishment of the leukemia clone and/or for the disease recurrence and responsible for responsiveness to therapy. From the clinical prospective, all these findings should be integrated into the clinic, providing new prognostic markers and novel targets for tailored therapies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Pui CH, Sandlund JT, Pei D, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children’s Research Hospital. Blood. 2004;104:2690–6.
Bassan R. Evolving strategies for the management of high-risk adult acute lymphoblastic leukemia. Haematologica. 2005;90:1299.
Andreasson P, Schwaller J, Anastasiadou E, et al. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genet Cytogenet. 2001;130:93–104.
Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2006;103:6688–93.
Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64.
Moorman AV, Harrison CJ, Buck GA, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109:3189–97.
A Collaborative Study of the Group Francais de Cytogenetique Hematologique. Cytogenetic abnormalities in adult acute lymphoblastic leukemia: correlations with hematologic findings outcome. Blood. 1996;87:3135–42.
Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–43.
Soverini S, Colarossi S, Gnani A, et al. Resistance to dasatinib in Philadelphia-positive leukemia patients and the presence or the selection of mutations at residues 315 and 317 in the BCR-ABL kinase domain. Haematologica. 2007;92:401–4.
Devaraj PE, Foroni L, Janossy G, et al. Expression of the E2A-PBX1 fusion transcripts in t(1;19)(q23;p13) and der(19)t(1;19) at diagnosis and in remission of acute lymphoblastic leukemia with different B lineage immunophenotypes. Leukemia. 1995;9:821–5.
Foa R, Vitale A, Mancini M, et al. E2A-PBX1 fusion in adult acute lymphoblastic leukaemia: biological and clinical features. Br J Haematol. 2003;120:484–7.
Romana SP, Poirel H, Leconiat M, et al. High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood. 1995;86:4263–9.
Raimondi SC, Pui CH, Hancock ML, et al. Heterogeneity of hyperdiploid (51–67) childhood acute lymphoblastic leukemia. Leukemia. 1996;10:213–24.
Harrison CJ, Moorman AV, Broadfield ZJ, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004;125:552–9.
Iacobucci I, Storlazzi CT, Cilloni D, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood. 2009;114:2159–67.
Kuiper RP, Schoenmakers EF, van Reijmersdal SV, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21:1258–66.
Iacobucci I, Lonetti A, Paoloni F, et al. The PAX5 gene is frequently rearranged in BCR-ABL1-positive acute lymphoblastic leukemia but is not associated with outcome A report on behalf of the GIMEMA Acute Leukemia Working Party. Haematologica. 2010;95:1683–90.
• Iacobucci I, Ferrari A, Lonetti A, et al. CDKN2A/B alterations impair prognosis in adult BCR-ABL1-positive acute lymphoblastic leukemia patients. Clin Cancer Res. 2011;17:7413–23. In adult BCR-ABL1 positive ALL CDKN2A/B deletions represent a frequent event and they are frequently acquired during leukemia progression. Deletions are significantly associated with poor outcomes in terms of overall survival, disease free-survival, and cumulative incidence of relapse.
Trageser D, Iacobucci I, Nahar R, et al. Pre-B cell receptor-mediated cell cycle arrest in Philadelphia chromosome-positive acute lymphoblastic leukemia requires IKAROS function. J Exp Med. 2009;206:1739–53.
Diouf B, Cheng Q, Krynetskaia NF, et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat Med. 2011;17:1298–303.
Georgopoulos K, Bigby M, Wang JH, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79:143–56.
Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–4.
Iacobucci I, Lonetti A, Messa F, et al. Expression of spliced oncogenic Ikaros isoforms in Philadelphia-positive acute lymphoblastic leukemia patients treated with tyrosine kinase inhibitors: implications for a new mechanism of resistance. Blood. 2008;112:3847–55.
Cazzaniga G, van Delft FW, Lo Nigro L, et al. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 deletions in monozygotic twins with Ph+acute lymphoblastic leukemia. Blood. 2011;118:5559–64.
