
Abstract
Patients suffering from familial hypercholesterolemia (FH) are characterized by increased plasma levels of low-density lipoprotein cholesterol (LDL-C) levels and are at increased risk for premature cardiovascular disease (CVD). Current guidelines emphasize the need to aggressively lower LDL-C in FH patients, and statins are the cornerstone in the current regimen. However, additional therapies are eagerly awaited, especially for those patients not tolerating statin therapy or not reaching the goals for therapy. Our understanding of LDL metabolism has improved over the last years and an increasing number of potential novel targets for therapy have been recently identified. Apart from novel targets, we have also been confronted with novel modalities of treatment, such as mRNA antisense therapy. Some of these emerging therapies have proven to be effective in lowering plasma LDL-C levels and are as such expected to have beneficial effects on CVD. Hopefully, they will enrich our armamentarium against the severe dyslipidemia observed in FH patients in the not too distant future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
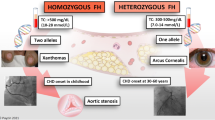
Familial hypercholesterolemia (FH) is an autosomal-dominant disorder characterized by elevated plasma low-density lipoprotein cholesterol (LDL-C) levels. Mutations in the gene encoding for the LDL receptor are the underlying molecular defect in the vast majority of FH patients [1], but mutations in APOB [2] and PCSK9 have also been shown to result in Mendelian forms of increased LDL-C levels [3]. FH patients are at sharply increased lifetime risk for cardiovascular disease (CVD) and, if left untreated, clinical symptoms of CVD typically manifest in men in their fourth decade and in women in their fifth decade of life [4]. Apart from the elevated LDL-C levels, other traditional CVD risk factors (ie, smoking, hypertension, diabetes) do add to the total risk in FH patients, and all modifiable risk factors should therefore be aggressively addressed. Current guidelines recommend lowering the LDL-C concentration to at least 50% from baseline. Statins are shown to safely lower LDL-C levels and are therefore the treatment of choice [5, 6]. Moreover, large clinical trials have provided us with overwhelming evidence that statins reduce cardiovascular mortality and morbidity [7••].
However, treatment goals are not achieved in a significant number of FH patients [8–10]. In such patients, and in case statin therapy is contraindicated or poorly tolerated, alternative lipid-lowering medications should be initiated. Ezetimibe, bile acid sequestrants, nicotinic acid, and fibrates are frequently prescribed as add-on therapy to initial treatment with statins [6].
In recent years, several novel promising therapeutic strategies for LDL-C lowering have been developed. In this review, we discuss the present and future treatment options for lipid lowering in FH patients, especially those medications that have been shown, or are anticipated, to result in LDL-C reduction.
Currently Approved Lipid-Lowering Therapy
Lifestyle Modification
In FH patients, lifestyle modification to lower LDL-C and reduce other CVD risk factors should be introduced, despite the modest and variable degree of LDL-C reduction (10%). A diet containing less than 7% saturated fat and less than 200 mg of cholesterol is to be advised. Additional use of plant sterol esters or plant stanol esters will reduce LDL-C levels, although trials showing a beneficial effect of these substances on CVD outcome are lacking [11, 12]. Patients should be encouraged to achieve and maintain a healthy body weight through physical activity and appropriate caloric intake. Alcohol consumption should be restricted and smoking should be discouraged, as it is strongly associated with CVD in patients with hypercholesterolemia [13, 14]. It should be kept in mind that lifestyle modification is rarely, if ever, sufficient to achieve the LDL-C treatment goal in patients with FH and drug therapy is therefore required in almost all patients.
Statins
Statins are 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (Table 1). They inhibit the rate-limiting step in cholesterol synthesis by reducing the conversion of HMG-CoA reductase to mevalonate. The consequently decreased intracellular cholesterol levels induce an upregulation of the LDL receptor, which leads to increased clearance of LDL-C and decreased plasma LDL-C concentrations [15]. Apart from the reduction in LDL-C, statins have been shown to improve endothelial function, stabilize atherosclerotic plaques, decrease oxidative stress and inflammation, and inhibit the thrombogenic response [16].
Statins have convincingly been shown to be safe and well-tolerated agents that reduce CVD morbidity and mortality in a wide range of patients [17]. Therefore, guidelines recommend these drugs as the first-line therapy in patients with FH.
Statins are the most commonly prescribed drugs in FH patients [18] and their impact on the natural cause of vascular disease in FH is large. Observational data from large FH cohorts suggest that long-term statin treatment removes the excess lifetime risk of CVD due to FH and reduced it to a level similar to that of the general population [19, 20].
