
Abstract
Like many eicosanoids, epoxyeicosatrienoic acids (EETs) have multiple biological functions, including reduction of blood pressure, inflammation, and atherosclerosis in multiple species. Hydration of EETs by the soluble epoxide hydrolase (sEH) is the major route of their degradation to the less bioactive diols. Inhibition of the sEH stabilizes EETs, thus, enhancing the beneficial effects of EETs. Human data show an association of sEH (Ephx2) gene polymorphisms with increased risk of atherosclerosis and cardiovascular diseases. These data suggest a potential therapeutic effect of sEH inhibitors (sEHI) in the treatment of atherosclerosis. Indeed, two laboratories reported independently that using different sEHIs in apolipoprotein E–deficient mice significantly attenuated atherosclerosis development and aneurysm formation. The antiatherosclerotic effects of sEHI are correlated with elevation in EET levels and associated with reduction of low-density lipoprotein and elevation of high-density lipoprotein cholesterols, as well as attenuation of expression of proinflammatory genes and proteins. In addition, the antihypertensive effects and improvement of endothelial function also contribute to the mechanism of the antiatherosclerotic effects of sEHI. The broad spectrum of biological action of EETs and sEHIs with multiple biological beneficial actions provides a promising new class of therapeutics for atherosclerosis and other cardiovascular diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is a chronic vascular inflammatory disease characterized by chronic inflammation and abnormalities in cholesterol transport leading to foam cell and plaque formation in the arterial wall. It is a major cause of cardiac death with several life-threatening cardiovascular complications, including myocardial infarction and stroke when the atherosclerotic plaque ruptures. Currently, the major pharmacologic intervention for atherosclerosis is to use statins alone or in combination with niacin and fibrates to lower low-density lipoprotein (LDL) and to increase high-density lipoprotein (HDL), in addition to modifications in diet. Statins inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in cholesterol synthesis, and increase LDL cholesterol clearance. Statins have been shown to be effective in reducing cholesterol and attenuation of atherosclerosis and cardiovascular risk in patients. Moreover, a new-generation statin, namely rosuvastatin, has been shown to attenuate coronary atherosclerosis [1]. In this clinical trial, known as A Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (ASTEROID), high-intensity rosuvastatin therapy not only dramatically decreased LDL cholesterol, but also attenuated atherosclerotic plaques in the coronary arteries [1]. Anti-inflammatory drugs, such as low-dose aspirin, and antihypertensive drugs, in particular angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, also have beneficial effects in the prevention of atherosclerosis and reducing the cardiovascular risk when used prophylactically or in combination with statins.
Several key enzymes and receptors in the arachidonic acid (AA) cascade are important targets for atherosclerosis [2]. A novel target is the soluble epoxide hydrolase (sEH). A major function of the sEH is to metabolize the epoxides of AA and linoleic acid that are the regioisomers of epoxyeicosatrienoic acids (EETs) and epoxyoctadecenoic acids (EpOMEs) to their corresponding diols, dihydroxyeicosatrienoic acid (DHET) and dihydroxyoctadecenoic acid (DiHOME), respectively. The epoxides of AA, EETs have protective effects on the vasculature, kidney, and the heart. Inhibitors of sEH reduce inflammation and prevent the development of atherosclerotic plaques, presumably via an increase in EETs and other epoxy lipids, as well as a decrease in the corresponding diols [3••, 4••].
The purpose of this review is to discuss the role of EETs and sEH in the pathogenesis of atherosclerosis as well as the preclinical and clinical evidence that supports the rationale to use sEHI as therapeutics for prevention and treatment of atherosclerosis and its complications.
Biological Activities of EETs and sEH and Potential Therapeutic Applications
AA can be metabolized by three major oxidative pathways: cyclooxygenase (COX), forming prostaglandins and related eicosanoids; lipoxygenase (LOX), forming leukotrienes and related compounds; and cytochrome P450 (CYP450) monooxygenase, forming epoxides and alcohols such as 20-HETE (Fig. 1). The COX and LOX pathways have been investigated extensively, and their eicosanoid products have been shown to play important roles in a variety of biological processes such as inflammation, cell proliferation, and intracellular signaling. However, the less studied “third pathway” of the AA cascade involving CYP450 enzymes is receiving increasing attention. EETs are synthesized from AA by oxidation reactions catalyzed by CYP450s. These regulatory lipid molecules have multiple biological functions, among which vasorelaxation has been studied most extensively. EETs and other bioactive fatty acid epoxides are formed in endothelial cells via stimulation of AA release from phospholipids and communicate with vascular smooth muscle cells (VSMC) through large conductance Ca2+ activated K (BK) channels via a putative G-protein–coupled receptor that signals via Gsα [5], leading to membrane hyperpolarization, hence causing vasorelaxation. EETs and other fatty acid epoxides thus are endothelium-derived hyperpolarizing factors (EDHFs) [6]. The EDHF effects of EETs dominate in microvessels and are independent of nitric oxide (NO) and prostacyclin (PGI2), which mainly act on conduit vessels. In the renal microcirculation, EETs contribute to vasorelaxation (leading to regulation of renal blood flow) and to ion transport (leading to inhibition of sodium reabsorption), thereby contributing to blood pressure lowering [7]. On the contrary, another CYP450 metabolite in the AA cascade, 20-HETE, has been shown to partially antagonize the effects of EETs by acting as a BK channel antagonist in the renal microcirculation, thereby increasing blood pressure [8].

