
Abstract
Ab initio density-functional theory calculations were carried out to investigate the energetics and structure of key Fe-containing intermetallic phases that precipitate from cast Al-Si alloys. These results were compared with published experimental information and used to provide insight into developing improved models to describe the thermodynamic properties of these phases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The control of harmful impurities such as Fe in cast Al-Si alloys is becoming ever more important as the demand for the use of scrap materials increases. Traditionally manganese is added to prevent the formation of the harmful β-AlFeSi phase. Therefore, an understanding of phase equilibria in the Al-Fe-Mn-Si system, the solubilities of elements in various intermetallic phases and the temperatures at which they form are prerequisites for any successful control of the microstructure and the phases formed during solidification. The calculation of phase diagrams from critically assessed thermodynamic data provides an invaluable tool for providing such an understanding.
Critically assessed thermodynamic data are derived through careful analysis of all the experimental studies of thermodynamic properties and phase diagram information for a given system. Additionally, today such assessments should also consider the results of ab initio calculations, which may provide results with an accuracy equivalent to the best experimental work. Ab initio calculations may also be used to explore composition ranges of phases that are unstable and therefore inaccessible for study by experiment. In this way they can be used not just to provide key values for the critical assessment of data, but also assist in the selection of an appropriate thermodynamic model for a particular phase.
In this article, we considered in detail certain key phases of Al6Mn, Al13Fe4 and α-Al(Fe,Mn)Si, which may form during the solidification of commercial aluminium alloys.1,2,3 This included a thorough survey of published information on the thermodynamic properties, phase equilibria and crystal structures for these phases coupled to the calculation of the lattice parameters and enthalpies of formation using ab initio density-functional theory (DFT). These results allowed us to make key decisions related to the thermodynamic models used for these phases. An overview of the crystal structures of these phases is given in Table I.
Thermodynamic Modelling and Structural Properties
Despite its importance technologically, the phase diagram, thermodynamic properties and crystal structure of key intermetallic phases in the Al-Fe-Mn-Si system are not completely understood. All of the published critical assessments of data for the system are largely based on the results of the COST507 European research programme.8,9,10,11 However, most of this work was carried out before reliable software and computer power made it possible to carry out detailed calculations on complex crystal structures in multicomponent systems using ab initio DFT.
Reliable thermodynamic modelling must be based on an understanding of the known crystal structure of a phase, e.g., the number of sublattices, the occupation of elements on the crystallographic sites and the energetics associated with vacancy formation. Some simplification is however sometimes necessary to minimise the number of parameters necessary to describe the thermodynamics of a phase. Ab initio calculations now provide an invaluable tool by calculating the energetics of different crystallographic configurations to identify which features are key to guide such simplification. Furthermore, as many of the measurements of thermodynamic properties were carried out years ago, ab initio calculations provide the means to check the validity of such data. In this work such calculations have been employed to calculate the enthalpies of the formation, crystal structure and energetics associated with defect formation and the solution of additional elements onto the crystal lattice.
Ab Initio Calculations
For the calculations reported here, the plane wave method using VASP, Vienna Ab initio Simulation Package, was used.12,13 The spin-polarized generalized gradient approximation (SP-GGA-PBE)14 within the projector-augmented wave (PAW) method15 was employed for the exchange and correlation energy terms because the spin-polarized generalized gradient approximation describes the 3d transition metals such as Fe better than the (spin-polarized) local density approximation (LDA).16,17,18,19 The cut-off energy of the wave functions was set at 550 eV and the cut-off energy of the augmentation functions was 700 eV to describe the rather localized Fe 3d orbitals. The electronic wave functions were sampled on dense k grids of the Brillouin zones of the unit cells.20 To obtain the ground state of the crystals, we performed calculations for different inputs. This avoided the possibility of our results falling into metastable solutions.16,17,18 Different k-meshes and cut-off energies were used for the waves and augmentation waves, respectively. Tests showed good convergence (< 1 meV/atom).
