
Abstract
Cardiovascular disease remains the major cause of worldwide morbidity and mortality. Its pathophysiology is complex and multifactorial. Because the phenotype of cardiovascular disease often shows a marked heritable pattern, it is likely that genetic factors play an important role. In recent years, large genome-wide association studies have been conducted to decipher the molecular mechanisms underlying this heritable and prevalent phenotype. The emphasis of this review is on the recently identified 17 susceptibility loci for coronary artery disease. Implications of their discovery for biology and clinical medicine are discussed. A description of the landscape of human genetics in the near future in the context of next-generation sequence technologies is provided at the conclusion of this review.
Similar content being viewed by others


Introduction
Cardiovascular disease (CVD) remains the major cause of worldwide morbidity and mortality despite currently available therapy. Full appreciation of all risk factors and the pathogenesis of this disease are inevitably required to identify additional targets for therapy. Atherosclerosis, the major underlying cause of CVD, is present in all humans at advancing age and progresses over a life time, but its extent and progression is dependent on factors such as dyslipidemia, hypertension, obesity, diabetes, and smoking [1]. In addition, genetic factors play an important role. Twin studies indicate that heritability of CVD, defined as the proportion of the inter-individual differences resulting from genetic factors, is 30% to 60% [2]. This translates to the fact that apart from the classical risk factors, a positive family history for premature coronary artery disease (CAD) in a first-degree relative also constitutes an additional and independent risk [3].
A large number of studies have aimed to decipher the molecular mechanisms underlying the heritability of CVD; however, in recent years the approach has seen major changes. In early attempts, case–control studies focused on the differences in carrier frequency of genetic variations in “candidate genes” among cases and controls. Genes were marked as “candidate” based on the fact that they were known to influence traditional risk factors (ie, genes involved in lipid metabolism) or were part of pathways with a putative role in atherogenesis.
Later, a whole genome approach was applied to find genetic variations without a prior hypothesis. Initial attempts included classical linkage studies in families with premature CAD using microsatellite markers and later single nucleotide polymorphism (SNP) arrays for linkage [4]. These early family-based studies have thus far resulted in the identification of two interesting candidate genes, MEF2A and LRP6.
Through linkage analysis in an extended kindred that displayed an autosomal dominant pattern of CAD, a large locus containing 93 genes was identified on 15q26 [5]. Within this locus, the gene for myocyte enhancer factor 2 (MEF2A) was considered the best candidate for further investigation, because of its previously described effect on myocardial development and vascular morphogenesis [5]. Interestingly, a number of genetic variants in MEF2A were identified in other small cohorts of patients with CAD, suggesting that MEF2A may be actively involved in atherosclerosis [6]. Recently, however, additional studies have not revealed any evidence for a role of MEF2A as a susceptibility gene for CAD, thereby refuting the original hypothesis [7, 8]. Novel missense mutations in MEF2A were identified, but in vitro expression of the mutant proteins did not show a difference in transcriptional activity. Thus, the support for a role for MEF2A in CVD remains limited [7].
Linkage studies have also identified a locus on 12p13 in a kindred with the combination of extensive CVD and elevated plasma low-density lipoprotein (LDL) cholesterol levels and osteoporosis. The linkage interval contained six annotated genes, including LDL-receptor related protein 6 (LRP6). The latter is an integral component of the Wnt signaling pathway, which is involved in vascular development and remodelling but also plays a role in the regulation of glucose and lipid metabolism [9]. Common variants in LRP6 have been associated with LDL metabolism in humans [10, 11].
To increase the power to detect more commonly occurring shared genetic loci, linkage analysis has been performed with data derived from several families. These studies have resulted in the identification of genetic variations in three genes involved in atherosclerosis: arachidonate 5-lipoxygenase-activating protein (ALOX5AP) and leukotriene A4 hydrolase (LTA4H), both of which are pro-inflammatory cytokines, and LDL receptor-related protein 8 (LRP8), also known as apoE receptor 2 (APOER2), which is mainly expressed in the brain [12, 13]. The relative contribution of these loci to atherosclerosis in replication studies remains modest, with typical odds ratios no larger than 1.5 [12].
