
Abstract
Helicobacter pylori infection remains common worldwide and is significantly associated with gastric adenocarcinoma and gastric mucosa-associated lymphoid tissue (MALT)lymphoma. This article reviews recent developments in the field of H. pylori with an emphasis on mechanisms of carcinogenesis, and the bacterial, environmental and host factors that may alter risk of developing gastric cancer or gastric MALT lymphoma. The topic of eradication of H. pylori to prevent the development of malignancy and the possibility of a vaccine against H. pylori are also explored.
Similar content being viewed by others
Introduction
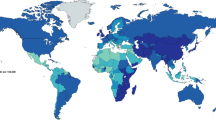
Helicobacter pylori haa been implicated in numerous gastrointestinal diseases after its association with chronic gastritis was established in 1983 by Warren and Marshall [1]. The spiral-shaped Gram-negative bacteria remains the most common chronic bacterial infection in humans worldwide. The prevalence of the infection varies greatly throughout different parts of the world, with an increased prevalence in developing countries and places with lower socioeconomic status compared to developed nations, where the frequency of infection has fallen substantially. There is as much as an 80 % or greater prevalence rate in developing countries as opposed to 20–50 % in developed countries [2] (Fig. 1). Gastric cancer is largely attributable to H. pylori infection, and it is the second most common cause of cancer death worldwide.

Color-coded map depicting the worldwide prevalence of H. pylori infection. More than three-quarters of the population in certain areas of Africa and Asia are infected with H. pylori, whereas the infection is less prevalent in Europe and North America. (Modified from the figure in “The Human Gastric Pathogen Helicobacter pylori and Its Association with Gastric Cancer and Ulcer Disease”, Ulcers, 2011) [69]
Of the various disorders that are associated with H. pylori, the relationship between H. pylori and gastrointestinal malignancy has become one of the best studied in the last three decades. Gastric malignancies that are most commonly associated with H. pylori are gastric adenocarcinoma and gastric mucosa-associated lymphoid tissue (MALT) lymphoma. As early as 1994, the International Agency for Research declared H. pylori a Group I human carcinogen for gastric adenocarcinoma [1].
Diagnosis and Classification of Gastric Adenocarcinoma and MALT Lymphoma
Gastric adenocarcinoma accounts for more than 90 % of cancers of the stomach and can be divided into two distinct types, intestinal and diffuse. The diagnosis of gastric adenocarcinoma continues to be made through endoscopy and tissue biopsy. A single biopsy of a suspicious lesion has 70 % sensitivity for diagnosing gastric cancer; however, increasing this number to 7 biopsies of the ulcer margin and base increase the sensitivity to 98 % [3].
There are two major classifications systems for gastric adenocarcinoma. The Japanese classification is based upon specific anatomic locations, focusing on the location of affected lymph nodes [4]. The more commonly used system in the United States was developed by the American Joint Committee on Cancer (AJCC) and the International Union Against Cancer (IUAC). As outlined in Table 1, this system is based on 3 major components of carcinoma, tumor (T), nodal involvement (N), and metastases (M) [5]. Tumor classification is based on layer of invasion of the primary tumor. Nodal classification reflects the number and location of the lymph nodes that are involved in the disease. Metastases classification is simply based on whether or not distant metastases are present.
Mucosa-associated lymphoid tissue lymphoma, or MALT lymphoma, accounts for up to 3 % of all gastric malignancies [6]. The diagnosis of MALT lymphoma is based on tissue histology from endoscopy, along with immunocytochemistry for B lymphocyte markers. Histological findings including lymphoepithelial changes, polymorphic cellular content, and centrocyte-like cells, impact the grade of the lymphoma and depends on the number of large blast cells present, with greater than 20 % leading to classification as high grade lymphoma [7].
Helicobacter pylori Infection and Gastric Adenocarcinoma
There is a substantial body of data that links H. pylori infection to gastric adenocarcinoma. A recent review indicates that 2 million cases of cancer each year are attributable to infection with H. pylori being a key infectious agent leading to gastric cancer worldwide [8•].
The EUROGAST study observed diverse populations and found a 6-fold increase in likelihood of developing gastric adenocarcinoma in patients with evidence of in H. pylori infection in comparison to those without infection [9]. Additionally, there is a much greater risk of developing adenocarcinoma in H. pylori infected individuals younger than 30 years of age[10].