The arachidonic acid (AA) cascade. AA is metabolized by three major oxidative pathways: 1) cyclooxygenase (COX), forming prostaglandins and related eicosanoids; 2) lipoxygenase (LOX), forming leukotrienes and related compounds; and 3) CYP450, forming epoxides (2C/2J) and 20-HETEs (4A/4F). Epoxyeicosatrienoic acids (EET)s are vasodilatory and anti-inflammatory, whereas 20-HETE antagonizes these effects of EETs. Soluble epoxide hydrolase (sEH) degrades EETs to their less bioactive corresponding dihydroxyeicosatrienoic acid (DHETs), thereby reducing beneficial effects of EETs. Inhibitors of sEH stabilize EETs, and prolong the duration of action of EETs, thus, enhancing the effects of reducing hypertension, inflammation, and pain
These biochemically short-lived EETs are quickly degraded by sEH into their corresponding diols or DHETs, which generally are less bioactive. The sEH thus reduces the beneficial effects of EETs [9]. Epoxide hydration is the dominant pathway of EETs degradation, but EETs are also metabolized by β oxidation, chain elongation, hydroxylation, incorporation into the cell membrane phospholipids, and/or even via other enzymes in the arachidonate cascade [10]. Early sEH inhibitors were competitive substrates or substrates that were slowly turned over. These compounds were of limited use in vivo. The first transition state mimic inhibitors potent enough for in vivo use were based on urea, carbamate, and amide pharmacophores [9, 11]. Inhibitors of sEH can enhance these effects by stabilizing EETs and other lipid epoxides and by reducing some proinflammatory diols [12]. Thus, inhibition of sEH has been proposed as a therapeutic target to treat hypertension and its complications [13]. Indeed, a variety of sEH inhibitors have been shown to decrease blood pressure in a number of rodent models of hypertension [7, 14, 15]. In addition to vasorelaxation, EETs have also been shown to be anti-inflammatory, analgesic, and mildly angiogenic [7, 10]. The efficacy of sEH inhibitors has been demonstrated in animal models to reduce cardiac hypertrophy [16], attenuate sepsis-induced inflammation [12], and decrease inflammatory pain [17]. The biological effects seen in these studies can be attributed to inhibition of sEH activity evidenced by an increase in plasma EET to DHET ratio. Furthermore, sEH is widely expressed in the liver, kidney, heart, brain, and other tissues; thus the anti-inflammatory effects of sEHI offer novel treatment options in a range of disease models, including atherosclerosis, pain, stroke, and diabetes [3••, 4••, 17–20].
sEHIs have been shown to reverse cardiac hypertrophy in several animal models. In a mouse model of angiotensin II (Ang II)-induced cardiac hypertrophy, treatment with a potent sEHI, 1-(1-methanesulfonyl-piperidin-4-yl)-3-(4-trifluoromethoxy-phenyl)-urea (TUPS), significantly reduced cardiac hypertrophy and improved cardiac contractility [21]. The in vitro experiment in neonatal cardiac myocytes treated with Ang II revealed that the antihypertrophic effect of sEHI is concentration dependent and associates with downregulation of COX-2 via an increase in the ratio of EET to DHET, indicating that antihypertrophic efficacy of sEHI acts directly on cardiomyocytes, independent of blood pressure regulation [22]. In a murine model of cardiac hypertrophy induced by thoracic aortic banding (TAC), the sEHI blocked nuclear factor–κB (NF-κB) activation, thereby reducing inflammation, and decreased the gene expression of the cardiac hypertrophy markers, including atrial natriuretic factor, skeletal actin, and major histocompatibility complex (MHC), compared with that in vehicle control group [22]. Later on, more insight in the role of sEH in cardiac hypertrophy came with another study in two different rat models: spontaneously hypertensive rats (SHRs) and Ang II-infused Wistar rats [16]. This study revealed that Ang II dose-dependently upregulated cardiac sEH expression involving AP-1 transcription factor activation, which could be reversed by losartan, an angiotensin type 1 (AT-1) receptor antagonist. In a murine model of coronary artery ligation, sEHIs are also found to be protective against heart failure and cardiac arrhythmias after myocardial infarct by reducing infarct size and preventing cardiac remodeling [23]. In this study, at 3 weeks after ligation, the ratios of EET to DHET and EpOME to DiHOME were increased, along with a decrease in cytokines in the sEHI-treated compared with the sEHI-untreated group. Because the sEHI had cardioprotective effects in variety animal models of cardiac hypertrophy and heart failure, including non–Ang II-induced models, multiple mechanisms may be involved in the mode of action.
sEH Polymorphism and Atherosclerosis
sEH is expressed in many organisms, including plants, nematodes, and humans. It was reported in the 1970s that laboratory rats have by far the lowest levels of hepatic sEH among the mammals studied. Increased sEH gene expression and activity along with renal eicosanoid metabolism have been reported to participate in the pathogenesis of hypertension in SHRs [14]. It has been observed in many laboratories that increased sEH is associated with a lowering of blood pressure in several but not all strains of SHR [24, 25]. Investigating the role of sEH in hypertensive animals, Fornage et al. [26] reported that sub-strains of SHR and its wild-type control WKY rats obtained from different sources (eg, Heidelberg vs Charles River) show differences in the level of Ephx2 (sEH) gene expression, activity, and protein abundance. In order to understand the underlying mechanisms of differences in sEH in these rat strains, the authors hypothesized that single nucleotide polymorphisms (SNPs) may be responsible for the differences in sEH as well as blood pressure in these animals. The genotyping of the renal cortical preparations from SHR and WKY revealed four SNPs, which create two Ephx2 alleles. These variants included G405A, G560A, C780T, and G1465A (NCBI NM_022936, reference sequence). The SHR Heidelberg sequence was found to be identical to that of Sprague-Dawley rats.