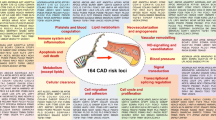
In recent years, numerous genome-wide association (GWA) studies with CVD as the outcome have been published. These studies primarily focus on populations, rather than on families, and are carried out under the assumption that common variants are shared by subjects with a pre-specified phenotype. In this review, we provide an update of the loci identified in GWA studies for CVD. Of note, a total of nine loci identified in the six GWA studies focusing on stroke have been poorly replicated and are not discussed. Only the 9p21 locus has been found in all the stroke GWA studies. The other eight loci (2q33, 4q25, 8q12, 11q12, 12q13, 14q22, 16q22, and 22q13) show inconsistent results between the different studies and warrant further validation [14]. To date, no GWA study on peripheral artery disease (PAD) has been conducted. For these reasons, this review focuses on loci discovered in GWA studies with CAD as outcome (Fig. 1). In addition, we discuss the possible current implications for biology and clinical medicine as a result of the discovery of these loci.

Proposed sites of action for the proteins involved in the pathogenesis of atherosclerosis: liver, vessel wall, plaque and thrombus
GWA Studies
The rationale behind GWA studies is the “common disease, common variant” hypothesis in which a limited number of genetic variants with a high frequency (typically above 5%) in the general population contribute to susceptibility for disease [15]. GWA studies became possible because of two major advances in genetics and technology. First, there was the identification of almost 10 million common SNPs with a minimum minor allele frequency of 5% in the HapMap project and the notion that a large number of these SNPs are in linkage disequilibrium (LD) [15, 16]. Because of these LD blocks, a reduced number of SNPs provide complete whole genome coverage [16]. Second, the development of DNA microarrays for genotyping large numbers of SNPs and the great reduction in their price have facilitated the investigation of large cohorts of patients and controls. The first-generation GWA studies used a microsatellite marker approach covering the genome by 400 unique microsatellite markers [4]. Currently, microarrays are available with more than 1 million SNPs. In addition, microarrays were developed based on a candidate gene approach, such as the IBC CardioChip (Illumina, San Diego, CA) [17]. These have the advantage of a denser SNP profile per locus, facilitating fine mapping, but the disadvantage is that these often fail to find signals in loci not previously associated with the phenotype.
Major GWA Studies in CVD and Implications for Vascular Biology
Until recently, GWA studies with CAD as the major outcome variable comprising more than 28,000 CAD cases and 65,000 controls had identified 13 unique loci [18••, 19]. The recently published Global Lipids Consortium study in 24,607 cases with CAD and 66,197 controls added another four unique loci associated with CAD [20••]. These 17 loci are summarized in Table 1. As expected, the effect sizes are modest and in the same order of magnitude as the results obtained in GWA studies for other disorders.
The 9p21 locus was the first novel locus associated with CAD risk in a large case–control study in six independent samples comprising more than 23,000 participants from four white populations [21]. Two copies of the 9p21 risk alleles are present in about 25% of the population and convey a 60% increased risk for CAD as compared to the common genotype [21, 22]. The minor risk alleles of SNPs in the 9p21 region are found at a higher frequency in patients with early compared to late onset CAD [21, 23–25]. Variations in the 9p21 locus might mediate their effect through modulation of platelet reactivity and vascular stiffness [26, 27]. Additionally, the locus has also been found to be associated with other diseases associated with CVD, such as stroke, abdominal aneurysms, and type 2 diabetes [28]. However, variants in this locus are also associated with ovarian cancer, acute lymphoblastic leukemia, poor physical function in the elderly, glioma, malignant melanoma, breast cancer, and pancreatic cancer, suggesting an interesting chromosomal area harboring genes involved in a number of different physiologic processes [28, 29].