Tonnelle C, Dijon M, Moreau T, et al. Stage specific over-expression of the dominant negative Ikaros 6 reveals distinct role of Ikaros throughout human B-cell differentiation. Mol Immunol. 2009;46:1736–43.
Virely C, Moulin S, Cobaleda C, et al. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia. Leukemia. 2010;24:1200–4.
Kuiper RP, Waanders E, van der Velden VH, et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24:1258–64.
Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol. 2009;27:5202–7.
Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–80.
Waanders E, van der Velden VH, van der Schoot CE, et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79 % of relapses in pediatric acute lymphoblastic leukemia. Leukemia; 2010.
Yang YL, Hung CC, Chen JS, et al. IKZF1 deletions predict a poor prognosis in children with B-cell progenitor acute lymphoblastic leukemia: a multicenter analysis in Taiwan. Cancer Sci. 2011;102:1874–81.
Venn NC, van der Velden VH, de Bie M, et al. Highly sensitive MRD tests for ALL based on the IKZF1 Delta3-6 microdeletion. Leukemia; 2011.
Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–34.
Busslinger M. Transcriptional control of early B cell development. Annu Rev Immunol. 2004;22:55–79.
Familiades J, Bousquet M, Lafage-Pochitaloff M, et al. PAX5 mutations occur frequently in adult B-cell progenitor acute lymphoblastic leukemia and PAX5 haploinsufficiency is associated with BCR-ABL1 and TCF3-PBX1 fusion genes: a GRAALL study. Leukemia; 2009.
Nebral K, Denk D, Attarbaschi A, et al. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23:134–43.
Faderl S, Estrov Z, Kantarjian HM, et al. The incidence of chromosome 9p21 abnormalities and deletions of tumor suppressor genes p15(INK4b)/p16(INK4a)/p14(ARF) in patients with acute lymphoblastic leukemia. Cytokines Cell Mol Ther. 1999;5:159–63.
Heyman M, Rasool O, Borgonovo Brandter L, et al. Prognostic importance of p15INK4B and p16INK4 gene inactivation in childhood acute lymphocytic leukemia. J Clin Oncol. 1996;14:1512–20.
Notta F, Mullighan CG, Wang JC, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469:362–7.
Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114:2688–98.
Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243–6.
Hertzberg L, Vendramini E, Ganmore I, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010;115:1006–17.
Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2010;107:252–7.
Liu YJ. TSLP in epithelial cell and dendritic cell cross talk. Adv Immunol. 2009;101:1–25.
Levin SD, Koelling RM, Friend SL, et al. Thymic stromal lymphopoietin: a cytokine that promotes the development of IgM+B cells in vitro and signals via a novel mechanism. J Immunol. 1999;162:677–83.
Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115:5312–21.
Cario G, Zimmermann M, Romey R, et al. Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood. 2010;115:5393–7.
Harrison CJ, Moorman AV, Barber KE, et al. Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol. 2005;129:520–30.
Rand V, Parker H, Russell LJ, et al. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2011;117:6848–55.
Moorman AV, Richards SM, Robinson HM, et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21). Blood. 2007;109:2327–30.
Attarbaschi A, Mann G, Panzer-Grumayer R, et al. Minimal residual disease values discriminate between low and high relapse risk in children with B-cell precursor acute lymphoblastic leukemia and an intrachromosomal amplification of chromosome 21: the Austrian and German acute lymphoblastic leukemia Berlin-Frankfurt-Munster (ALL-BFM) trials. J Clin Oncol. 2008;26:3046–50.
Soverini S, Vitale A, Poerio A, et al. Philadelphia-positive acute lymphoblastic leukemia patients already harbor BCR-ABL kinase domain mutations at low levels at the time of diagnosis. Haematologica. 2011;96:552–7.
Zhang J, Mullighan CG, Harvey RC, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118:3080–7.