Helicobacter pylori infection has been associated with an increase in both intestinal and diffuse types of gastric adenocarcinoma [10, 11]. However, there may be a difference in the location of gastric cancer found in H. pylori-infected patients. Distal gastric cancer is much more likely to occur in H. pylori-infected patients than gastroesophageal junction adenocarcinoma [12].
Despite the well-established and clear association between persistent H. pylori infection and gastric adenocarcinoma, only a small percentage of infected individuals will develop malignancy. This is likely due to a myriad of external or environmental factors that are believed to affect the disease course and progression. Factors that promote development of malignancy include certain dietary influences, such as high salt diet, red and processed meat, and nitrosamines, while other factors including diets high in fresh foods and vegetables may reduce the risk [13, 14]. Recently, an association between an elevated white blood cell count and an increased risk for development of gastric cancer has been reported in Japanese subjects [15]. The greatest risk was in patients with H. pylori infection and an elevated white blood cell count.
Obesity has been reported to be associated with gastric cardia adenocarcinoma [16]. This association may be related to H. pylori infection as there is an apparent increased prevalence of H. pylori infection in obese patients [17]. A mechanism explaining this phenomenon has not yet been proven, and the observation may not directly reflect obesity per se. This may relate to reports suggesting that hyperglycemia is a factor increasing the risk of developing gastric cancer [18]. Collectively, these studies lead to the possibility that eradication of H. pylori in conjunction with weight loss or better glycemic control might decrease risk of gastric cancer.
Mechanisms of Carcinogenesis and Helicobacter pylori Infection
Despite the extensive data linking H. pylori and gastric cancer, the exact mechanisms by which gastric cancer develops as a result of infection have not been established; however, data support the role of host factors modulating the immune and epithelial response, environmental factors such as diet, and, to a certain extent, bacterial factors. Emerging information about the human microbiome may reveal further insights into why only subsets of infected individuals develop gastric cancer.
Epithelial Response to Infection
A key aspect of the chronic infection involves establishing infection of the mucosal layer, which reflects bacterial properties including expression of flagella, adhesion molecules and other established virulence factors. A newly proposed mechanism of H. pylori infectivity involves the interaction between H. pylori and protein kinase C isozymes. H. pylori have been reported to induce phosphorylation of protein kinase C. This, in turn, activates matrix mettaloproteinase-1 (MMP-1) expression and secretion. MMP-1 is an interstitial collagenase, which, in increased quantities, facilitates H. pylori cellular invasion [19].
H. pylori infection causes hyperproliferation of epithelial cells and increased rates of apoptosis in antral cells in H. pylori-infected individuals, which return to normal once eradication of the infection occurs [20]. There are multiple proposed pathways for how H. pylori leads to dysregulation of apoptosis and, eventually, to gastric cancer. A proposed pathophysiology is that H. pylori infection induces increased expression of the FAS receptor on gastric epithelium cells. This causes increased activation of apoptosis through the FAS death receptor pathway [21]. Hyperproliferation of gastric epithelial cells may occur due to the fact that dysplastic gastric proliferating cells may produce increased levels of Bcl-2, an antiapoptotic protein, and thus are more resistant to apoptosis [22, 23]. A recent report indicates that c-Src and c-Abl kinases sequentially phosphorylate CagA [24]. The two phosphorylation events need not occur on the same CagA molecule but are both required for the biological effects of CagA. Another recent study demonstrated that vacuolating cytotoxin and variants in Atg16L1 disrupt autophagy and promote H. pylori infection in humans. As autophagy protects against infection with H pylori, this could contribute to inflammation and eventual carcinogenesis [25].
The bacterial virulence factor CagA is a possible culprit in the activation of the apoptosis pathway. A recent article about H. pylori found a relationship between CagA and the p53 tumor suppressor gene. CagA interacts with an apoptosis-stimulating protein of p53 and leads to degradation of p53. This can lead to unsuppressed cell growth and replication [26]. Cellular inhibitor of apoptosis protein 2 (clAP2) may be a potential gene to target in H. pylori-induced carcinogenesis. In mice with H. pylori-induced gastric cancer, knocking out of clAP2 in cancer cells resulted in a 30 % decrease in cellular proliferation and 20 % increase in apoptosis [27]. The majority of human gastric cancer cells are thought to have higher amounts of clAP2 and it is hypothesized that targeting clAP2 in humans could actually be more effective than in the reported mice trials. A mechanism for the interaction with H. pylori and cIAP2 has not yet been explored.