The homozygous ancestral alleles were identified to be present in the SHR (Heidelberg) and WKY (Charles River), whereas the variant alleles were identified in the SHR (Charles River) and WKY (Heidelberg) strains. A strong correlation was found between the inheritance of these alleles and the gene expression, protein abundance (confirmed by semi-quantitative Western blot analysis), and sEH activity (in renal cortex) but not blood pressure in the F2 progeny obtained from crossing the SHR and WKY Heidelberg strains. The positive findings in the F2 progeny were also supported by the semi-quantitative Western blot analyses showing high Ephx2 gene expression in the renal cortex of the parental WKY (Heidelberg) but not in SHR (Heidelberg). Overall, a strong association exists between gene sequence, protein abundance, and enzyme activity, apart from the hypertensive state.
Later in this study, when the SHR and WKY rats from Charles River were administered an sEHI, a small drop in blood pressure was observed in both groups. However, the absence of a dramatic reduction in blood pressure with an sEHI in SHRs was inconsistent with previous reports obtained with tail cuff measurements in SHRs. In the end, this study shows that it is very crucial to know the genotype of the animals studied and whether they carry the ancestral or variant Ephx2 alleles in the research of sEH and hypertension. Although the authors consider other possible allelic influences, their observation in the F2 progeny showing that there is no association between the inherited Ephx2 alleles and blood pressure contrasts with previous reports on the role of sEH in hypertension.
The association between sEH gene (Ephx2) polymorphisms and cardiovascular diseases has also been evaluated in humans. sEH gene is located on chromosome 8p21-p12, enclosing 19 exons. The polymorphisms in CYP2J2, a major CYP450 enzyme generating EETs from AA in the vasculature, are associated with low levels of EETs and an increased risk of coronary artery disease [27]. In another study investigating the role of Ephx2 polymorphisms in atherosclerosis in African Americans and whites from the Coronary Artery Risk Development in young Adults (CARDIA) study of 18- to 30-year olds, where coronary artery plaque calcification (CAC) measured by tomography was used as a noninvasive indicator of atherosclerosis, several gene polymorphisms have been identified, some of which change the activity and the stability of the sEH enzyme [28]. The results of the CARDIA study showed that the sEH gene polymorphisms associated with CAC correlate most highly among cigarette smokers, consistent with previous findings that cigarette smoking upregulates sEH in the vasculature [29]. The Atherosclerosis Risk in Communities (ARIC) study, which includes15,792 men and women 45–64 years of age, revealed 10 nonsynonymous genetic variations in both the coding and noncoding regions of Ephx2 gene [30]. Among those polymorphisms, Lys55Arg (K55R) polymorphism in exon 2 has been found to be an important risk factor for the development of coronary heart disease (CHD) in whites with high frequency (20.8%) compared with that in controls (15.3%). In functional studies, K55R increases sEH activity, whereas another common polymorphism, Arg287Gln (R287Q), reduces sEH activity. Thus, the K55R polymorphism, in both the heterozygous (A/G) and homozygous (G/G) states, correlates with low ratio of plasma 12, 13- EpOME to DiHOME, which provides a good in vivo biomarker of increased sEH activity associated with K55R polymorphism and CHD. This study also evaluated the significance of these polymorphisms with reconstructed Ephx2 haplotypes and the incidence of CHD. Indeed, the presence of six haplotypes differed significantly between the CHD and control cases, suggesting the importance of sEH gene polymorphisms in cardiovascular diseases, and in particular atherosclerosis. An association has also been found between the Ephx2 sequence variation, Glu287Arg, and plasma cholesterol levels in familial hypercholesterolemia using intra-familial correlation analysis [31].
In contrast to these studies, sEH mRNA transcript levels were found to be five times lower leading to lower sEH activity in the brain of stroke -prone SHRs rather than stroke-resistant SHRs due to sequence variations in the sEH gene promoter [32]. The low sEH activity did not result in high levels of EETs in stroke-prone SHRs as would be expected. When the gene expression of the P450 enzymes synthesizing these epoxides were looked at, the data were found to be consistent with EET levels, indicating differences in the mediators other than EETs in the mechanism of the end organ injury. Although Ephx2 gene polymorphisms have been shown to be associated with stroke, the allelic differences in the Ephx2 in the SHRs did not predict blood pressure levels [26]. Similarly, the study in stroke-prone versus stroke-resistant SHRs revealed that the sequence variations in the Ephx2 promoter somewhat relates to the enzymatic activity, but fails to directly correlate these polymorphisms with the risk for stroke in vivo due to the complexity of the conditions [32]. A study in African Americans also found no association between the epoxygenase gene SNPs (CYP2C8, 2C9, 2J2, and sEH) and hypertension [33]. Thus, several studies suggest an association among the enzymes that produce and degrade EETs and cardiovascular disease; however, the association appears complex.