• Mullighan CG, Zhang J, Kasper LH, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. Mutations of CREBBP, occurring 8.3 % of relapse B-ALL, are either present at diagnosis or acquired at relapse, and resulted in truncated alleles or deleterious substitutions in conserved residues of the histone acetyltransferase domain that impair histone acetylation and transcriptional regulation of CREBBP targets, including glucocorticoid responsive genes.
Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84.
Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29:3185–93.
Lilljebjorn H, Rissler M, Lassen C, et al. Whole-exome sequencing of pediatric acute lymphoblastic leukemia. Leukemia; 2011.
Iacobucci I, Ferrarini A, Sazzini M, et al. Application of the whole transcriptome shotgun sequencing approach to the study of philadelphia-positive acute lymphoblastic leukemia. Blood Cancer Journal; 2012.
Carroll AJ, Crist WM, Link MP, et al. The t(1;14)(p34;q11) is nonrandom and restricted to T-cell acute lymphoblastic leukemia: a Pediatric Oncology Group study. Blood. 1990;76:1220–4.
Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75–87.
van Oostveen J, Bijl J, Raaphorst F, et al. The role of homeobox genes in normal hematopoiesis and hematological malignancies. Leukemia. 1999;13:1675–90.
Soulier J, Clappier E, Cayuela JM, et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood. 2005;106:274–86.
Speleman F, Cauwelier B, Dastugue N, et al. A new recurrent inversion, inv(7)(p15q34), leads to transcriptional activation of HOXA10 and HOXA11 in a subset of T-cell acute lymphoblastic leukemias. Leukemia. 2005;19:358–66.
Bernard OA, Busson-LeConiat M, Ballerini P, et al. A new recurrent and specific cryptic translocation, t(5;14)(q35;q32), is associated with expression of the Hox11L2 gene in T acute lymphoblastic leukemia. Leukemia. 2001;15:1495–504.
Tycko B, Smith SD, Sklar J. Chromosomal translocations joining LCK and TCRB loci in human T cell leukemia. J Exp Med. 1991;174:867–73.
Clappier E, Cuccuini W, Cayuela JM, et al. Cyclin D2 dysregulation by chromosomal translocations to TCR loci in T-cell acute lymphoblastic leukemias. Leukemia. 2006;20:82–6.
Karrman K, Kjeldsen E, Lassen C, et al. The t(X;7)(q22;q34) in paediatric T-cell acute lymphoblastic leukaemia results in overexpression of the insulin receptor substrate 4 gene through illegitimate recombination with the T-cell receptor beta locus. Br J Haematol. 2009;144:546–51.
Rowe JM. Prognostic factors in adult acute lymphoblastic leukaemia. Br J Haematol. 2010;150:389–405.
Van Vlierberghe P, van Grotel M, Tchinda J, et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood. 2008;111:4668–80.
Zipfel PA, Zhang W, Quiroz M, et al. Requirement for Abl kinases in T cell receptor signaling. Curr Biol. 2004;14:1222–31.
Hagemeijer A, Graux C. ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2010;49:299–308.
Flex E, Petrangeli V, Stella L, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205:751–8.
Otsubo K, Kanegane H, Eguchi M, et al. ETV6-ARNT fusion in a patient with childhood T lymphoblastic leukemia. Cancer Genet Cytogenet. 2010;202:22–6.
Lahortiga I, De Keersmaecker K, Van Vlierberghe P, et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet. 2007;39:593–5.
Hebert J, Cayuela JM, Berkeley J, et al. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood. 1994;84:4038–44.
Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13:1203–10.
Maser RS, Choudhury B, Campbell PJ, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–71.
O’Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–24.
Ferrando A. NOTCH mutations as prognostic markers in T-ALL. Leukemia. 2010;24:2003–4.
Balgobind BV, Van Vlierberghe P, van den Ouweland AM, et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood. 2008;111:4322–8.