Al6Mn
The most recent review as well as assessment of data for the Al-Fe-Mn system is from the work of Lin and Selleby.21 There are two invariant reactions in the Al-rich corner of the Al-Fe-Mn system, a eutectic reaction:
at 654°C and a probable ternary peritectic reaction at 730°C:
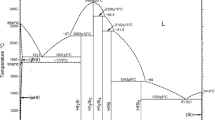
Figure 1 shows the calculated liquidus projection for the Al-rich corner of the system with key experimental values superimposed. The phase labelled “fcc” is the aluminium-based solid solution in which iron and manganese and, in the Al-Fe-Mn-Si system, silicon dissolve in small amounts. Al13Fe4 is the most aluminium-rich intermetallic phase in the Al-Fe system and often can precipitate from an aluminium-rich liquid containing relatively small amounts of iron. Other elements, particularly manganese, have appreciable solubility in this phase. Al6Mn is the most aluminium-rich intermetallic in the Al-Mn system that is in equilibrium with the liquid phase. Of particular interest is the very high solubility of iron in this phase, which extends nearly all the way across the system to the Al-Fe system. The equivalent compound Al6Fe has been observed experimentally, but is considered to be metastable. Al6Mn has a bottom-centred orthorhombic (BCO) lattice with space group Cmcm (No. 63). There are three crystallographically different species of Al with 28 atoms per unit cell.

Calculated liquidus project for the Al-rich corner of the Al-Fe-Mn system from the assessed data of Lindahl and Selleby21
Figure 2 shows a calculated isopleth in the Al-Fe-Mn system from Al6Mn to the composition of Al6Fe. It shows the appreciable solubility of Fe in the phase. Experimental phase equilibrium data are superimposed.

Calculated isopleth between Al6Mn and Al6Fe using the data of Lindahl and Selleby21
Ab initio DFT calculations have been carried out to explore the crystal structure of the Al6Mn in detail to identify which crystallographic site iron atoms are likely to replace aluminium and to consider the possibility of solution of silicon in the phase. Figure 3a shows that the substitution of Mn by Fe is predicted to be accompanied by a heat release of 2.8 kJ mol−1 per formula unit for substitution of half of the Mn by Fe. Figure 3b shows the excellent agreement between the calculated lattice parameters and the experimental values.29 The potential for Si substituting for Al in the Al6Mn lattice was also explored and shows that this is accompanied by a large enthalpy increase, i.e., substitution is not favoured.

(a) Calculated formation energy (ΔE, meV/atom) associated with the solution of Fe in Al6Mn and Mn in Al13Fe4 and (b) the calculated lattice parameters for the solution of Fe in Al6Mn. The dashed green lines in a indicate the averaged formation energy between the intermetallic compounds on the Al-Fe and Al-Mn systems. The filled spheres in (b) represent the experimental data29
Al13Fe4
Figures 1 and 2 show that Al6Mn is in equilibrium with Al13Fe4 in the Al-Fe-Mn system. Just as Fe substitutes for Mn in the Al6Mn phase, the Al13Fe4 phase is capable of dissolving substantial amounts of Mn. However, in the binary Al-Fe system the Al13Fe4 phase exists only over a relatively narrow range of homogeneity (maximum 4 at.%) that, according to recent experimental work,30 becomes smaller as the temperature is reduced.
The crystal structure of stoichiometric Al13Fe4 is now well defined.5,31,32,33,34,35 It has a total of 102 atomic sites per unit cell, 78 occupied by Al and 24 by Fe. If all lattice sites are occupied, this gives the composition Al78Fe24, i.e., Al13Fe4, and this represents the most Al-rich composition of the phase according to the phase diagram. There appear to have been no detailed crystal structure determinations on compositions away from stoichiometry to indicate where defects arise (from either vacancy formation or substitution). So far, the thermodynamic models for the phase have all been based on the assumption of vacancy formation on some of the aluminium sites. This has been successful in modelling the thermodynamic properties to calculate the phase diagram for the Al-Fe system, but, because it is not based on any firm foundation, is not able to provide a good basis for representing data for ternary systems such as the Al-Fe-Mn system where substitution of Mn is complex.