Inhibition of sEH in Animal Models of Atherosclerosis
Although the human data demonstrate that the sEH (Ephx2) gene polymorphisms are associated with increased risk for atherosclerotic vascular diseases [30, 34–36], exploration of the in vivo effects of sEH inhibition in animal models of atherosclerosis only was reported recently. Ulu et al. [4••] reported antiatherosclerotic effects of an sEH inhibitor using an established murine model of atherosclerosis in 5-month-old apolipoprotein E (apoE)-deficient mice. In this model, atherosclerosis development was accelerated in mice by feeding the Paigen atherogenic diet and subcutaneously infusing Ang II (1.44 mg/kg per day) for 4 weeks. An sEH inhibitor, AEPU (1-adamantan-3-(5-(2-(2-ethylethoxy) ethoxy) pentyl) urea) (Fig. 2), was given in drinking water (90 mg/L) for 8 weeks starting 4 weeks prior to the initiation of atherogenic diet and Ang II and continued to the end of the study. AEPU treatment reduced the atherosclerotic lesion area in the descending aortas by 53% compared with that in control mice treated with drinking water. The antiatherosclerotic effect of AEPU was accompanied by an inhibition of sEH activity measured by a significant increase in linoleic and AA epoxides, as well as the ratios of EETs to DHETs. The reduction in the atherosclerotic lesion area was inversely correlated with the ratios of 11, 12-EET/DHET and 14, 15-EET/DHET, suggesting that the efficacy corresponds to the inhibition of sEH.

Representative structures of soluble epoxide hydrolase (sEH) inhibitors. The most useful sEH inhibitors to date are competitive transition state inhibitors of sEH [9, 11]. There are a variety of sEH inhibitors available for use in animal models. The commercial compounds such as AUDA are very potent, but hard to formulate and rapidly metabolized. Increases in oral bioavailability and improved physicochemical properties were obtained with AEPU and APAU, with some loss of potency. Compounds such as TPPU and t-TUCB have higher potency and much longer half-lives in vivo. The potency of the piperidine compounds (APAU, TPPU) tends to be over-estimated by roughly 20-fold compared with other inhibitors when a common fluorescent assay is used. The Merck compound (Merck & Co., Whitehouse Station, NJ) combines good potency and good rodent pharmacokinetics [70]. Sorafenib is a commercial anti-cancer drug. Its potency and pharmacokinetics indicate that it should inhibit the sEH in humans at a fraction of the doses used therapeutically for cancer [69]. A caution is that some but not all sEH inhibitors are active across multiple species. For example, the activity of the piperidines (APAU, TPPU) is often orders of magnitude lower in feline or canine models
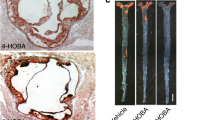
In parallel, Wang and Cassis [37] reported more in-depth observations in a similar model in which 6-month-old apoE-deficient mice on normal rodent diet were infused with Ang II (1.44 mg/kg per day) for 4 weeks to accelerate atherosclerosis development and abdominal aortic aneurysm formation. In addition, the left common carotid artery was ligated at its bifurcation to induce vascular remodeling, including neointima formation and adventitia development, which resembles the pathogenesis of restenosis after surgical angioplasty [38, 39•, 40, 41]. The mice had free access to 5% hydroxypropyl-β-cyclodextrin in drinking water containing an sEH inhibitor, AR9276 (1-(1-nicotinoylpiperidin-4-yl)-3-(4-(trifluoromethoxy) phenyl) urea) at a concentration of 1.5 g/L for 4 weeks starting 1 day before the Ang II infusion [3••]. Treatment with AR9276 completely abolished whole blood sEH activity measured by monitoring the conversion rate of 14, 15-EET to 14, 15-DHET. Treatment with AR9276 not only reduced atherosclerotic lesion area in the right carotid artery and aortic arch by 33% and 39%, respectively, but also decreased the incidence of abdominal aortic aneurysm formation by 72% and the average diameters of abdominal aorta by 32% compared with control mice drinking HPBCD water only (Fig. 3). However, treatment of AR9276 did not affect ligation-induced vascular remodeling in the right carotid artery.

Treatment of a soluble epoxide hydrolase (sEH) inhibitor reduces atherosclerotic lesions in the aorta (left) and carotid artery (middle) and attenuates abdominal aortic aneurysm formation (right) in apolipoprotein E–deficient mice. An sEH inhibitor, AR9276, was administrated in drinking water (1.5 g/L) for 4 weeks in apolipoprotein E–deficient mice (6-months of age) chronically infused with angiotensin II (1.44 mg/kg per day) to accelerate atherosclerosis development and induce abdominal aneurysm formation
These two independent reports provide direct and compelling evidence that sEH plays an important role in the pathogenesis of atherosclerosis development and aneurysm formation, suggesting that inhibition of sEH may be a promising therapeutic intervention in these indications. The exact mechanism by which sEH inhibitors exert therapeutic benefits is still not clear, to which the following aspects may contribute.