Although the chromosomal region of 9p21 contains a number of genes such as cyclin-dependent kinase inhibitor 2A and 2B (CDKN2A, CDNK2B), methylthioadenosine phosphorylase (MTAP), and ANRIL, a long non-coding RNA, the disease-associated SNPs are located far away from these genes, which makes it more difficult to find the causal gene responsible for the observed signal in CVD [30]. CDKN2A, CDKN2B, and MTAP are involved in the regulation of the cell cycle by interfering with cell cycle progression, but additionally they are key players in physiologic processes including replicative senescence, apoptosis, and stem cell renewal. Direct sequencing of the chromosomal region did not reveal any causal variants that could explain the observed association. Interestingly, no association could be detected between the causal SNPs and differential expression of these genes [31]. CDKN2A, CDKN2B, and MTAP are expressed in human vasculature and atherosclerotic plaques. In mice, deletion of the risk interval of 9p21 was shown to result in decreased expression of CDKN2A and CDKN2B [32]. Moreover, aortic smooth muscle cells harvested from mice deficient in 9p21 were found to have increased proliferative capacity [32]. Another finding was that the 9p21 knockout mice gained significantly more weight than their control littermates [32]. An association between the 9p21 risk variant and body weight has not been observed in humans. Also, the burden of atherosclerosis measured by fatty lesion formation was similar in mutant and wild-type animals.
Risk variants in the 9p21 locus associate with gene expression levels of ANRIL, a non-coding RNA that can alter gene expression of neighboring genes by RNA interference or gene silencing [33]. Whole blood RNA expression revealed a positive association between expression of ANRIL with CDKN2A and CDKN2B [30]. ANRIL is expressed in eight different splice variants depending on the tissue type, which complicates the analysis of the 9p21 locus [30]. In conclusion, the 9p21 locus illustrates the complexity of gene identification studies using the GWA approach.
Another highly significant signal associated with both cholesterol levels and the occurrence of CVD is locus 1p31 [34••]. Although several SNPs in this region were associated with these traits, fine mapping of the locus identified the rs12740374, located in the intergenic noncoding region, as the causative SNP [20••]. Individuals who are homozygous carriers of the major allele have up to 16 mg/dL (0.4 mmol/L) higher LDL cholesterol plasma levels as well as a 40% increased risk of myocardial infarction when compared to carriers homozygous for the minor allele [23, 24]. Three genes, sortilin1 (SORT1), cadherin (CELSR2), and proline-serine rich coiled coil1 (PCSR1), are located in the 1p31 region. Tissue expression analysis reveals a strong association with hepatic SORT1 and PSRC1 expression. Rs12740374 creates a predicted binding site for C/EBPalpha transcription factors [33]. As a consequence, the hepatic gene expression of SORT1 will be altered. SORT1 encodes for the sortilin protein, also known as neurotensin receptor, a protein that functions as a multi-ligand sorting receptor. Two studies have been published recently investigating the role of sortilin1 in hepatic lipoprotein metabolism using different mouse models with conflicting results. In one study, Sort1- deficient mice showed a reduction in the secretion of apolipoprotein B100 (apoB100)-containing lipoproteins from the liver and a reduction in atherosclerosis, whereas overexpression of SORT1 increased plasma cholesterol levels [35]. In the second study, a different approach using specific hepatic overexpression of SORT1 in the liver using adenovirus-mediated gene transfer in humanized mouse models revealed a marked decrease in plasma cholesterol levels by 73%, change in LDL size toward larger particles, and consequently a reduction in atherosclerosis [34]. A second approach using small interfering RNA technology revealed similar results. Currently, there is no explanation for the conflicting laboratory data observed.
In the GWA studies, highly significant signals at 19p13 at the LDL receptor (LDLR) and 1p32 at the proprotein convertase subtilisin/kexin type 9 (PCSK9) were found. Both well-known proteins play an important role in LDL metabolism and are strongly associated with atherosclerosis risk and demonstrate that GWA studies are indeed capable of detecting genes that are related to CVD [36, 37]. Rare variants in LDLR and PCSK9 result in the Mendelian forms of familial hypercholesterolemia (FH). Clinically, FH manifests in lipid accumulation in tendons (xanthomas), the cornea (arcus lipoides), and arterial wall, resulting in accelerated atherosclerosis and premature CVD [38, 39]. In untreated heterozygous FH patients, about 50% of males and 30% of females will develop CVD before the age of 60 years [40].