The enthalpy of formation of Al13Fe4 has been measured calorimetrically36,37,38 and calculated using ab initio DTF at 0 K.39,40 The calculated values are very close but significantly more negative than the calorimetric data.
We also studied the energetics (Fig. 4) associated with defect formation on the various sites of the Al13Fe4 lattice.40 The formation of vacancies was shown to be energetically highly unfavourable, which is in contrast to the commonly used thermodynamic model for the phase, which draws on the experimental work of Grin et al.5 for the stoichiometric composition. Further calculations showed that substitution of Fe in three aluminium sites is much more favourable energetically and led to the development of a new thermodynamic model for the phase:
where the Roman numerals IX, VII and V indicate the appropriate crystallographic site. This allows for a wider range of homogeneity than is necessary for the Al-Fe system itself but would be necessary to model the extent of the homogeneity range of the phase in the Al-Fe-Mn system.26,27 The calculations also show that the substitution of Fe onto Al sites induces magnetism. The nature of this magnetic ordering is quite complicated depending on the nature of the site or sites in which the Fe atoms substitute. On the whole, the ordering is predicted to be ferromagnetic but certain arrangements of atoms are predicted to give rise to antiferromagnetic or ferrimagnetic ordering. Further calculations were also carried out to investigate the effect of substituting Fe by Mn (Fig. 3a).

Calculated formation energies (eV/cell) at 0 K associated with the introduction of intrinsic defects into the unit cell of Al13Fe4. The vertical dotted blue line represents the stoichiometric composition. The dotted red curve connects the most stable configurations
α-Al(Fe,Mn)Si
The cubic α-Al(Fe,Mn)Si phase (also known as Al15(Fe,Mn)3Si2) forms as a large crystal of the primary intermetallic phase on solidification of many commercial Al alloys. It also forms with a “Chinese script” morphology during a subsequent eutectic reaction. The exact nature of this phase has been the subject of much uncertainty over the years and this has not been helped by the confusing and inconsistent terminology adopted for the various intermetallic phases in the quaternary system. To this end, in this article we will refer to Al7.4Fe2Si (sometimes referred to as the α-τ5 phase and α-AlFeSi) and Al9Mn2Si (sometimes referred to as α-AlMnSi). In their pioneering work, Phillips and Varley41 did not distinguish between Al9Mn2Si and the Al7.4Fe2Si and assumed that they formed a cubic solid solution phase across the system. Munson42 showed that while Al9Mn2Si is cubic, Al7.4Fe2Si is hexagonal and therefore the two phases could not form a continuous solid solution. Other work29,43,44,45 showed that the cubic α-Al(Fe,Mn)Si emanates from Al9Mn2Si by substantial substitution of Mn by Fe but that it did not extend to the Al-Fe-Si system. They also showed that the phase had a broad range of homogeneity with respect to the Si content. On the other hand, others have identified quaternary phases.46,47,48,49
The confusion has been partially resolved by Krendelsberger.50,51,52 As part of a comprehensive study of the Al-Fe-Si, Al-Mn-Si and Al-Fe-Mn-Si systems, she studied the section between Al9Mn2Si and Al7.4Fe2Si at 700°C. She carried out a full structure analysis of single-phase samples corresponding to Al9Mn2Si and an α-Al(Fe,Mn)Si containing approximately equal amounts of Fe and Mn. She was able to confirm that both samples had close to an identical cubic crystal structure with 138 atoms per unit cell. However, the Al9Mn2Si sample showed a simple primitive cubic lattice while the α-Al(Fe,Mn)Si sample had a bcc structure in which some of the peaks had disappeared. It is still unclear whether this indicates that a first- or second-order phase transformation takes place on the addition of Fe to Al9Mn2Si.
Ab initio calculations have been carried out to help understand this issue. Based on our experience with the calculations on the Al13Fe4 phase and the experimental crystal structure model for α-Al(Fe,Mn)Si, we built a framework for DFT calculations with 24 Fe atoms occupying the two 12j sites and the remaining nine Wyckoff sites occupied by Al. This unit cell contains 24 Fe and 114 Al atoms. As expected, the calculated formation energy is noticeably higher than that of Al13Fe4. Then, in subsequent calculations, we replaced Fe by Mn and Al by Si in a systematic way. Further calculations were carried out to examine the effect of substitution of Al by Fe or Mn.