It has been established that elevation of circulating LDL cholesterol and reduction of HDL play a causal role in atherosclerosis development and progression [42]. Atherosclerotic lesions start with increased delivery and oxidation of LDL cholesterol in the intima layer of the arterial wall that initiates LDL uptake by monocytes and macrophages to form foam cells, whereas HDL removes cholesterol from the vascular wall. In addition, LDL tends to destabilize platelet membrane activity and disrupt normal functions of macrophages, endothelium, and VSMC, whereas HDL tends to reverse these abnormalities [42]. An important mechanism by which statins prevent atherosclerosis and exert therapeutic effects in coronary artery disease is by lowering LDL in conjunction with minor increase in HDL [43]. The current clinical application of statins in combination with niacin to increase HDL levels has been shown more effective than monotherapy. Zhang et al. [3••] demonstrated that sEH inhibition lowered circulating LDL by 30%, elevated HDL by 43%, and elevated the HDL to LDL ratio by 96%, which could contribute, at least partially, to the antiatherosclerotic effects of sEH inhibitors. In sEH-deficient mice and sEH-expressing mammalian cell lines, low cholesterol levels have been found. Farnesyl phosphate and pyrophosphate are key intermediates in steroid biosynthesis and prenylation and they are excellent substrate for the phosphatase component of sEH. Thus, the sEH phosphatase activity has been proposed as possibly responsible for this effect [44]. In contrast to the lipid-lowering effect of AR9276 reported by Zhang et al. [3••], in the study by Ulu et al. [4••], AEPU did not affect blood cholesterol levels. This discrepancy between the two laboratories could result from the use of different sEH inhibitors. Alternatively, the use of different diets or sources of animals in the two studies may also impact the profile of responses.
Vascular inflammation characterized by infiltration of monocytes/macrophages and lymphocytes into the arterial wall is a crucial pathogenic event in atherosclerosis and aneurysm formation [45–47]. Cholesterol accumulation and oxidative LDL in the vascular wall lead to expression of adhesion molecules and release of chemotactic factors. This process triggers the release of cytokines and VSMC migration. which secretes extracellular matrix metalloproteinases, resulting in chronic inflammation [48]. Moreover, Ang II is not only a vasoconstrictor, but it also is a proinflammatory factor that promotes atherosclerosis development and vascular remodeling via stimulating proinflammatory factors in vascular endothelium. including vascular adhesion molecule, chemokines, cytokines, and growth factors that further facilitate monocyte/macrophage and T-cell infiltration and VSMC migration into the subendothelial space [49–51]. Ang II is also a potent inducer of the she. which may account for some of its inflammatory actions [7, 21]. These inflammatory cells take up lipids, becoming foam cells that coalesce into atherosclerotic lesions [52]. Tedgui and Mallat [53] demonstrated that treatment with AR9276 reduced serum inflammatory markers, including interleukin (IL)-1α, IL-6, and KC, and murine IL-8, all of which were reported to be involved in the progression of atherosclerosis. In agreement with this, Fang et al. [54, 55] reported that sEH inhibition increased 10, 11-epoxy-16:2 generation from 14, 15-EET, which not only inhibited tumor necrosis factor (TNF)-α–mediated generation of the proinflammatory cytokine IL-8 in human coronary endothelial cells, but also enhanced dilation of coronary microvessels [54, 55]. This observation raises the possibility that epoxides of a variety of fatty acids, including chain lengthened and shortened metabolites of AA, omega-3 lipids and other fatty acids, and epoxides from COX and LOX products, may both be biologically active and stabilized by sEHI. Indeed, Galkina and Ley [56] further showed that treatment with AR9276 decreased proinflammatory gene expression in the arterial wall, especially vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), which are critical in initiation and development of atherosclerosis. Furthermore, direct application of EETs in human endothelial cells inhibited TNF-α–induced VCAM-1 and ICAM-1 expression [10]. Taken together, these data support the concept that anti-inflammatory effects may be another important mechanism by which sEH inhibitor attenuates Ang II–accelerated atherosclerosis and aneurysm formation.
High blood pressure can cause vascular injury, thereby, facilitating the development of atherosclerosis. It has been established that treatment with sEH inhibitors reduces blood pressure in several animal models of hypertension [7, 14, 15]. Thus, blood pressure reduction can also contribute to the antiatherosclerotic effects of sEH inhibitors. One of the important functions of endothelium is to protect the vessel from injury. EETs are generated from endothelium and play a critical role in regulation of endothelial-derived vascular function. Endothelial dysfunction, manifest as impaired endothelium-dependent vasorelaxation, is a hallmark of, and plays a pivotal role in, pathogenesis of cardiometabolic diseases, including atherosclerosis, type 2 diabetes, obesity, and hypertension. Multiple studies show that treatment with sEHIs improves endothelial functions in a number of animal models of hypertension, obesity, and diabetes [3••]. Thus, improvement of endothelial function can also contribute to the antiatherosclerotic effects of sEH inhibitors.