Another highly significant signal in GWA studies was on locus 6q25.3-26. This region harbors the gene lipoprotein(a) (LPA), which was also previously shown to be implicated in atherosclerosis, and also the solute carrier family 22 member 3 (SLC22A3) and lipoprotein(a)-like 2 (LPAL2) genes [41]. Increased plasma Lp(a) levels are causally related to CVD [42]. Lp(a) is a cholesterol-rich LDL particle with one apoprotein(a) molecule attached to the apoB100 protein [43]. Due to homology of the repeated kringle structure of apo(a) with plasminogen, it has been suggested that Lp(a) intervenes with the process of atherosclerosis through involvement of fibrinolysis and, as a consequence, the atherothrombotic processes. Plasma levels of Lp(a) are strongly genetically determined by the number of kringle IV repeats in the apo(a) protein, which has a heritability between 21% and 27% [44]. SNPs identified by GWA studies are strongly associated to the kringle IV repeats and thus to plasma Lp(a) levels [45]. A recent meta-analysis revealed that plasma Lp(a) concentration is a stronger biomarker for risk than the gene score based on above-mentioned SNPs [45].
Other loci that have been identified in the GWA studies are less frequently studied. In some cases there is suggestive evidence for a role in atherosclerosis. In the next section we discuss the genes located in proximity of these identified loci.
The gene of interest on locus 10q11 is CXCL12/SDF1 (chemokine [C-X-C-motif] ligand 12/stromal cell-derived factor-1). In patients with stable and unstable angina, plasma levels of CXCL12 are lower compared to healthy controls [46]. Interestingly, CXCL12 is expressed in atherosclerotic lesions and is of importance for vascular repair and remodelling [46]. Furthermore, it has a role in neo-intimal formation after injury [47]. Blocking its receptor CXCR4 in Apoe −/− or Ldlr −/− mice aggravates diet-induced atherosclerosis by affecting neutrophil recruitment machinery [48]. CXCL12 may promote atherothrombosis by its effect on platelets. It stimulates platelet adhesion to immobilized collagen and fibrinogen and induces the release of various platelet chemokines, in particular P-selectin. P-selectin plays an essential role in the initial recruitment of leukocytes to the site of injury during inflammatory processes such as atherosclerosis [49]. Additional evidence that genetic variation in CXCL12 is associated with CVD risk is eagerly awaited.
The locus on 3q22 encodes the Ras-related protein M-Ras (MRAS) gene. The lead SNP, rs9818870, is located in the 3′ untranslated region of MRAS in close proximity to a cluster of miRNA binding sites [50]. MRAS is widely expressed in all tissues, with high expression in the cardiovascular system and brain [50, 51]. MRAS is a member of the RAS family of small molecular-weight GTP binding molecules and is involved in the phosphoinositide kinase-3/mitogen-activated protein kinase (PI3K-MAPK) signaling pathways. The authors who reported the locus suggest a role for MRAS in adhesion signaling, which is important in the atherosclerotic process. No direct clues can be found in the literature to bolster this statement. Recently, rs9818870 was shown to be significantly associated with MRAS gene expression in aortic tissue of patients, confirming the previous findings [50, 52]. Additional functional studies are lacking.
Phosphatase and actin regulator 1 (PHACTR1) is an inhibitor of protein phosphatase 1 (PP1), an enzyme that dephosphorylates serine and threonine residues on a range of proteins and resides near 6q26–27 [53]. PP1 is a ubiquitously expressed protein and involved in a number of physiologica processes such as protein kinase-A (PKA) and protein kinase C (PKC) phosphorylation [53]. It is interesting that in an independent GWA study for coronary artery calcification in more than 10,000 participants from six prospective cohort studies, PHACTR1 SNPs (along with chromosome 9p21 SNPs) were associated with coronary artery calcification at genome-wide significance (C.J. O'Donnell, NHBL Institute, personal communication).