Inflammatory Response to Infection and Oxidative Stress
Activation of neutrophils in response to H. pylori infection appears to be an important aspect of gastric carcinogenesis. Neutrophils produce nitric oxide synthase, which leads to the formation of nitric oxide and reactive oxygen metabolites, including superoxides and hydroxyl ions. These compounds lead to significant DNA damage, mutations, and eventually malignancy. This DNA damage can be in the form of DNA demethylation which is reported in multiple studies to be a key factor in H. pylori-induced carcinogenesis. Demethylation of the Sat α repetitive gene sequence has in particular been correlated with H. pylori-positive gastric cancer. This increase is observed predominantly in patients under the age of 45 [28].
Chronic H. pylori infection can lead to atrophic gastritis, intestinal metaplasia, and the loss of parietal cells, with an increase in stomach pH levels and decreased amounts of ascorbic acid in the stomach. This provides a receptive environment for nitrate-reducing bacteria to thrive and to produce nitrates and free radicals. Normally, ascorbic acid blocks this nitrosation reaction by scavenging nitrates and radicals. However, in cases of intestinal metaplasia, the decreased parietal cell mass leads to reduced levels of ascorbic acid and, thus, increased damaging products in the gastric lumen. A recent study suggests that, by inducing spermine oxidase which metabolizes the polyamine spermine into spermidine and generates H2O2, H pylori CagA generates cells with oxidative DNA damage, rendering a subpopulation of these cells resistant to apoptosis and, thus, at high risk for malignant transformation [29].
Regulation of Host Response to Infection
Apurinic-apyrimidinic endonuclease (APE)-1 is a rate-limiting DNA base excision DNA repair enzyme. The molecule is also known as redox factor 1 (ref-1) for its transcriptional regulatory activity. APE-1’s function and expression can be altered in the setting of H. pylori infection. APE-1 expression is increased in gastric cancer and other malignancies and it is increased in gastric epithelial cells by H. pylori infection. These results suggest that it plays a role in carcinogenesis [30] and, interestingly, APE-1 is known to regulate oxidative stress, chemokine expression, and apoptosis [31].
Runt-related transcription factor (RUNX3) is a candidate tumor suppressor gene whose deficiency has been seen with gastric cancer. A recent study found that abnormal methylation of the gene along with H. pylori infection was associated with an increased risk of gastric cancer in patients with chronic atrophic gastritis [32], implicating RUNX3 as a potential biomarker in early gastric cancer.
One very intriguing genetic polymorphism that has been found to be protective against H. pylori carcinogenesis in mice is vitamin D3 upregulated protein 1 (VDUP1). This leads to the possibility of a link between vitamin D deficiency and propensity for H. pylori infection to progress into gastric cancer in patients [33].
Helicobacter pylori Virulence Factors
Cag A is the most studied of H. pylori virulence factors. There is now evidence that Cag A may be an oncogene, regardless of the presence of gastritis [34]. There have been studies that show transgenic expression of Cag A leads to cancer in mice, despite the absence of gastritis. However, to date, there have been no human studies like this. Additionally, there may be variations in propensity to develop gastric cancer in H. pylori-infected patients based on the presence of distinct virulence factors. In an Iranian study, the presence of the homB gene was found to be significantly higher in H. pylori patients with gastric cancer as opposed to peptic ulcers or gastritis [35].
The combination of specific bacterial virulence factors and specific genetic polymorphisms can significantly potentiate disease risks. For example, patients infected with the vacA s1 genotype (vacuole cytotoxin), who possessed a particular IL-1B polymorphism (IL-1B-511), were found to have a significantly increased risk of developing gastric cancer with an odds ratio of 87 [36]. However, despite the data for multiple virulence factors, the updated Maastricht guidelines still state that no particular bacterial virulence marker can be used for clinical practice at this time [37•].