Smooth muscle cell migration and proliferation are some of the major pathologic processes evident in the Ang II–accelerated vascular remodeling in the carotid artery ligation model [39, 41]. EETs have been shown to attenuate the migration of VSMC stimulated with platelet-derived growth factor in rat aortas [57]. Recently, Davis et al. [58, 59] reported that sEH inhibitors attenuated VSMC proliferation; however, they later showed that this effect is compound chemical-structure specific and independent of sEH inhibition. Consistent with the latter report, Simpkins et al. [60] observed that treatment with AR9276 had no effect on ligation-induced vascular remodeling. Furthermore, a very recent article reported that sEH inhibition or Ephx2 gene deletion antagonizes neointimal formation in vivo by mechanisms that are endothelium dependent [60]. In contrast, Kullmann et al. [61] reported that the Ephx2 K55R polymorphism is not associated with restenosis after percutaneous coronary intervention in human. Taken together, it appears that the effects of sEH inhibition on VSMC proliferation and migration and on neointimal formation after vascular injury are controversial.
Conclusions
The preclinical observations of the beneficial effects of EETs and sEH inhibitors, including vasodilation, antihypertension, antiinflammation, improving endothelial function, and lipid regulation, along with the clinical observation of an association of the sEH (Ephx2) gene polymorphisms with increased risks of atherosclerotic vascular disease, provide a strong rationale to target sEH in the treatment of atherosclerosis. In support of this rationale are the observations that treatment with sEH inhibitors in apoE-deficient mice significantly attenuated atherosclerosis development and aneurysm formation. The pathogenesis of atherosclerosis is complex, involving multiple mechanisms. Although translation of preclinical efficacy in animal models to human disease remains to be demonstrated in clinical trials, the numerous positive effects of sEHI in cardiovascular diseases render the sEH a promising therapeutic target [13, 62••].
In addition to monotherapy, sEH inhibitors may also be combined with other existing drugs for atherosclerotic vascular disease. Even though the current antiatherosclerotic therapy is effective in reducing cholesterol and cardiovascular risk, it targets the disease beyond the underlying processes. For example, atorvastatin possesses anti-inflammatory properties similar to 15-epi-lipoxin A4 (15-epi- LXA4), which resides in the lipoxygenase branch of the AA cascade and acts to resolve inflammation [63, 64]. Lovastatin has been shown to reduce acute mucosal inflammation in polymorphonuclear leukocytes and airway epithelial cells by a mechanism involving the generation of 15-epi-LXA4 and 14, 15- EET [65]. At the cellular level, 14, 15- EET has been shown to induce the generation of 15- epi- LXA in this study. Given these data, an advanced therapy to reduce the underlying causes of atherosclerosis such as inflammation, by using both a statin and a sEH inhibitor to reduce cholesterol and induce anti-inflammation via EETs and 15-epi-LXA4, remains to be elucidated. Furthermore, aspirin, nonsteroidal anti-inflammatory drugs, and LOX5 inhibitors have been shown to synergize dramatically with sEHI in several indications, including inflammation [20, 66]. This synergy might be applicable to treating atherosclerosis. sEHI and peroxisome proliferator–activated receptor (PPAR)-based drugs are also synergistic when used preclinically as a combination therapy [67].
Drugs targeting inflammation and other mechanisms underlying atherosclerosis, such as VSMC proliferation and migration, endothelial dysfunction, and activation of adhesion molecules, offer promise in prevention and treatment of atherosclerosis and in reduction of cardiac events. The combination of sEHI with existing therapies may synergistically enhance the lipid lowering, anti-inflammatory, and antihypertensive benefits, or may reduce the adverse effects by lowering the doses of existing drugs. The broad spectrum of sEH inhibitors with multiple biological beneficial actions provides a promising new class of therapeutics for atherosclerosis and other cardiovascular diseases.
However, the broad spectrum of biological activities of EETs may render concerns of potential undesired effects for sEHIs. For example, EETs have been reported to promote angiogenesis via growth factors and cell signaling mechanisms involving Janus-activated STAT signaling [68]. Such angiogenic effects may lead to metastasis in various types of cancers. In contrast, the angiogenesis is beneficial in promoting wound healing. and tissue repair. Thus, the exact impact of sEH inhibitors on therapeutic benefits versus potential adverse effects remains to be examined.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Nissen SE, Nicholls SJ, Sipahi I, et al.: Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006, 295:1556–1565.
Wang M, Song WL, Cheng Y, Fitzgerald GA: Microsomal prostaglandin E synthase-1 inhibition in cardiovascular inflammatory disease. J Intern Med 2008, 263:500–505.
•• Zhang LN, Vincelette J, Cheng Y, et al.: Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb Vasc Biol 2009, 29:1265–1270. This article investigated the anti-inflammatory mechanisms involved in the anti-atherosclerotic effects of sEH inhibitors.
•• Ulu A, Davis BB, Tsai HJ, et al.: Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol 2008, 52:314–323. This is the first article to demonstrate the antiatherosclerotic effects of sEH inhibitors and correlation of these effects with plasma epoxide to diol levels.
Li PL, Campbell WB: Epoxyeicosatrienoic acids activate K + channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ Res 1997, 80:877–884.
Fleming I: Cytochrome P450 epoxygenases as EDHF synthase(s). Pharmacol Res 2004, 49:525–533.
Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD: An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 2005, 46:975–981.
Fleming I: Cytochrome p450 and vascular homeostasis. Circ Res 2001, 89:753–762.
Morisseau C, Goodrow MH, Dowdy D, et al.: Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci U S A 1999, 96:8849–8854.
Node K, Huo Y, Ruan X, et al.: Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285:1276–1279.
Morisseau C, Hammock BD: Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol 2005, 45:311–333.