Statins reduce LDL-C levels in a dose-dependent manner [7••], and the rationale to treat FH patients with high dosages of these therapeutics is based on clinical trials showing benefit in terms of reductions of cardiovascular events and death [21]. Although trials with events as primary outcome are lacking in FH patients, the ASAP trial showed a beneficial effect of intensified therapy on carotid intima-media thickness (cIMT), a surrogate marker of atherosclerosis [22]. FH patients should initially be treated with more potent statins, which have been shown to reduce LDL-C levels by 50% to 60% at their maximum approved doses [23]. Initial concerns about the safety profile of statins, especially in children, have been refuted by a number of clinical trials [24–26].
Other Currently Available Treatment Options
In many FH patients, LDL-C treatment goals cannot be reached with the maximum available or tolerated dose of a statin. In such cases, adding ezetimibe, a bile acid binding resin, fibric acid derivates, or nicotinic acid should be considered. In homozygous FH individuals or in heterozygous FH patients at extreme risk (which is not uniformly specified), LDL apheresis has been shown to be an effective means to reduce LDL-C as well.
Ezetimibe selectively inhibits the intestinal absorption of both dietary and biliary cholesterol by blocking the Niemann-Pick C1-like 1 (NPC1L1) protein transporter, which reduces the delivery of intestinal cholesterol to the liver (Table 1). As a result, the LDL receptor expression is upregulated and clearance of LDL-C from plasma is increased. In patients with FH, ezetimibe can be safely coadministrated with statins [27]. Ezetimibe reduces LDL-C by approximately 15% to 20% [28, 29•]. The clinical benefit of adding ezetimibe to statin therapy has not been proven. In fact, in a large clinical trial in which adult FH patients were randomized to statin or a combination of statin and ezetimibe, no effect was found on the extent of atherosclerosis assessed by cIMT, despite a significant reduction in LDL-C in the combination therapy arm [29•]. A possible explanation for this rather counterintuitive finding is the fact that the baseline cIMT measured in the enrolled patients was not as severely affected as anticipated. The potential beneficial effect of ezetimibe is demonstrated in a recent analysis, in which ezetimibe was shown in FH patients to not only result in significant reductions in LDL-C but also in other CVD-associated plasma markers in FH patients [30]. The SHARP trial showed a beneficial effect on CVD risk (risk reduction, 17%; P = 0.002) of simvastatin combined with ezetimibe compared with placebo in a large cohort (> 9000) patients suffering from chronic kidney disease. In this study, the combination therapy was not associated with altered risk of myopathy or cancer during the 4.9 years of follow-up [31]. Although there is no direct clinical evidence that ezetimibe would result in a beneficial vascular outcome, it is reasonable to consider it as add-on therapy to statins in FH patients given its tolerability and safety.
Bile acid binding resins act by binding to bile acids in the intestinal lumen. This interrupts the enterohepatic circulation of bile acids, leading to increased conversion of cholesterol into bile in the liver. The resulting decreased cholesterol levels in the hepatocytes induce a hepatic upregulation of LDL receptor activity, causing an increased clearance of LDL-C from the circulation by up to 20% [32]. Because bile acid binding sequestrants act in the intestinal lumen and are not systemically absorbed, they are considered to be safer than other lipid-lowering drugs. However, cholestyramine and colestipol are associated with significant adverse gastrointestinal side effects, drug-drug interactions, and poor patient compliance. The second-generation bile acid sequestrant colesevelam can be used at a lower dose and is associated with less gastrointestinal side effects, and is therefore currently the recommended bile acid sequestrant for use in patients with FH in combination with statins [33].
The mechanism of action of fibric acid derivates is complex and largely unknown, but is commonly thought to be mediated via a peroxisome proliferator-activated receptor-α regulated mechanism. Treatment with fibrates results in decreased production of very low density lipoprotein cholesterol (VLDL-C) and an increased clearance of triglycerides. Fibrates have also shown to lower total cholesterol and LDL-C and elevate HDL-C to some extent (Table 1). Adverse reactions are to some extent similar to statins (Table 1); however, the combination of fibrates, most notably gemfibrozil, with statins will increase the risk of myopathy or rhabdomyolysis [34], and therefore fibrates are not recommended in FH patients without elevated triglyceride levels.