Van Vlierberghe P, Meijerink JP, Stam RW, et al. Activating FLT3 mutations in CD4+/CD8- pediatric T-cell acute lymphoblastic leukemias. Blood. 2005;106:4414–5.
Paietta E, Ferrando AA, Neuberg D, et al. Activating FLT3 mutations in CD117/KIT(+) T-cell acute lymphoblastic leukemias. Blood. 2004;104:558–60.
Kleppe M, Lahortiga I, El Chaar T, et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42:530–5.
Remke M, Pfister S, Kox C, et al. High-resolution genomic profiling of childhood T-ALL reveals frequent copy-number alterations affecting the TGF-beta and PI3K-AKT pathways and deletions at 6q15–16.1 as a genomic marker for unfavorable early treatment response. Blood. 2009;114:1053–62.
Van Vlierberghe P, Palomero T, Khiabanian H, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42:338–42.
•• Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. Zhang and colleagues performed whole genome sequencing of 12 ETP ALL cases identifying activating mutations in genes regulating cytokine receptor and RAS signaling (67 %), inactivating lesions disrupting hematopoietic development (58 %), and histone-modifying genes (48 %). Gene expression profile of ETP ALL resulted similar to that of normal and myeloid leukemia hematopoietic stem cells, suggesting the possibility that myeloid-directed therapies might improve the poor outcome of ETP ALL.
Sulong S, Moorman AV, Irving JA, et al. A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood. 2009;113:100–7.
Mullighan CG, Williams RT, Downing JR, et al. Failure of CDKN2A/B (INK4A/B-ARF)-mediated tumor suppression and resistance to targeted therapy in acute lymphoblastic leukemia induced by BCR-ABL. Genes Dev. 2008;22:1411–5.
Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71.
Breit S, Stanulla M, Flohr T, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108:1151–7.
Asnafi V, Buzyn A, Le Noir S, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113:3918–24.
Baldus CD, Thibaut J, Goekbuget N, et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adult acute T-lymphoblastic leukemia. Haematologica. 2009;94:1383–90.
Gutierrez A, Sanda T, Grebliunaite R, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114:647–50.
Yamada Y, Hatta Y, Murata K, et al. Deletions of p15 and/or p16 genes as a poor-prognosis factor in adult T-cell leukemia. J Clin Oncol. 1997;15:1778–85.
Fizzotti M, Cimino G, Pisegna S, et al. Detection of homozygous deletions of the cyclin-dependent kinase 4 inhibitor (p16) gene in acute lymphoblastic leukemia and association with adverse prognostic features. Blood. 1995;85:2685–90.
Markaki EA, Stiakaki E, Zafiropoulos A, et al. Mutational analysis of the cell cycle inhibitor Kip1/p27 in childhood leukemia. Pediatr Blood Cancer. 2006;47:14–21.
Tosello V, Mansour MR, Barnes K, et al. WT1 mutations in T-ALL. Blood. 2009;114:1038–45.
Renneville A, Kaltenbach S, Clappier E, et al. Wilms tumor 1 (WT1) gene mutations in pediatric T-cell malignancies. Leukemia. 2010;24:476–80.
Gutierrez A, Sanda T, Ma W, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood. 2010;115:2845–51.
Kleppe M, Soulier J, Asnafi V, et al. PTPN2 negatively regulates oncogenic JAK1 in T-cell acute lymphoblastic leukemia. Blood. 2011;117:7090–8.
Acknowledgments
Supported by European LeukemiaNet, AIL, AIRC, Fondazione Del Monte di Bologna e Ravenna, FIRB 2006, Ateneo RFO grants, Project of integreted program (PIO), Programma di Ricerca Regione—Università 2007–2009.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Iacobucci, I., Papayannidis, C., Lonetti, A. et al. Cytogenetic and Molecular Predictors of Outcome in Acute Lymphocytic Leukemia: Recent Developments. Curr Hematol Malig Rep 7, 133–143 (2012). https://doi.org/10.1007/s11899-012-0122-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-012-0122-5