Schmelzer KR, Kubala L, Newman JW, et al.: Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A 2005, 102:9772–9777.
Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD: The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol 2007, 50:225–237.
Yu Z, Xu F, Huse LM, et al.: Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res 2000, 87:992–998.
Jung O, Brandes RP, Kim IH, Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 2005, 45:759–765.
Ai D, Pang W, Li N, et al.: Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci U S A 2009, 106:564–569.
Inceoglu B, Jinks SL, Schmelzer KR, et al.: Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci 2006, 79:2311–2319.
Zhang W, Koerner IP, Noppens R, et al.: Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab 2007, 27:1931–1940.
Ohtoshi K, Kaneto H, Node K, et al.: Association of soluble epoxide hydrolase gene polymorphism with insulin resistance in type 2 diabetic patients. Biochem Biophys Res Commun 2005, 331:347–350.
Inceoglu B, Jinks SL, Ulu A, et al.: Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A 2008, 105:18901–18906.
Ai D, Fu Y, Guo D, et al.: Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc Natl Acad Sci U S A 2007, 104:9018–9023.
Xu D, Li N, He Y, et al.: Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A 2006, 103:18733–18738.
Li N, Liu JY, Timofeyev V, et al.: Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J Mol Cell Cardiol 2009, 47:835–845.
Olearczyk JJ, Quigley JE, Mitchell BC, et al.: Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin Sci (Lond) 2009, 116:61–70.
Dorrance AM, Rupp N, Pollock DM, et al.: An epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol 2005, 46:842–848.
Fornage M, Hinojos CA, Nurowska BW, et al.: Polymorphism in soluble epoxide hydrolase and blood pressure in spontaneously hypertensive rats. Hypertension 2002, 40:485–490.
Spiecker M, Darius H, Hankeln T, et al.: Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation 2004, 110:2132–2136.
Wei Q, Doris PA, Pollizotto MV, et al.: Sequence variation in the soluble epoxide hydrolase gene and subclinical coronary atherosclerosis: interaction with cigarette smoking. Atherosclerosis 2007, 190:26–34.
Maresh JG, Xu H, Jiang N, et al.: Tobacco smoke dysregulates endothelial vasoregulatory transcripts in vivo. Physiol Genomics 2005, 21:308–313.
Lee CR, North KE, Bray MS, et al.: Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet 2006, 15:1640–1649.
Sato K, Emi M, Ezura Y, et al.: Soluble epoxide hydrolase variant (Glu287Arg) modifies plasma total cholesterol and triglyceride phenotype in familial hypercholesterolemia: intrafamilial association study in an eight-generation hyperlipidemic kindred. J Hum Genet 2004, 49:29–34.
Corenblum MJ, Wise VE, Georgi K, et al.: Altered soluble epoxide hydrolase gene expression and function and vascular disease risk in the stroke-prone spontaneously hypertensive rat. Hypertension 2008, 51:567–573.
Dreisbach AW, Japa S, Sigel A, et al.: The Prevalence of CYP2C8, 2C9, 2J2, and soluble epoxide hydrolase polymorphisms in African Americans with hypertension. Am J Hypertens 2005, 18:1276–1281.
Fornage M, Boerwinkle E, Doris PA, et al.: Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: the Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation 2004, 109:335–339.
Fornage M, Lee CR, Doris PA, et al.: The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet 2005, 14:2829–2837.
Przybyla-Zawislak BD, Srivastava PK, Vazquez-Matias J, et al.: Polymorphisms in human soluble epoxide hydrolase. Mol Pharmacol 2003, 64:482–490.
Wang YX, Cassis LA: Angiotensin II-induced aortic aneurysms. In A Handbook of Mouse Models for Cardiovascular Diseases. Edited by Xu Q. Chichester, England: Life and Medical Sciences, John Wiley & Sons; 2006:125–136.
Zhang LN, da Cunha V, Martin-McNulty B, et al.: Monocyte chemoattractant protein-1 or macrophage inflammatory protein-1alpha deficiency does not affect angiotensin II-induced intimal hyperplasia in carotid artery ligation model. Cardiovasc Pathol 2007, 16:231–236.
• Zhang LN, Parkinson JF, Haskell C, Wang YX: Mechanisms of intimal hyperplasia learned from a murine carotid artery ligation model. Curr Vasc Pharmacol 2008, 6:37–43. This review article discussed a mouse model of carotid artery ligation-induced vascular remodeling used in the cited study.
Zhang LN, Velichko S, Vincelette J, et al.: Interferon-beta attenuates angiotensin II-accelerated atherosclerosis and vascular remodeling in apolipoprotein E deficient mice. Atherosclerosis 2008, 197:204–211.
Zhang LN, Wilson DW, da Cunha V, et al.: Endothelial NO synthase deficiency promotes smooth muscle progenitor cells in association with upregulation of stromal cell-derived factor-1alpha in a mouse model of carotid artery ligation. Arterioscler Thromb Vasc Biol 2006, 26:765–772.
Teramoto T, Sasaki J, Ueshima H, et al.: Goals of dyslipidemia management. J Atheroscler Thromb 2007, 14:209–212.
Vaughan CJ, Gotto AM Jr, Basson CT: The evolving role of statins in the management of atherosclerosis. J Am Coll Cardiol 2000, 35:1–10.