Management of Gastric Adenocarcinoma and H. pylori Infection
There are multiple options for the treatment of gastric adenocarcinoma. They include endoscopic resection, surgical gastrectomy, adjuvant chemotherapy, and H. pylori eradication. There are specific criteria for eligibility for endoscopic resection which takes in to account tumor size, histology, nodal involvement, and level of invasion [38, 39]. The latest Maastricht guidelines have made a grade A recommendation that H. pylori infection is the most consistent risk factor for gastric cancer and that its elimination is the most promising strategy to reduce the incidence of gastric cancer [37•].
Treatment of H. pylori has been proven to decrease the risk of developing gastric cancer and decreases recurrence following treatment of gastric cancer [40, 41]. A recent study once again demonstrated that, after H. pylori eradication, the risk of developing gastric atrophy and intestinal metaplasia are all significantly decreased. This can lead to a reduction in gastric cancer in previously H. pylori-infected people[40]. In contrast, a study of mass eradication of H. pylori infection starting in 2004 in Taiwan demonstrated a reduction of gastric atrophy, but not intestinal metaplasia, when compared to the 5-year period before the study started [42•]. There was an associated decrease in gastric cancer and peptic ulcer disease and an increase in esophagitis. Further studies, including longer-term follow-up, are needed to confirm these findings.
Once intestinal metaplasia and gastric cancer are established, there are mixed data as to what the eradication of H. pylori actually accomplishes. In some studies, H. pylori eradication has been observed to aid prevention of the reoccurrence of early gastric cancer (EGC) even after endoscopic resection [41, 43]. This was studied in a randomized control trial comparing standard endoscopic resection of EGC versus the addition of triple therapy for H. pylori eradication. The group receiving triple therapy was observed to have a lower risk of metachronous gastric cancer over a 3-year period from diagnosis [41, 43]. However, recently, a retrospective study showed that there was no significant difference in development of metachronous gastric cancer in patients who underwent endoscopic gastric cancer resection, whether or not they received H. pylori eradication treatment, when analyzed after 5 years post-eradication [44•].
There may be a difference in outcomes of eradication of the disease based upon which portions of the stomach are affected. A meta-analysis of 12 studies, including over 2,600 patients, found that eradication of H. pylori significantly improves corpus gastric atrophy, but not antral gastric atrophy. This same study reported no effect of eradication on gastric intestinal metaplasia [45]. Whether eradication of infection once premalignant changes are established is beneficial with regard to the development of cancer remains unclear and warrants further study.
It has been proposed that measurement of serum pepsinogen I may predict whether H. pylori eradication will be effective in preventing development of gastric cancer. In a pooled analysis of 6,695 patients, only patients with normal pepsinogen levels were seen to have a significant reduction in gastric cancer incidence [46]. This suggests that cancer which arises after H. pylori eradication is due to the presence of extensive atrophic gastritis prior to eradication and that eradication is most beneficial in patients with absent or mild atrophy. Serum pepsinogen has been proposed as a possible screening test for gastric cancer, due to the ability to detect severe atrophy [47].
Helicobacter Pylori and Gastric Lymphoma
Chronic H. pylori infection leads to chronic gastritis which involves both T cells and B cells arising from the mucosal-associated lymphoid tissue (MALT). The recognition of the H. pylori antigen by the immune system leads to T cell activation, lymphoid follicle formation, and B cell proliferation. The gastric follicle that forms has three layers: an inner center of centroblasts and centrocytes and two outer layers comprising of B cells, the middle mantle layer and the outer marginal layer. It is thought that the H. pylori antigen-presenting cells interact with CD4-expressing cells. This CD4 cell then binds to a B cell in the marginal zone causing hyperproliferation of B cells [48, 49]. In this milieu, it is believe that chronic stimulation leads to the development of B cell lymphoma, also known as a MALT lymphoma or maltoma.
When H. pylori is present in patients with gastric MALT lymphoma, eradication of H. pylori has been an effective initial method of treatment of localized early stage gastric MALT lymphoma with anywhere from 50 to 80 % rates of complete histological remission [50–56, 57•]. However, recent studies have further stratified this by looking at outcomes by stage. H. pylori eradication led to remission in greater than 75 % of patients in with stage I gastric MALT lymphoma and about 55 % in patients with stage II disease [58]. Additionally, the location of the lymphoma may make a difference in remission rates. There has been at least one study that has shown that patients with more superficial tumors located in the distal stomach were more likely to achieve complete remission [59]. Complete remission has been documented as early as 5 months and as long as 3 years from time of treatment [50–56, 57•]. The chromosomal translocation, t(11;18)(q21;q21), and gene expression, nuclear BCL, have been found to be resistant to H. pylori eradication [60].