The mechanism of action of nicotinic acid or niacin is not fully understood. Niacin is a water-soluble B vitamin and favorably affects VLDL, LDL-C, and increases HDL-C. The adverse effects of niacin (Table 1), mainly flushing due to vasodilatation, are considered a major drawback, but by combining niacin with a prostaglandin D2 inhibitor (laropiprant), this side effect was significantly decreased [35–37]. A meta-analysis has shown cardiovascular risk benefit of niacin in terms of reduction of cardiovascular events and atherosclerosis [38], but a more definitive answer regarding CVD reduction of niacin will be provided by the HPS-2-Thrive study. As for all other agents, however, no conclusions can be drawn regarding the benefit of niacin added to statin therapy in FH patients. The AIM-HIGH trial investigated whether adding extended-release niacin to statin treatment in high-risk patients would be beneficial in risk reduction. This trial has recently been stopped prematurely by the National Heart, Lung, and Blood Institute after 18 months, because no effect was shown in the interim analysis (http://public.nhlbi.nih.gov/newsroom/home/GetPressRelease.aspx?id=2792).
LDL apheresis may be considered for homozygous FH patients or heterozygous FH patients who require intensification of therapy because of high LDL-C levels despite a maximal dose of statins and/or multiple other risk factors for CVD. LDL apheresis selectively removes apo B–containing lipoprotein particles from the circulation with extracorporeal precipitation through different techniques, resulting in an LDL-C reduction of approximately 60% [39]. Furthermore, it reduces lipoprotein (a) levels by more than 50%. The procedure is time consuming, must be repeated every 1 to 2 weeks, and is costly. However, several clinical trials have shown that LDL apheresis delays the progression of CVD [40].
Future Lipid-Lowering Therapy
Antisense Oligonucleotides to Inhibit Apolipoprotein B Production
Apolipoprotein B (apo B) is mainly expressed in the liver and is regarded as an essential protein at core and on the surface of atherogenic lipoproteins. It is crucial for the production of VLDL (the precursor of LDL) and following secretion by the liver, apo B is bound to its lipoprotein particle. Apo B is also pivotal for the subsequent clearance of cholesterol transported by lipoproteins; upon binding to the LDL receptor, cholesterol is withdrawn from the plasma pool [41]. In FH patients apo B levels are invariably increased, and large prospective studies have shown that apo B levels are directly associated with CVD risk [42]. In line with this, patients with extremely low levels of apo B (< 5th percentile) due to familial hypobetalipoproteinemia seem to be protected against CVD [43, 44]. Based on these observations apo B is conceptually an attractive target to reduce CVD risk. Mipomersen (formerly known as ISIS-301012), a second-generation apo B synthesis inhibitor, is the first agent available for human use to directly target apo B100 production. This subcutaneously administered short single-stranded synthetic oligonucleotide is complementary to apo B100 mRNA, and upon binding to the mRNA, degradation by endogenous RNase-H takes place. This subsequently results in inhibition of synthesis of the apo B protein, and a decrease in VLDL and LDL levels [45].
The initial phase 1 study [46] showed a dose-dependent effect on apo B and LDL as well as on all other atherogenic particles. The highly significant dose reductions in LDL-C (Table 2) were confirmed in a number of studies, including trials in FH patients [47–50]. The effect on plasma lipids was shown not to be influenced by coadministration of other lipid-lowering medication [47, 50]. In addition to its beneficial effect on LDL-C, mipomersen also lowered serum levels of apo B, triglycerides, and lipoprotein (a). A dose of 200 mg of mipomersen once weekly administered subcutaneously was selected for further evaluation in phase 3 clinical trials. Reductions in LDL-C lasted up to 4 weeks after the last dose and pharmacokinetic studies showed no clinically relevant interactions of mipomersen with the disposition and clearance of simvastatin or ezetimibe [51], which is pivotal for the role of mipomersen as additive medication.
In a recently published double-blind trial, 51 homozygous FH patients, treated with maximum tolerated dosages of lipid-lowering medications, were randomly assigned to mipomersen, 200 mg, subcutaneously every week or placebo. After 26 weeks of treatment, a mean reduction in LDL-C of 25% was observed in the mipomersen-treated group versus 3% in placebo-treated patients (P < 0.001). In addition, patients treated with mipomersen experienced a 27% reduction in apo B and a 21% reduction in total cholesterol. No correlation was found between the LDL receptor mutation and response to therapy [50].
Similar reductions in LDL-C levels (28%) were shown in a study conducted in 124 patients with heterozygous FH, who were on maximally tolerated statin therapy and had a history of coronary heart disease. It is of note that 45% of these high-risk patients treated with mipomersen reached the treatment goal of LDL-C below 100 mg/dL [52].
Mipomersen is well tolerated and commonly described adverse events include injection site reactions, flu-like symptoms, and increases in alanine aminotransferase. The latter were shown not to be directly related to increased steatosis, as being measured in a study using magnetic resonance spectroscopy [53].