Rare variants in hepatocyte nuclear factor 1 homeobox A (HNF1A) on 12q24 locus result in Mendelian forms of maturity onset diabetes of the young (MODY) [54]. In a recent GWA study searching for loci predictive for type 2 diabetes, rs7957197 in HNF1A reached genome-wide significance. HNF1A is transcription factor involved in expression of genes involved in innate immunity, blood coagulation and lipid and glucose metabolism [55].
For insulin receptor substrate 1 gene (IRS1) and SH2B adaptor protein 3 (SH2B3), the evidence for a role in atherosclerosis is much weaker. The rs2972146 SNP on chromosome 2 is close to IRS1, a gene that has been shown to be essential in insulin signaling. Genetic variants in IRS1 impaired insulin signaling in endothelial cells. As a consequence, nitric oxide (NO) release by these cells is decreased [56]. However, a relation between endothelial dysfunction and genetic variants of IRS1 has not been published so far. SH2B3 located near 12q24 is expressed in human vascular endothelial cells, where it promotes inflammation. The lead SNP, rs3184504, which results in a missense change, could contribute to the progression of plaque formation in coronary arteries, leading to myocardial infarction by its induction of a pro-inflammatory state. SH2B3 has previously been shown to be associated with type 1 diabetes and celiac disease [57].
Finally, more evidence has to be generated to further identify the suspected candidate genes at the following loci and their association with atherosclerosis: 1q41 locus, melanoma inhibitory activity family member 3 (MIA3); 2q33 locus, WD repeat domain 12(WDR12); 7q32 locus, Kruppel-like factor 14 (KLF14); 8p32 locus, N-acetyltransferase 2 (NAT2) and 21q22 locus potassium voltage-gated channel subfamily H member (KCNH2) [58].
Implications for Clinical Medicine
The identification of the current 17 common variants associated with CVD risk has so far had little impact on routine clinical medicine. However, unravelling the biological role of the proteins in the process of atherosclerosis, as has been done for the 1p13 locus, will probably lead to a better understanding of the pathways involved in the pathophysiology of CVD. In addition, these pathways might be targets for development of future therapeutic compounds.
The findings so far cannot be directly translated into a risk profile for an individual patient. In a risk score comprising nine GWA study SNPs associated with early-onset MI, including a 9p21 SNPs and three SNPs associated with LDL cholesterol, a twofold difference in risk for MI between extreme quintiles of scores was found [23]. However, in a prospective cohort, there was no improvement in risk discrimination by addition of the risk alleles of 9p21 in men or women [59, 60].
Conclusions
In the past years, efforts to disentangle the genetics of CVD based on GWA studies have resulted in the identification of several common variants with moderate effects. This has resulted in the identification of 17 loci. Still, only part of the heritability of CVD is currently explained. More of the missing heritability could be discovered by efforts to identify novel SNPs and efforts to fine map candidate loci [61]. Identification of novel SNPs will occur through future GWA studies and meta-analyses of the current studies. At least two large GWA studies (Cardiogram and C4D) on CVD are near completion and fine mapping of existing candidate loci is ongoing.
Nevertheless, even with the outcome of these efforts only a modest part of the risk for CVD will be explained. We expect that a substantial part of the missing heritability will be explained by rare variants with severe effects on the phenotype. Some rare variants causing CVD could even be de novo mutations, as recently found in mental retardation [62]. Others might be detected by studying families with severe disease (defined by a combination of very early-onset CVD and multiple affected individuals per family), possibly by a combination of exclusion linkage studies and next-generation sequencing.
References
Papers of particular interest, published recently, have been highlighted as: •• of major importance
Berenson GS, Srinivasan SR, Bao W, et al. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa heart study. N Engl J Med. 1998;338:1650–6.
Marenberg ME, Risch N, Berkman LF, et al. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–6.
Sivapalaratnam S, Boekholdt SM, Trip MD et al. Family history of premature coronary heart disease and risk prediction in the EPIC-Norfolk prospective population study. Heart 2010
Aouizerat BE, Allayee H, Cantor RM, et al. A genome scan for familial combined hyperlipidemia reveals evidence of linkage with a locus on chromosome 11. Am J Hum Genet. 1999;65:397–412.
Wang L, Fan C, Topol SE, et al. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science. 2003;302:1578–81.