H. pylori eradication is not thought to be effective in patients with metastases, lymphadenopathy, or diffuse large B cell lymphomas [61–63]. However, a recent study showed that, in early stage gastric diffuse large B-cell lymphomas both with and without features of mucosa-associated lymphoid tissue, H. pylori eradication resulted in complete pathologic remission in a significant percentage of cases [64].
Alternative therapies such as radiation therapy have been found to have just as effective, if not more effective, in achieving complete remission as eradication of infection, and have also been found to have lower recurrence rates than following H. pylori treatment. However, there are significantly more complications with radiation treatment as well as with gastrectomy than eradication of infection.
Future Developments: Vaccination?
Research on the development of a vaccine for the prevention or treatment of H. pylori dates back to the early 1990s, but unfortunately, successes have been few and far between. Immunity has rarely been achieved in animal models and no consensus opinion has been reached on adjuvants, antigens, or method of delivery. There have been a few clinical trials that for the most part have been unsuccessful, but there is some hope for the future. Much of the current research focuses on Th1, Th17, and regulatory T cell-based immunity including exploration of the role of Th17 cells and the role of IL-17 in H. pylori infection and vaccination [65, 66].
T cell epitopes have been studied as a target of H. pylori vaccination [67]. In a recent study, conserved DNA sequence encoding was used to develop HLA-2 epitopes and administered via an intranasal vaccine [67]. This vaccine led to about 25 % (5/19) sterile immunity at 32 weeks in mice [67]. Although many barriers still remain, this approach did provide some promising results and a possible method for further studies of vaccine development. H. pylori urease has been extensively studied as a target for vaccination, given its integral role in H. pylori colonization. However, developing epitopes that induce immunity has been challenging, and some researchers have proposed that combination with other antigens may provide more promising results, including genetic fusion of multiple proteins to develop a multivalent vaccine [68]. Other antigens have been explored, such as Omp18, TonB, superoxide dismutase, and protein-conjugated LPS, but the results are not conclusive. Human vaccination still faces numerous obstacles, both in development and in safety; however some progress has been made, and research on this topic is ongoing.
Conclusions
Helicobacter pylori infection remains the most common chronic bacterial infection of humans, but, due to improved socioeconomic status of the developed world, rates are declining in these nations. Given that gastric malignancies result from longstanding infection, the burden of gastric cancer is still high in areas of the world where H. pylori infection is common, such as the Far East, Central and South America, and eastern Europe. First generation immigrants from these at-risk areas of the world will also have an increased risk of gastric cancer. Early and effective eradication of infection continues to remain the mainstay of treatment or prevention of gastric malignancy at this time. When, in the sequence of progression to malignancy, it is best to eradicate infection requires additional study. Prevention of H. pylori has been studied, but socioeconomic advances are the major reason for decreasing rates of infection. Since complete elimination of H pylori infection worldwide is unlikely to happen for many generations, if ever, research is needed to identify which infected individuals will go on to develop gastrointestinal malignancy, and potentially target those populations for treatment to prevent gastric cancer. Strategies to modulate infection and the ensuing inflammatory response to reduce the likelihood of developing cancer are another area for further investigation.
Reference
Papers of particular interest, published recently, have been highlighted as: • Of importance
McColl KEL. Helicobacter pylori infection. NEJM. 2010;362:1597–604.
Pounder RE, Ng D. The prevalence of Helicobacter pylori infection in different countries. Aliment Pharmacol Ther. 1995;9(Supp 2):33.
Graham DY, Schwartz JT, Cain GD, Gyorkey F. Prospective evaluation of biopsy number in the diagnosis of esophageal and gastric carcinoma. Gastroenterology. 1982;82(2):228.
Japanese Research Society for Gastric Cancer: The General Rules for the Gastric Cancer Study in Surgery and Pathology. Kanahara Shuppan 1993.
Edge SB, Byrd DR, Compton CC, et al.: American Joint Committee on Cancer Staging Manual 2010. 117.