Mipomersen should be considered a potential novel treatment modality in FH, given its lipid-lowering effect, relatively easy mode of administration, and lack of interactions with other lipid-modifying drugs.
PCSK9 Targeted Therapy
After the initial report that gain-of-function mutations in the gene encoding for proprotein convertase subtilisin/kexin type 9 (PCSK9) cause Mendelian hypercholesterolemia [3], PCSK9 has gained large interest as a target for lipid lowering. PCSK9 has been shown to be a pivotal regulator of LDL-C metabolism by virtue of its role in lysosomal degradation of the LDL receptor within hepatocytes. The notion that loss-of-function PCSK9 mutations confer an 80% CVD risk reduction [54] has further substantiated the role of PCSK9 as a potential target. Although PCSK9 could be anticipated to act as a protease on other substrates as well, it is of importance to note that subjects with lifelong half normal activity of PCSK9 due to loss-of-function mutations were not characterized by other untoward clinical features.
A number of strategies to specifically lower PCSK9 activity are currently in different stages of development and testing: antisense nucleotide-based therapy (similar to mipomersen described above) [55], monoclonal antibodies binding to the catalytic site of PCSK9 (eg, AMG 145, 1D05-IgG2, and REGN727), and small interfering RNAs.
Although animal studies have shown beneficial effects of use of these novel compounds (with LDL lowering up to 80%) [56, 57], no human studies on the effect of either of these different strategies have been published thus far. The finding, however, that statins and fibrates induce increased PCSK9 expression [58] further underlines that PCSK9 inhibition could induce robust LDL-C reductions as add-on therapy and a number of phase 2 and phase 3 trials will likely be initiated soon.
Microsomal Triglyceride Transfer Protein Inhibitors
Microsomal triglyceride transfer protein (MTP) plays an important role in the hepatic assembly of plasma lipoproteins, by mediating the transfer of triglycerides to VLDL [59, 60]. MTP mutation carriers are characterized by hypobetalipoproteinemia [61] and one could therefore anticipate an MTP-lowering therapy to result in a decrease of VLDL and LDL-C levels (Table 2). Cuchel et al. [62] showed that MTP inhibition by means of BMS-201038 gave rise to approximately 50% reductions of plasma LDL-C levels in the highest dosage (0.1 mg/kg/day). However, this trial, performed in six homozygous FH patients [62], also showed that MTP inhibition induced an increase in hepatic steatosis. This finding raised serious concerns, and the drug is therefore only studied at its higher dosages in homozygous FH patients where LDL-C reduction is considered to outweigh potential steatosis. The maximum studied dose in a subsequent trial in 10 homozygous patients was 60 mg/day, and this regimen resulted in a 44% reduction in LDL-C levels, over and above the effect already achieved by coadministered other lipid-modifying medication [63]. The extent of steatosis in this trial was reduced compared with the initial trial.
A low-dose regimen of MTP inhibition was studied by Samaha et al. [64]. The 84 patients with hypercholesterolemia were randomized to ezetimibe, 10 mg, daily (n = 29); MTP inhibition by lomitapide (also known as AEGR-733 and BMS −201038) in increasing dosages (5.0, 7.5, and 10 mg daily for each consecutive 4 weeks [n = 28]); or ezetimibe, 10 mg daily, and lomitapide administered with the dose titration described above (n = 28). Ezetimibe therapy resulted in an expected LDL-C reduction of 20%. Lomitapide was shown to induce a lowering of LDL-C levels in a dose-dependent manner: 19%, 26%, and 30% in the 5-, 7.5-, and 10-mg dosing regimens, respectively. Combined therapy produced similar but larger dose-dependent decreases (35%, 38% and 46%, respectively). Mild transaminase elevations (n = 9) and diarrhea were the primary cause for discontinuations from lomitapide (Table 2). Despite the fact that MTP inhibition induced steatosis in high dosages, one might consider MTP an attractive candidate for lipid lowering in FH patients if administered in lower dosages.
Thyroid Mimetics
The notion that hyperthyroidism results in sharply decreased LDL-C levels reduced has driven the attempts to mimic this by administration of thyroid hormone analogues. As seen in hyperthyroidism, however, these therapies (D-thyroxine and tiratricol) failed, due to the associated cardiac and bone-related side effects.