Wang Q, Rao S, Topol EJ. Miscues on the "lack of MEF2A mutations" in coronary artery disease. J Clin Invest. 2005;115:1399–400.
Guella I, Rimoldi V, Asselta R, et al. Association and functional analyses of MEF2A as a susceptibility gene for premature myocardial infarction and coronary artery disease. Circ Cardiovasc Genet. 2009;2:165–72.
Lieb W, Mayer B, Konig IR, et al. Lack of association between the MEF2A gene and myocardial infarction. Circulation. 2008;117:185–91.
Mani A, Radhakrishnan J, Wang H, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278–82.
Sarzani R, Salvi F, Bordicchia M et al. Carotid artery atherosclerosis in hypertensive patients with a functional LDL receptor-related protein 6 gene variant. Nutr Metab Cardiovasc Dis. 2009
Tomaszewski M, Charchar FJ, Barnes T, et al. A common variant in low-density lipoprotein receptor-related protein 6 gene (LRP6) is associated with LDL-cholesterol. Arterioscler Thromb Vasc Biol. 2009;29:1316–21.
Crosslin DR, Shah SH, Nelson SC, et al. Genetic effects in the leukotriene biosynthesis pathway and association with atherosclerosis. Hum Genet. 2009;125:217–29.
Shen GQ, Li L, Girelli D, et al. An LRP8 variant is associated with familial and premature coronary artery disease and myocardial infarction. Am J Hum Genet. 2007;81:780–91.
Lanktree MB, Dichgans M, Hegele RA. Advances in genomic analysis of stroke: what have we learned and where are we headed? Stroke. 2010;41:825–32.
Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–76.
Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9.
Keating BJ, Tischfield S, Murray SS, et al. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS ONE. 2008;3:e3583.
•• Musunuru K and Kathiresan S. Genetics of coronary artery disease. Annu. Rev. Genomics Hum. Genet. 2010, 11: 91–108. This is an excellent review on the current status of genetic studies with CVD as the endpoint.
Schunkert H, Erdmann J, Samani NJ. Genetics of myocardial infarction: a progress report. Eur Heart J. 2010;31:918–25.
•• Teslovich TM, Musunuru K, Smith AV et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466: 707–13. This article uses a combination of GWA study meta-analyses and gene expression data to proof the importance of novel candidate genes for lipid metabolism. A novel gene, GALNT2, has been described in more detial in a novel mouse model.
McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–91.
Samani NJ, Schunkert H. Chromosome 9p21 and cardiovascular disease: the story unfolds. Circ Cardiovasc Genet. 2008;1:81–4.
Kathiresan S, Voight BF, Purcell S et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009
Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–53.
Samani NJ, Deloukas P, Erdmann J, et al. Large scale association analysis of novel genetic loci for coronary artery disease. Arterioscler Thromb Vasc Biol. 2009;29:774–80.
Musunuru K, Post WS, Herzog W, et al. Association of single nucleotide polymorphisms on chromosome 9p21.3 with platelet reactivity: a potential mechanism for increased vascular disease. Circ Cardiovasc Genet. 2010;3:445–53.
Bjorck HM, Lanne T, Alehagen U, et al. Association of genetic variation on chromosome 9p21.3 and arterial stiffness. J Intern Med. 2009;265:373–81.
Cunnington MS, Santibanez KM, Mayosi BM, et al. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. 2010;6:e1000899.
Helgadottir A, Thorleifsson G, Magnusson KP, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–24.
Folkersen L, Kyriakou T, Goel A, et al. Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS ONE. 2009;4:e7677.
McPherson R. Chromosome 9p21 and coronary artery disease. N Engl J Med. 2010;362:1736–7.
Visel A, Zhu Y, May D, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–12.
Jarinova O, Stewart AF, Roberts R, et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. 2009;29:1671–7.
•• Musunuru K, Strong A, Frank-Kamenetsky M et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 2010, 466: 714–19. This article describes the approach to identify a GWA signal and translate it into a functional mechanism using mouse models, in vitro studies, and also large cohorts to identify the QTL signal.
Kjolby M, Andersen OM, Breiderhoff T, et al. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12:213–23.