Freeman C, Berg JW, Cutler SJ. Occurrence and prognosis of extranodal lymphomas. Cancer. 1972;29(1):252.
Isaacson PG: Gastrointestinal lymphomas and lymphoid hyperplasias. Neoplastic Haematolpathology 1992:953.
• de Martel C, Ferlay J, Franceschi S, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13(6):607–15. This is an interesting report indicating that of the 12.7 million new cancers occurring in 2008, the population attributable fraction due to infection was over 16% with H. pylori, hepatitis B and C and human papilloma virus responsible for 1.9 million cases. The fraction was higher (almost 23%) in the less developed world.
The EUROGAST Study Group. An international association between Helicobacter pylori infection and gastric cancer. Lancet. 1993;341(8857):1359.
Huang JQ, Sridhar S, Chen Y, et al. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology. 1998;114(6):1169.
Hansson LR, Engstrand L, Nyrén O, et al. Prevalence of Helicobacter pylori infection in subtypes of gastric cancer. Gastroenterology. 1995;109(3):885.
Kamada T, Kurose H, Yamanka Y, et al. Relationship between Gastroesophageal Junction Adenocarcinoma and Helicobacter pylori Infection in Japan. Digestion. 2012;85(4):256–60.
Tsugane S, Tei Y, Takahashi T, et al. Salty food intake and risk of Helicobacter pylori infection. Jpn J Cancer Res. 1994;85:474.
González CA, López-Carrillo L. Helicobacter pylori, nutrition and smoking interactions: their impact in gastric carcinogenesis. Scand J Gastroenterol. 2010;45:6.
Iida M, Ikeda F, Ninomiya T, et al. White blood cell count and risk of gastric cancer incidence in a general Japanese population: the Hisayama study. Am J Epid. 2012;175(6):504–10.
Cho Y, Lee DH, Oh HS, et al.: Higher Prevalence of Obesity in Gastric Cardia Adenocarcinoma Compared to Gastric Non-Cardia Adenocarcinoma. Digestive Diseases and Sciences 2012.
Li Q, Zhang J, Zhou YN, Qiao L. Obesity and gastric cancer. Front biosci: j virtual library. 2012;17:2383–90.
Ikeda F, Doi Y, Yonemoto K, et al. Hyperglycemia increases risk of gastric cancer posed by Helicobacter pylori infection: a population-based cohort study. Gastroenterology. 2009;136(4):1234–41.
Sokolova O, Vieth M, Naumann M: Protein kinase C isozymes regulate matrix metalloproteinase-1 expression and cell invasion in Helicobacter pylori infection. Gut 2012
Moss SF, Calam J, Agarwal B, et al. Induction of gastric epithelial apoptosis by Helicobacter pylori. Gut. 1996;38:498.
Jones NL, Day AS, Jennings HA, et al. Helicobacter pylori induces gastric epithelial cell apoptosis in association with increased Fas receptor expression. Infect Immun. 1999;67:4237.
Correa P, Miller MJ. Helicobacter pylori and gastric atrophy–cancer paradoxes. J Natl Cancer Inst. 1995;87:1731.
Lauwers GY, Scott GV, Hendricks J. Immunohistochemical evidence of aberrant bcl-2 protein expression in gastric epithelial dysplasia. Cancer. 1994;73:2900.
Müller A. Multistep activation of the Helicobacter pylori effector CagA. J Clin Invest. 2012;122(4):1192–5.
Raju D, Hussey S, Ang M, et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology. 2012;142(5):1160–71.
Buti L, Spooner E, Van der Veen AG, et al. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc Natl Acad Sci USA. 2011;108(22):9238–43.
Li Z, Chen J, Chan KW, et al. A possible role of cIAP2 in Helicobacter pylori-associated gastric cancer. Cancer Lett. 2011;313(2):192–200.
Saito M, Suzuki K, Maeda T, et al. The accumulation of DNA demethylation in Sat α in normal gastric tissues with Helicobacter pylori infection renders susceptibility to gastric cancer in some individuals. Oncology Rep. 2012;27(6):1717–25.
Chaturvedi R, Asim M, Romero-Gallo J, et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology. 2011;141(5):1696–708.