Recently, the differential molecular mechanisms underlying the “beneficial” (mainly via the thyroid receptor-β [TRβ] route; this receptor is mainly expressed in the liver) and “deleterious” aspects (mediated by TRα [expressed in brain and heart]–induced processes) of hyperthyroidism have been elucidated. Eprotirome (KB2115; Karo Bio AB, Stockholm, Sweden), sobetirome (QRX-431/GC-1; formerly owned by QuatRx Pharmaceuticals, Ann Arbor, MI), and MB07811 (Ligand Pharmaceuticals, La Jolla, CA) are selective TRβ agonists and they are currently in different stages of investigation.
A recent study showed the effects of adding eprotirome to standard statin therapy [65]. In this 12-week trial, Ladenson et al. [65] enrolled 184 patients, who were treated with placebo or eprotirome on top of a statin (simvastatin ≤ 40 mg and atorvastatin ≤ 20 mg daily). Randomization to placebo or eprotirome in three dosages (25, 50 and 100 ug daily) resulted in a decrease of LDL-C level by 7%, 22%, 28%, and 32%, respectively (Table 2). Eprotirome was not associated with adverse events on heart (arrhythmia) or bone (serum markers of bone turnover) [65]. A large phase 3 trial will start in FH patients in the near future.
Sobetirome and MB07811 have not been tested for their effect on dyslipidemia in humans, but the beneficial effect of eprotirome on LDL-C levels in non-FH patients holds promise for FH patients.
Cholesterol Ester Transfer Protein Inhibitors
The HDL-bound enzyme cholesterol ester transfer protein (CETP) mediates the process of transfer of cholesteryl esters from HDL particles to apolipoprotein B–containing particles [66]. Elevated CETP levels were shown to be associated with an increased risk for coronary artery disease in apparently healthy subjects [67], and inhibition of CETP in rabbit models of atherosclerosis dramatically reduced the extent of the disease [68]. Two approaches to inhibit CETP activity have been described; a vaccine-based strategy and a small molecule inhibitor-mediated method.
CETi-1 (Avant Immunotherapeutics, Needham, MA) is a synthetic peptide that includes residues of the human CETP protein. Upon administration, this vaccine raises an immune response and production of autoantibodies against CETP. Initial animal studies showed the efficacy of this vaccine to increase HDL-C levels and reduce aortic atherosclerosis [69]. However, the phase 1 trial in humans showed a relatively poor response in terms of the presence of autoantibodies (in 1 out of 23 patients) and the effect on plasma lipid levels was negligible [70].
Torcetrapib (Pfizer, New York, NY), anacetrapib (MK-0895; Merck, White House Station, NJ), dalcetrapib (formerly known as JTT-705; Roche, Basel, Switzerland), and evacetrapib (LY2484595; Eli Lilly; Indianapolis, IN) are molecules that antagonize CETP activity by binding to the protein. The reason why these primarily HDL-C–increasing medications are mentioned in this review is the fact that torcetrapib is tested in FH patients and that all CETP inhibitors have been shown to beneficially affect LDL-C levels.
The RADIANCE I trial, in which over 800 patients with FH were enrolled, showed that addition of torcetrapib to atorvastatin did not result in reduction of atherosclerosis, as assessed by intima-media thickness [71], despite a significant reduction in LDL-C (21%) and increase in HDL-C (52%). These findings are in line with the ILLUMINATE trial, which was prematurely terminated because of unexpected increased mortality and morbidity in patients treated with atorvastatin combined with torcetrapib [72]. The exact mechanism underlying this counterintuitive finding is not fully elucidated, but a recent chemical systems biology analysis shed light on this topic. The study suggested the presence of off-target effects of torcetrapib, and these might partially explained by the blood pressure increase induced by torcetrapib [73]. The fact that the other CETP inhibitors do not show an effect on blood pressure further confirms a molecule-specific off-target effect.
The two remaining CETP inhibitors in phase 3 development, anacetrapib and dalcetrapib, have been shown to be effective lipid modifiers [74, 75], but the cardiovascular outcome trials (DAL-outcomes I and II and HPS3/REVEAL) are eagerly awaited. Once shown to benefit mild hypercholesterolemic patients and other patients at CVD risk, CETP inhibition is likely to benefit patients with FH.
Conclusions
During the last decade we have been confronted with an increase in our understanding of human lipid biology. This knowledge has given great impetus to the identification of novel strategies to inhibit specific pathways in dyslipidemia. Upon approval of efficacy in CVD reduction, these agents will be beneficial for all patients at risk, such as FH patients.