Goldstein JL, Sobhani MK, Faust JR, Brown MS. Heterozygous familial hypercholesterolemia: failure of normal allele to compensate for mutant allele at a regulated genetic locus. Cell. 1976;9:195–203.
Kotowski IK, Pertsemlidis A, Luke A, et al. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78:410–22.
Huijgen R, Vissers MN, Defesche JC, et al. Familial hypercholesterolemia: current treatment and advances in management. Expert Rev Cardiovasc Ther. 2008;6:567–81.
van Aalst-Cohen ES, Jansen AC, Tanck MW, et al. Diagnosing familial hypercholesterolaemia: the relevance of genetic testing. Eur Heart J. 2006;27:2240–6.
Stone NJ, Levy RI, Fredrickson DS, Verter J. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation. 1974;49:476–88.
Tregouet DA, Konig IR, Erdmann J, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–5.
Kamstrup PR. Lipoprotein(a) and ischemic heart disease–a causal association? A review. Atherosclerosis. 2010;211:15–23.
Utermann G. Genetic architecture and evolution of the lipoprotein(a) trait. Curr Opin Lipidol. 1999;10:133–41.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–9.
Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–28.
Zernecke A, Shagdarsuren E, Weber C. Chemokines in atherosclerosis: an update. Arterioscler Thromb Vasc Biol. 2008;28:1897–908.
Gleissner CA, von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol. 2008;28:1920–7.
Zernecke A, Bot I, Djalali-Talab Y, et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–17.
van Gils JM, da Costa Martins PA, Mol A, et al. Transendothelial migration drives dissociation of plateletmonocyte complexes. Thromb Haemost. 2008;100:271–9.
Erdmann J, Grosshennig A, Braund PS, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41:280–2.
Nunez RN, Lee IN, Banno A, et al. Characterization of R-ras3/m-ras null mice reveals a potential role in trophic factor signaling. Mol Cell Biol. 2006;26:7145–54.
Folkersen L, van’t Hooft F, Chernogubova E, et al. Association of genetic risk variants with expression of proximal genes identifies novel susceptibility genes for cardiovascular disease. Circ Cardiovasc Genet. 2010;3:365–73.
Allen PB, Greenfield AT, Svenningsson P, et al. Phactrs 1–4: a family of protein phosphatase 1 and actin regulatory proteins. Proc Natl Acad Sci USA. 2004;101:7187–92.
Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971–80.
Armendariz AD, Krauss RM. Hepatic nuclear factor 1-alpha: inflammation, genetics, and atherosclerosis. Curr Opin Lipidol. 2009;20:106–11.
Prudente S, Morini E, Trischitta V. Insulin signaling regulating genes: effect on T2DM and cardiovascular risk. Nat Rev Endocrinol. 2009;5:682–93.
Gudbjartsson DF, Bjornsdottir US, Halapi E, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. 2009;41:342–7.
Tsai CT, Lai LP, Hwang JJ, et al. Molecular genetics of atrial fibrillation. J Am Coll Cardiol. 2008;52:241–50.
Paynter NP, Chasman DI, Buring JE, et al. Cardiovascular disease risk prediction with and without knowledge of genetic variation at chromosome 9p21.3. Ann Intern Med. 2009;150:65–72.
Talmud PJ, Cooper JA, Palmen J, et al. Chromosome 9p21.3 coronary heart disease locus genotype and prospective risk of CHD in healthy middle-aged men. Clin Chem. 2008;54:467–74.
Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53.
Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–12.
Acknowledgments
S. Maiwald received financial support from NETSIM Marie Curie Initial Training Network, EC (Project Number 215820). M.M. Motazacker was supported by a grant of the European Community (FP6-2005-LIFESCIHEALTH-6; STREP contract number 037631).
Disclosure
The authors report no potential conflicts of interest relevant to this article.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sivapalaratnam, S., Motazacker, M.M., Maiwald, S. et al. Genome-Wide Association Studies in Atherosclerosis. Curr Atheroscler Rep 13, 225–232 (2011). https://doi.org/10.1007/s11883-011-0173-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11883-011-0173-4