The results clearly show that mixing of Fe and Mn on the two 12j sites is favoured although one set of sites seemed more favourable for substitution than the other. More calculations are currently underway and it is hoped, that they will shed more light on the chemical compositions and related symmetry, crystal structure, energetics and electronic properties of the α-Al(Fe,Mn)Si compound.
Conclusion
Experimental phase diagram, thermodynamic and crystal structure data for three key Fe-based intermetallic phases that precipitate from cast Al-Si alloys were reviewed. Ab initio DFT calculations were employed to better understand the non-stoichiometry in these phases in terms of the defects. In all of the phases studied there was a marked tendency for Fe and Mn to mix on the same crystallographic sites with a negative enthalpy of mixing relative to the pure constituent compounds (Fig. 3a). For the Al13Fe4 phase it was shown that the relatively narrow range of homogeneity in the Al-Fe system is due to the substitution of Fe atoms on three specific Al sites. This is in contrast to the accepted thermodynamic description, which introduced vacancies. Our calculations show that this is highly unfavourable energetically.
References
Z.P. Que, Y.P. Zhou, Y. Wang, and Z. Fan, Trans. Indian Inst. Met. 68, 1167 (2015).
Z.P. Que, Y. Wang, Y.P. Zhou, and Z. Fan, Mater. Sci. Forum 828–829, 53 (2015).
Z.P. Que, Y. Wang, and Z. Fan, Metall. Mater. Trans. A 49A, 2173 (2018).
W.B. Pearson, A handbook of lattice spacings and structures of metals and alloys, Vol. 2 (Oxford: Pergamon Press, 1967).
J. Grin, U. Burkhardt, M. Ellner, and K. Peters, Z. Kistallogr. 209, 479 (1994).
M. Cooper, Acta Cryst. 23, 1106 (1967).
M. Cooper and K. Robinson, Acta Cryst. 20, 614 (1966).
I. Ansara, A.T. Dinsdale, and M.H. Rand, eds., COST507, Thermochemical database for light metal alloys, Vol. 1 (Luxembourg: Office for official publications of the European Communities, 1998).
Y. Du, Y.A. Chang, S. Liu, B. Huang, F.-Y. Xie, Y. Yang, and S.-L. Chen, Z. Metallkde. 96, 1351 (2005).
J. Lacaze, L. Eleno, and B. Sundman, Metall. Mater. Trans. A 41, 2208 (2010).
E. Balitchev, T. Jantzen, I. Hurtado, and D. Neuschutz, CALPHAD 27, 275 (2003).
G. Kresse and J. Hafner, Phys. Rev. B 49, 14251 (1994).
G. Kresse and J. Furthmüller, Comput. Mater. Sci. 6, 15 (1996).
J.P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996).
P.E. Blöchl, Phys. Rev. B 50, 17953 (1994).
C.M. Fang, M.H.F. Sluiter, M.A. van Huis, and H.W. Zandbergen, Phys. Rev. Lett. 105, 055503 (2010).
C.M. Fang and M.A. van Huis, Heliyon 3, e00408 (2017).
C.M. Fang, M.A. van Huis, and M.H.F. Sluiter, Acta Mater. 103, 273 (2016).
C. Amador, W.R.L. Lambrecht, and B. Segall, Phys. Rev. B 46, 1870(R) (1992).
H.J. Monkhorst and J.D. Pack, Phys. Rev. B 13, 5188 (1976).
B.B. Lindahl and M. Selleby, CALPHAD 43, 86 (2013).
E. Degischer, Alum Arch. 18, 5 (1939).
H.W.L. Phillips, J. Inst. Metals 69, 275 (1943).
W.T. Denholm, J.D. Esdaile, N.G. Siviour, and B.W. Wilson, Metall. Trans. A 15, 1311 (1984).
G.V. Raynor, J. Inst. Metal. 70, 531 (1944).