Abbreviations
- AIM-HIGH:
-
Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides and Impact on Global Health Outcomes
- ASAP:
-
Atorvastatin Versus Simvastatin on Atherosclerosis Progression
- HPS-2-Thrive:
-
Treatment of High-Density Lipoprotein to Reduce the Incidence of Vascular Events
- HPS3/REVEAL:
-
Randomized Evaluation of the Effects of Anacetrapib Through Lipid Modification
- ILLUMINATE:
-
Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events
- RADIANCE I:
-
Carotid B-Mode Ultrasound Study to Compare Anti-Atherosclerotic Effect of Torcetrapib/Atorvastatin to Atorvastatin Alone
- SHARP:
-
Study of Heart and Renal Protection.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Goldstein JL, Hobbs HH, Brown MS. Familial Hypercholesterolemia. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 2863–913.
Innerarity TL, Mahley RW, Weisgraber KH, et al. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J Lipid Res. 1990;31:1337–49.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6.
Wierzbicki AS, Humphries SE, Minhas R. Familial hypercholesterolaemia: summary of NICE guidance. BMJ. 2008;337:a1095.
Robinson JG, Goldberg AC. Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S18–29.
DeMott K, Nherera L, Shaw E, et al. Clinical guidelines and evidence review for familial hypercholesterolemias: the indentification and management of adults and children with familial hypercholesterolemia. London: National Collaborating Centre for Primary Care and Royal College of General Practitioners; 2008.
•• Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010;376:1670–81. This article describes the largest meta-analysis in patients on statin therapy so far and includes 26 different statin trials.
Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis. 2010;209:189–94.
Mata N, Alonso R, Badimon L, et al. Clinical characteristics and evaluation of LDL-cholesterol treatment of the Spanish Familial Hypercholesterolemia Longitudinal Cohort Study (SAFEHEART). Lipids Health Dis. 2011;10:94.
Leren TP, Berge KE. Subjects with molecularly defined familial hypercholesterolemia or familial defective apoB-100 are not being adequately treated. PLoS One. 2011;6:e16721.
Doggrell SA. Lowering LDL cholesterol with margarine containing plant stanol/sterol esters: is it still relevant in 2011? Complement Ther Med. 2011;19:37–46.
Shafiq N, Singh M, Kaur S et al. Dietary treatment for familial hypercholesterolaemia. Cochrane Database Syst Rev. 2010;CD001918.
Jansen AC, van Aalst-Cohen ES, Tanck MW, et al. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients. J Intern Med. 2004;256:482–90.
Alonso R, Mata N, Castillo S, et al. Cardiovascular disease in familial hypercholesterolaemia: influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis. 2008;200:315–21.
Endo A. The discovery and development of HMG-CoA reductase inhibitors. J Lipid Res. 1992;33:1569–82.
Lahera V, Goicoechea M, de Vinuesa SG, et al. Endothelial dysfunction, oxidative stress and inflammation in atherosclerosis: beneficial effects of statins. Curr Med Chem. 2007;14:243–8.
Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78.
Elis A, Zhou R, Stein EA. Effect of lipid-lowering treatment on natural history of heterozygous familial hypercholesterolemia in past three decades. Am J Cardiol. 2011;108:223–6.
Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423.
Neil A, Cooper J, Betteridge J, et al. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study. Eur Heart J. 2008;29:2625–33.
LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–35.
Smilde TJ, van Wissen S, Wollersheim H, et al. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet. 2001;357:577–81.
Jones PH, Davidson MH, Stein EA, et al. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003;92:152–60.
de Jongh S, Ose L, Szamosi T, et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized, double-blind, placebo-controlled trial with simvastatin. Circulation. 2002;106:2231–7.
Avis HJ, Vissers MN, Stein EA, et al. A systematic review and meta-analysis of statin therapy in children with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2007;27:1803–10.
Avis HJ, Hutten BA, Gagne C, et al. Efficacy and safety of rosuvastatin therapy for children with familial hypercholesterolemia. J Am Coll Cardiol. 2010;55:1121–6.
van der Graaf A, Cuffie-Jackson C, Vissers MN, et al. Efficacy and safety of coadministration of ezetimibe and simvastatin in adolescents with heterozygous familial hypercholesterolemia. J Am Coll Cardiol. 2008;52:1421–9.
Hamilton-Craig I, Kostner K, Colquhoun D, Woodhouse S. Combination therapy of statin and ezetimibe for the treatment of familial hypercholesterolemia. Vasc Health Risk Manag. 2010;6:1023–37.
• Kastelein JJ, Akdim F, Stroes ES, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–43. This is a large clinical trial combining statin and statin-ezetimibe therapy. This resulted in significant LDL-C lowering but did not show additional effects on atherosclerosis assessed by cIMT.