EnayetAllah AE, Luria A, Luo B, et al.: Opposite regulation of cholesterol levels by the phosphatase and hydrolase domains of soluble epoxide hydrolase. J Biol Chem 2008, 283:36592–36598.
Golledge J, Muller J, Daugherty A, Norman P: Abdominal aortic aneurysm: pathogenesis and implications for management. Arterioscler Thromb Vasc Biol 2006, 26:2605–2613.
Libby P, Ridker PM, Maseri A: Inflammation and atherosclerosis. Circulation 2002, 105:1135–1143.
Ross R: Atherosclerosis is an inflammatory disease. Am Heart J 1999, 138(5 Pt 2):S419–S420.
Daugherty A, Cassis LA: Mechanisms of abdominal aortic aneurysm formation. Curr Atheroscler Rep 2002, 4:222–227.
Martin-McNulty B, Tham DM, da Cunha V, et al.: 17 Beta-estradiol attenuates development of angiotensin II-induced aortic abdominal aneurysm in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2003, 23:1627–1632.
Tham DM, Martin-McNulty B, Wang YX, et al.: Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiol Genomics 2002, 11:21–30.
Wang YX, Martin-McNulty B, Freay AD, et al.: Angiotensin II increases urokinase-type plasminogen activator expression and induces aneurysm in the abdominal aorta of apolipoprotein E-deficient mice. Am J Pathol 2001, 159:1455–1464.
Nobuhiko A, Suganuma E, Babaev VR, et al.: Angiotensin II amplifies macrophage-driven atherosclerosis. Arterioscler Thromb Vasc Biol 2004, 24:2143–2148.
Tedgui A, Mallat Z: Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev 2006, 86:515–581.
Fang X, Kaduce TL, Weintraub NL, et al.: Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem 2001, 276:14867–14874.
Fang X, Weintraub NL, Oltman CL, et al.: Human coronary endothelial cells convert 14,15-EET to a biologically active chain-shortened epoxide. Am J Physiol Heart Circ Physiol 2002, 283:H2306–2314.
Galkina E, Ley K: Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol 2007, 27:2292–2301.
Sun J, Sui X, Bradbury JA, et al.: Inhibition of vascular smooth muscle cell migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ Res 2002, 90:1020–1027.
Davis BB, Thompson DA, Howard LL, et al.: Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A 2002, 99:2222–2227.
Davis BB, Morisseau C, Newman JW, et al.: Attenuation of vascular smooth muscle cell proliferation by 1-cyclohexyl-3-dodecyl urea is independent of soluble epoxide hydrolase inhibition. J Pharmacol Exp Ther 2006, 316:815–821.
Simpkins AN, Rudic RD, Schreihofer DA, et al.: Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol 2009, 174:2086–2095.
Kullmann S, Binner P, Rackebrandt K, et al.: Variation in the human soluble epoxide hydrolase gene and risk of restenosis after percutaneous coronary intervention. BMC Cardiovasc Disord 2009, 9:48.
•• Imig JD, Hammock BD: Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 2009, 8:794–805. This is the most recent review article on sEH as a novel therapeutic target for cardiovascular disease.
Birnbaum Y, Ye Y, Lin Y, et al.: Augmentation of myocardial production of 15-epi-lipoxin-a4 by pioglitazone and atorvastatin in the rat. Circulation 2006, 114:929–935.
Levy BD: Myocardial 15-epi-lipoxin A4 generation provides a new mechanism for the immunomodulatory effects of statins and thiazolidinediones. Circulation 2006, 114:873–875.
Planaguma A, Pfeffer MA, Rubin G, et al.: Lovastatin decreases acute mucosal inflammation via 15-epi-lipoxin A(4). Mucosal Immunol 2010 (in press).
Liu JY, Yang J, Inceoglu B, et al.: Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem Pharmacol 2010, 79:880–887.
De Taeye BM, Morisseau C, Coyle J, et al.: Expression and regulation of soluble epoxide hydrolase in adipose tissue. Obesity (Silver Spring) 2010, 18:489–498.
Cheranov SY, Karpurapu M, Wang D, et al.: An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis. Blood 2008, 111:5581–5591.
Strumberg D: Preclinical and clinical development of the oral multikinase inhibitor sorafenib in cancer treatment. Drugs Today (Barc) 2005, 41:773–784.
Shen HC, Ding F-X, Wang S, et al.: Discovery of a highly potent, selective, and bioavailable soluble epoxide hydrolase inhibitor with excellent ex vivo target engagement. J Med Chem 2009, 52:5009–5012.
Acknowledgment
Partial support for this work came from grants R01 ES002710 and P42 ES004699 from NIEHS and R01 HL059699 from NIH/NHLBI. The authors would like to thank Dr. Euan MacIntyle for critically reviewing this manuscript.
Disclosure
Y-X Wang and L-N Zhang were employees of Arête Therapeutics, Inc., a pharmaceutical company developing new class of sEH inhibitors for human therapeutics. BD Hammock and A Ulu are employees of the University of California, which has intellectual property in this area.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wang, YX.J., Ulu, A., Zhang, LN. et al. Soluble Epoxide Hydrolase in Atherosclerosis. Curr Atheroscler Rep 12, 174–183 (2010). https://doi.org/10.1007/s11883-010-0108-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11883-010-0108-5