S. Balanetskyy, D. Pavlyuchkov, and T. Velikanova, B. Grushko. J. Alloys Compounds 619, 211 (2015).
F. Weitzer, P. Rogl, and M. Bohn, COST507, Thermochemical Database For Light Metal Alloys, Vol. 1 (Luxembourg: Office for Official Publications of the European Communities, 1998), p. 53.
M.C. Thornton and P.V. Evans, Alcan International. Unpublished research referred to by A. Jansson, T.G. Chart, COST507, Thermochemical Database For Light Metal Alloys, vol 1, (Office for Official Publications of the European Communities, Luxembourg 1998), p. 257
J.G. Barlock and L.F. Mondolfo, Z. Metallkde. 66, 605 (1975).
K. Han, I. Ohnuma, and R. Kainuma, J. Alloys Compounds 668, 97 (2016).
P.J. Black, Acta Crystallogr. 8, 43 (1955).
P.J. Black, Acta Crystallogr. 8, 175 (1955).
R.C. Hudd and W.H. Taylor, Acta Cryst. 15, 441 (1962).
A. Griger, V. Sterfaniay, and T. Turmezy, Z. Metallkde. 77, 30 (1986).
T.I. Yanson, N.B. Manyako, O.I. Bodak, R. Cêrny, R.E. Gladyshevskii, and K. Yvon, J. Alloys Compounds 219, 135 (1995).
W. Biltz, Z. Metallkde. 29, 73 (1937).
O. Kubaschewski and W.A. Dench, Acta Metall. 3, 339 (1955).
W. Oelsen and W. Middel, Mitt. KW Inst. Eisenforschung 19, 1 (1937).
M. Mihalkovič and M. Widom, Phys. Rev. B 85, 014113 (2012).
C.M. Fang, A.T. Dinsdale, Z.P. Que, and Z. Fan, J. Phys. Mater. 2, 15004 (2019).
H.W.L. Phillips and P.C. Varley, J. Inst. Met. 69, 317 (1943).
D. Munson, J. Inst. Met. 95, 217 (1967).
C.Y. Sun and L.F. Mondolfo, J. Inst. Met. 95, 384 (1967).
G. Davignon, A. Serneels, B. Verlinden, and L. Delaey, Metall. Mater. Trans. A 27A, 3357 (1996).
J.E. Tibbals, J.A. Horst, and C.J. Simensen, J. Mater. Sci. 36, 937 (2001).
A.M. Zakharov, I.T. Gul’din, A.A. Arnol’d, and Y.A. Matsenko, Izvest. Akad. Nauk SSSR, Metally 4, 214 (1989); Russ. Metall. 4, 209 (1989)
A. Flores-Valdes, M.I. Pech-Canul, M. Mendez-Nonell, and M. Sukiennik, Scr. Metall. Mater. 30, 435 (1994).
A. Flores-Valdes, M. Sukiennik, A.H. Castillejos-Escobar, F.A. Acosta-Gonzalez, and M. Mendez-Nonell, Arch. Metall. 41, 294 (1996).
M. Sukiennik, A. Flores-Valdes, A.H. Castillejos-Escobar, F.A. Acosta-Gonzalez, and M.I. Pech-Canul, Arch. Metall. 42, 25 (1997).
N. Krendelsberger, in Ph.D.Thesis (Innovative Materials Group, Universitat Wien, Vienna, 2001)
N. Krendelsberger, F. Weitzer, and J.C. Schuster, Metall. Mater. Trans. A 33A, 3311 (2002).
N. Krendelsberger, F. Weitzer, and J.C. Schuster, Metall. Mater. Trans. A 38A, 1681 (2007).
Acknowledgements
The study was funded by Engineering and Physical Sciences Research Council (Grant No. EP/N007638/1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dinsdale, A., Fang, C., Que, Z. et al. Understanding the Thermodynamics and Crystal Structure of Complex Fe Containing Intermetallic Phases Formed on Solidification of Aluminium Alloys. JOM 71, 1731–1736 (2019). https://doi.org/10.1007/s11837-019-03380-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-019-03380-4