Avellone G, Di Garbo V, Guarnotta V, et al. Efficacy and safety of long-term ezetimibe/simvastatin treatment in patients with familial hypercholesterolemia. Int Angiol. 2010;29:514–24.
Baigent C, Landray MJ, Reith C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2011;377:2181–92.
Shepherd J, Packard CJ, Bicker S, et al. Cholestyramine promotes receptor-mediated low-density-lipoprotein catabolism. N Engl J Med. 1980;302:1219–22.
Robinson DM, Keating GM. Colesevelam: a review of its use in hypercholesterolemia. Am J Cardiovasc Drugs. 2007;7:453–65.
Jacobson TA. Myopathy with statin-fibrate combination therapy: clinical considerations. Nat Rev Endocrinol. 2009;5:507–18.
Morgan JM, Capuzzi DM, Guyton JR. A new extended-release niacin (Niaspan): efficacy, tolerability, and safety in hypercholesterolemic patients. Am J Cardiol. 1998;82:29U–34U.
Wolfe ML, Vartanian SF, Ross JL, et al. Safety and effectiveness of Niaspan when added sequentially to a statin for treatment of dyslipidemia. Am J Cardiol. 2001;87:476–9. A7.
Paolini JF, Mitchel YB, Reyes R, et al. Effects of laropiprant on nicotinic acid-induced flushing in patients with dyslipidemia. Am J Cardiol. 2008;101:625–30.
Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010;210:353–61.
Thompson GR. Recommendations for the use of LDL apheresis. Atherosclerosis. 2008;198:247–55.
Thompsen J, Thompson PD. A systematic review of LDL apheresis in the treatment of cardiovascular disease. Atherosclerosis. 2006;189:31–8.
Davidson NO, Shelness GS. APOLIPOPROTEIN B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu Rev Nutr. 2000;20:169–93.
Walldius G, Jungner I, Holme I, et al. High apolipoprotein B, low apolipoprotein A-I, and improvement in the prediction of fatal myocardial infarction (AMORIS study): a prospective study. Lancet. 2001;358:2026–33.
Sankatsing RR, Fouchier SW, de Haan S, et al. Hepatic and cardiovascular consequences of familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2005;25:1979–84.
Schonfeld G, Lin X, Yue P. Familial hypobetalipoproteinemia: genetics and metabolism. Cell Mol Life Sci. 2005;62:1372–8.
Ito MK. ISIS 301012 gene therapy for hypercholesterolemia: sense, antisense, or nonsense? Ann Pharmacother. 2007;41:1669–78.
Kastelein JJ, Wedel MK, Baker BF, et al. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114:1729–35.
Akdim F, Visser ME, Tribble DL, et al. Effect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia. Am J Cardiol. 2010;105:1413–9.
Akdim F, Stroes ES, Sijbrands EJ, et al. Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy. J Am Coll Cardiol. 2010;55:1611–8.
Akdim F, Tribble DL, Flaim JD, et al. Efficacy of apolipoprotein B synthesis inhibition in subjects with mild-to-moderate hyperlipidaemia. Eur Heart J. 2011.
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006.
Yu RZ, Geary RS, Flaim JD, et al. Lack of pharmacokinetic interaction of mipomersen sodium (ISIS 301012), a 2′-O-methoxyethyl modified antisense oligonucleotide targeting apolipoprotein B-100 messenger RNA, with simvastatin and ezetimibe. Clin Pharmacokinet. 2009;48:39–50.
Genzyme Corp IPI: Mipomersen Data Presented at European Atherosclerosis Society Congress Press Release available at http://ir.isispharm.com/phoenix.zhtml?c=222170&p=irolnewsArticle_Print&ID=1580350&highlight=. Accessed July 2011.
Visser ME, Akdim F, Tribble DL, et al. Effect of apolipoprotein-B synthesis inhibition on liver triglyceride content in patients with familial hypercholesterolemia. J Lipid Res. 2010;51:1057–62.
Cohen JC, Boerwinkle E, Mosley Jr TH. Hobbs HH: Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Graham MJ, Lemonidis KM, Whipple CP, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763–7.
Frank-Kamenetsky M, Grefhorst A, Anderson NN, et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci USA. 2008;105:11915–20.
Chan JC, Piper DE, Cao Q, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci USA. 2009;106:9820–5.
Konrad RJ, Troutt JS, Cao G. Effects of currently prescribed LDL-C-lowering drugs on PCSK9 and implications for the next generation of LDL-C-lowering agents. Lipids Health Dis. 2011;10:38.
Burnett JR, Watts GF. MTP inhibition as a treatment for dyslipidaemias: time to deliver or empty promises? Expert Opin Ther Targets. 2007;11:181–9.