Silva-Fernandes IJ, da Silva TA, Agnez-Lima LF, et al. Helicobacter pylori genotype and polymorphisms in DNA repair enzymes: Where do they correlate in gastric cancer? J Surg Oncol. 2012;106(4):448–55.
Chattopadhyay R, Bhattacharyya A, Crowe SE. Dual regulation by apurinic/apyrimidinic endonuclease-1 inhibits gastric epithelial cell apoptosis during Helicobacter pylori infection. Cancer Res. 2010;70(7):2799–808.
Lu XX, Yu JL, Ying LS, et al.: Stepwise cumulation of RUNX3 methylation mediated by Helicobacter pylori infection contributes to gastric carcinoma progression. Cancer 2012
Kwon HJ, Won YS, Nam KT, et al. Vitamin D3 upregulated protein 1 deficiency promotes N-methyl-N-nitrosourea and Helicobacter pylori-induced gastric carcinogenesis in mice. Gut. 2012;61(1):53–63.
Ohnishi N, Yuasa H, Tanaka S, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105:1003.
Talebi B, Abadi A, Rafiei A, et al. Helicobacter pylori homB, but not cagA, is associated with gastric cancer in Iran. J Clin Microbiol. 2011;49(9):3191–7.
Figueiredo C, Machado JC, Pharoah P, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680e7.
• Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection–the Maastricht IV/ Florence Consensus Report. Gut. 2012;61(5):646–64. This important consensus arising from a meeting of 44 experts from 24 countries addresses treatment of H. pylori infection and strategies for reducing incidence of gastric adenocarcinoma secondary to H. pylori.
Gotoda T. Endoscopic resection of early gastric cancer: the Japanese perspective. Curr Opin Gastroenterol. 2006;22(5):561.
Soetikno R, Kaltenbach T, Yeh R, et al. Endoscopic mucosal resection for early cancers of the upper gastrointestinal tract. J Clin Oncol. 2005;23(20):4490.
Fuccio L, Zagari RM, Eusebi LH, et al. Meta-analysis: can Helicobacter pylori eradication treatment reduce the risk for gastric cancer? Ann Intern Med. 2009;151:121.
Uemura N, Mukai T, Okamoto S, et al. Effect of Helicobacter pylori eradication on subsequent development of cancer after endoscopic resection of early gastric cancer. Cancer Epidemiol Biomarkers Prev. 1997;6:639.
• Lee YC, Chen TH, Chiu HM, et al.: The benefit of mass eradication of Helicobacter pylori infection: a community-based study of gastric cancer prevention. Gut 2012. This unique study reports outcomes from two time periods 1995-2003 and 2004-208 before and after a national H. pylori eradication program in Taiwan.
Fukase K, Kato M, Kikuchi S, et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008;372:392.
• Maehata Y, Nakamura S, Fujisawa K, et al. Long-term effect of Helicobacter pylori eradication on the development of metachronous gastric cancer after endoscopic resection of early gastric cancer. Gastrointest Endosc. 2012;75(1):39–46. This retrospective multicenter study reports results from longer follow-up which showed a lower rate of new gastric cancers after endoscopic mucosal resection in the patients in whom infection was eradicated compared those in whom infection persisted but only up to 5 years. With longer-term follow-up the results were no longer significant.
Wang J, Xu L, Shi R, et al. Gastric atrophy and intestinal metaplasia before and after Helicobacter pylori eradication: a meta-analysis. Digestion. 2011;83:253e60.
Yanaoka K, Oka M, Ohata H, et al. Eradication of Helicobacter pylori prevents cancer development in subjects with mild gastric atrophy identified by serum pepsinogen levels. Int J Cancer. 2009;125:2697e703.
Miki K, Morita M, Sasajima M, et al. Usefulness of gastric cancer screening using the serum pepsinogen test method. Am J Gastroenterol. 2003;98:735e9.
Lydyard P, Grossi C: Secondary lymphoid organs and tissues. Immunology 1996
D'Elios MM, Amedei A, Manghetti M, et al. Impaired T-cell regulation of B-cell growth in Helicobacter pylori–related gastric low-grade MALT lymphoma. Gastroenterology. 1999;117:1105.
Roggero E, Zucca E, Pinotti G, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med. 1995;122:767.