Sharp D, Blinderman L, Combs KA, et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature. 1993;365:65–9.
Wetterau JR, Aggerbeck LP, Bouma ME, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001.
Cuchel M, Bloedon LT, Szapary PO, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–56.
Cuchel M, Meagher E, Marais AD, et al. Abstract 1077: A phase III study of microsomal triglyceride transfer protein inhibitor Lomitapide (AEGR-733) in patients with homozygous familial hypercholesterolemia: interim results at 6 months. Circulation. 2009;120:S441.
Samaha FF, McKenney J, Bloedon LT, et al. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2008;5:497–505.
Ladenson PW, Kristensen JD, Ridgway EC, et al. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med. 2010;362:906–16.
Barter PJ, Brewer Jr HB, Chapman MJ, et al. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–7.
Boekholdt SM, Kuivenhoven JA, Wareham NJ, et al. Plasma levels of cholesteryl ester transfer protein and the risk of future coronary artery disease in apparently healthy men and women: the prospective EPIC (European Prospective Investigation into Cancer and nutrition)-Norfolk population study. Circulation. 2004;110:1418–23.
Okamoto H, Yonemori F, Wakitani K, et al. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature. 2000;406:203–7.
Rittershaus CW, Miller DP, Thomas LJ, et al. Vaccine-induced antibodies inhibit CETP activity in vivo and reduce aortic lesions in a rabbit model of atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:2106–12.
Davidson MH, Maki K, Umporowicz D, et al. The safety and immunogenicity of a CETP vaccine in healthy adults. Atherosclerosis. 2003;169:113–20.
Kastelein JJ, van Leuven SI, Burgess L, et al. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007;356:1620–30.
Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
Xie L, Li J, Xie L, Bourne PE. Drug discovery using chemical systems biology: identification of the protein-ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput Biol. 2009;5:e1000387.
Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–15.
Stein EA, Stroes ES, Steiner G, et al. Safety and tolerability of dalcetrapib. Am J Cardiol. 2009;104:82–91.
Weng TC, Yang YH, Lin SJ, Tai SH. A systematic review and meta-analysis on the therapeutic equivalence of statins. J Clin Pharm Ther. 2010;35:139–51.
Sharma M, Ansari MT, Abou-Setta AM, et al. Systematic review: comparative effectiveness and harms of combination therapy and monotherapy for dyslipidemia. Ann Intern Med. 2009;151:622–30.
Knopp RH, Gitter H, Truitt T, et al. Effects of ezetimibe, a new cholesterol absorption inhibitor, on plasma lipids in patients with primary hypercholesterolemia. Eur Heart J. 2003;24:729–41.
Huijgen R, Abbink EJ, Bruckert E, et al. Colesevelam added to combination therapy with a statin and ezetimibe in patients with familial hypercholesterolemia: a 12-week, multicenter, randomized, double-blind, controlled trial. Clin Ther. 2010;32:615–25.
Bays HE, Davidson M, Jones MR, Abby SL. Effects of colesevelam hydrochloride on low-density lipoprotein cholesterol and high-sensitivity C-reactive protein when added to statins in patients with hypercholesterolemia. Am J Cardiol. 2006;97:1198–205.
Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: a meta-analysis of randomized controlled trials. J Am Coll Cardiol. 2005;45:185–97.
Loomba RS, Arora R. Prevention of cardiovascular disease utilizing fibrates–a pooled meta-analysis. Am J Ther. 2010;17:e182–8.
Berkenstam A, Kristensen J, Mellstrom K, et al. The thyroid hormone mimetic compound KB2115 lowers plasma LDL cholesterol and stimulates bile acid synthesis without cardiac effects in humans. Proc Natl Acad Sci USA. 2008;105:663–7.
Vergeer M, Stroes ES. The pharmacology and off-target effects of some cholesterol ester transfer protein inhibitors. Am J Cardiol. 2009;104:32E–8E.
Disclosure
Conflicts of interest: B. Sjouke: none; D.M. Kusters: none; J.J.P. Kastelein: has been a consultant for AstraZeneca, MSD, Novartis, Roche, Eli Lilly, Isis, Cerenis, Genzyme, Boehringer Ingelheim, Sanofi-Aventis, Regeneron, and Anthera; G.K. Hovingh: none.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sjouke, B., Kusters, D.M., Kastelein, J.J.P. et al. Familial Hypercholesterolemia: Present and Future Management. Curr Cardiol Rep 13, 527–536 (2011). https://doi.org/10.1007/s11886-011-0219-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-011-0219-9