Bayerdörffer E, Neubauer A, Rudolph B, et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection: MALT Lymphoma Study Group. Lancet. 1995;345:1591.
Steinbach G, Ford R, Glober G, et al. Antibiotic treatment of gastric lymphoma of mucosa-associated lymphoid tissue: an uncontrolled trial. Ann Intern Med. 1999;131:88.
Chen LT, Lin JT, Tai JJ, et al. Long-term results of anti-Helicobacter pylori therapy in early-stage gastric high-grade transformed MALT lymphoma. J Natl Cancer Inst. 2005;97:1345.
Bertoni F, Conconi A, Capella C, et al. Molecular follow-up in gastric mucosa-associated lymphoid tissue lymphomas: early analysis of the LY03 cooperative trial. Blood. 2002;99:2541.
Wündisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol. 2005;23:8018.
Stathis A, Chini C, Bertoni F, et al. Long-term outcome following Helicobacter pylori eradication in a retrospective study of 105 patients with localized gastric marginal zone B-cell lymphoma of MALT type. Ann Oncol. 2009;20:1086.
• Nakamura S, Sugiyama T, Matsumoto T, et al. Long-term clinical outcome of gastric MALT lymphoma after eradication of Helicobacter pylori: a multicentre cohort follow-up study of 420 patients in Japan. Gut. 2012;61:507. This recent multicenter Japanese study describes the follow-up of 420 patients with low-grade MALT lymphoma for three or more years after H. pylori eradication with a 77 % response.
Zullo A, Hassan C, Cristofari F, et al. Effects of Helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphoma. Clin Gastroenterol Hepatol. 2010;8(2):105–10.
Kim JS, Chung SJ, Choi YS, et al. Helicobacter pylori eradication for low-grade gastric mucosa-associated lymphoid tissue lymphoma is more successful in inducing remission in distal compared to proximal disease. Br J Cancer. 2007;96:1324.
Yepes S, Torres MM, Saavedra C, et al. Gastric mucosa-associated lymphoid tissue lymphomas and Helicobacter pylori infection: a Colombian perspective. World J Gastroenterol. 2012;18(7):685–91.
Chen LT, Lin JT, Shyu RY, et al. Prospective Study of Helicobacter pylori Eradication Therapy in Stage I(E) High-Grade Mucosa-Associated Lymphoid Tissue Lymphoma of the Stomach. J Clin Oncol. 2001;19:4245.
Fischbach W, Dragosics B, Kolve-Goebeler ME, et al. Primary gastric B-cell lymphoma: results of a prospective multicenter study: The German-Austrian Gastrointestinal Lymphoma Study Group. Gastroenterology. 2000;119:1191.
Ruskone-Fourmestraux A, Lavergne A, Aegerter PH, et al. Predictive factors for regression of gastric MALT lymphoma after anti-Helicobacter pylori treatment. Gut. 2001;48:297.
Kuo SH, Yeh KH, Wu MS, et al. Helicobacter pylori eradication therapy is effective in the treatment of early-stage H pylori-positivegastric diffuse large B-cell lymphomas. Blood. 2012;119(21):4838–44.
Velin D, Narayan S, Bernasconi E, et al. PAR2 promotes vaccine-induced protection against Helicobacter infection in mice. Gastroenterology. 2011;141(4):1273–82.
Zhang JY, Liu T, Guo H, et al. Induction of a Th17 cell response by Helicobacter pylori Urease subunit B. Immunobiology. 2011;216(7):803–10.
Moss SF, Moise L, Lee DS, et al. HelicoVax: Epitope-based therapeutic H. pylori vaccination in a mouse model. Vaccine. 2011;29(11):2085–91.
Wang L, Liu XF, Yun S, et al. Protection against Helicobacter pylori infection by a trivalent fusion vaccine based on a fragment of urease B-UreB414. J Microbiol (South Korea). 2010;48(2):223–8.
Bauer B, Meyer TF: The Human Gastric Pathogen Helicobacter pylori and Its Association with Gastric Cancer and Ulcer Disease. Ulcers 2011. doi:10.1155/2011/340157.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bhandari, A., Crowe, S.E. Helicobacter pylori in Gastric Malignancies. Curr Gastroenterol Rep 14, 489–496 (2012). https://doi.org/10.1007/s11894-012-0296-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11894-012-0296-y