
Abstract
In patients with inflammatory bowel disease (IBD), intestinal activation of the transcription factor NF-κB as well as intercellular adhesion molecule (ICAM)-1 expression, which is involved in recruiting leukocytes to the side of inflammation is increased. Moreover, colonic arachidonic acid (ARA) proportions are increased and oleic acid (OA) proportions are decreased. Fish oils are protective in IBD patients however, a side-by-side comparison between effects of fish oils, ARA and OA has not been made. We therefore, compared effects of eicosapentaenoic acid (EPA) versus ARA and OA on ICAM-1 expression in Caco-2 enterocytes. To validate our model we showed that dexamethasone, sulfasalazine and PPARα (GW7647) or PPARγ (troglitazone) agonists significantly lowered ICAM-1 expression. ICAM-1 expression of non-stimulated and cytokine stimulated Caco-2 cells cultured for 22 days with ARA was significant higher as compared to EPA and OA. Furthermore, ARA increased NF-κB activation in a reporter cell-line as compared to EPA. Antibody array analysis of multiple inflammatory proteins particularly showed an increased monocyte chemotactic protein (MCP)-1 and angiogenin production and a decreased interleukin (IL)-6 and IL-10 production by ARA as compared to EPA. Our results showed that ARA but not EPA and OA activates NF-κB and elevates ICAM-1 expression in Caco-2 enterocytes. It suggests that replacement of ARA by EPA or OA in the colon mucosa might have beneficial effects for IBD patients. Finally, we suggest that the pro-inflammatory effects of ARA versus EPA and OA are not related to PPARγ activation and/or eicosanoid formation.
Similar content being viewed by others


Introduction
Epidemiological studies have shown a low incidence of inflammatory bowel disease (IBD) in Eskimo’s as compared to West-European populations [1] and increasing incidences of IBD in Japan [2]. These findings suggest that an increased dietary intake of n-6 polyunsaturated fatty acids (PUFA) and a lower intake of n-3 PUFAs contribute to the development of IBD. Thus, n-3 fish oil PUFAs may have anti-inflammatory effects as compared to n-6 PUFA [3]. Indeed, elevated proportions of the n-6 PUFA arachidonic acid (ARA) in colon mucosa of both ulcerative colitis (UC) and Crohn's disease (CD) patients as compared to those of control subjects have been shown [4–7]. Although the proportion of the n-3 PUFA eicosapentaenoic acid (EPA) in the mucosa was in most studies not statistically different between IBD patients and healthy controls, one study reported a tendency towards lower EPA proportions in IBD patients [4]. In contrast to EPA, the proportion of the fish oil PUFA docosahexaenoic acid (DHA) was elevated in colonic mucosa of IBD patients as compared to control subjects [4, 5, 7]. Interestingly, the proportion of the n-9 monounsaturated fatty acid (MUFA) oleic acid (OA) was lower in colon mucosa of IBD patients [4, 7]. Because of these observations, and because EPA is a more important precursor of eicosanoids than DHA [8], we decided to compare the effects of EPA versus ARA and OA in an in vitro model of intestinal inflammation. OA is already the most abundant fatty acid present both in our diet [9] as well as in colon mucosa [4]. Therefore, and because of differences in ability to compete with ARA for incorporation in tissue phospholipids [10, 11], it is probably easier to lower mucosal ARA levels by increasing fish oil intake than by increasing OA intake. Indeed, by increasing their intake of fish oil, ARA in the colon mucosa of IBD patients was replaced by EPA and DHA [12], which was associated with significantly reduced corticosteroid requirements [12] and lower relapse rates [13]. It should be noted however, that not all intervention studies using fish oils were that positive, although the overall conclusion is that fish oil supplementation shows at least minor protective effects [14].
The intercellular adhesion molecule (ICAM)-1 plays an important role in the pathology of IBD. In IBD patients intestinal ICAM-1 expression [15] and plasma levels of soluble ICAM-1 (sICAM) are increased [16], and IBD is associated with polymorphisms in the gene encoding for ICAM-1 [17]. Moreover, animal models [18, 19] and a human intervention study [20] have shown that ICAM-1 blocking inhibited intestinal inflammation. The transcription factor NF-κB is a key regulator of the inflammatory response and activation of NF-κB seems to play a critical role in the initiation and perpetuation of intestinal inflammation in IBD [21, 22]. NF-κB activity in the colon is increased during active episodes in IBD patients and certain anti-inflammatory drugs commonly used for IBD appear to inhibit NF-κB [23–27]. In animal models, NF-κB blockade abolished experimental colitis [28, 29]. A side-by-side comparison of the n-3 PUFA EPA, the n-6 PUFA ARA and the n-9 MUFA OA on ICAM-1 expression and NF-κB activation of intestinal cells has, as far as we are aware of, never been performed. Therefore the aim of the present study was to compare the effects of EPA, ARA and OA on ICAM-1 expression and NF-κB activation in the human intestinal epithelial Caco-2 cell line in vitro.
Materials and Methods
Reagents
Bovine serum albumin (BSA; endotoxin and fatty acid-free), sulfasalazine, dexamethasone, GW7647, oleic acid (OA), arachidonic acid (ARA), eicosapentaenoic acid (EPA) and indomethacin were obtained from Sigma Chemical Company (St Louis, MO). Troglitazone was purchased from Biomol (Plymouth Meeting, PA). Recombinant human IL-1β and interferon (IFN)γ were purchased from Roche Molecular Biochemicals (Mannheim, Germany). DMEM, trypsin, penicillin streptomycin (PS), sodium pyruvate (SP) and non-essential amino acids (NEAA) were obtained from Invitrogen Corporation (Paisley, UK). Fetal calf serum (FCS; South-American) was obtained from Greiner Bio-one (Frickenhausen, Germany).
Intestinal Cell Cultures
The human cell line Caco-2 was purchased from the American Tissue Type Collection (ATTC). Caco-2 cells were cultured in DMEM supplemented with 10% heat-inactivated FCS and 1% penicillin streptomycin (PS), 1% sodium pyruvate (SP) and 1% non-essential amino acids (NEAA). Cells were cultured at 37 °C in a 5% CO2 humidified atmosphere, refreshed every second day and separated by trypsin–0.03% EDTA, when they had reached 70–90% confluence. To evaluate the immune-modulating effects of different interventions, Caco-2 cells were plated in six well tissue culture plates at an initial density of 0.5 × 106 cells/mL in a total volume of 1.5 mL. Medium was replaced every other day for 24 days. After 24 days Caco-2 wells were fully differentiated into small intestinal enterocytes [30]. First, effects of immune-suppressive pharmacological compounds (sulfasalazine, dexamethasone and troglitazone) were tested to validate the model. Although these pharmacologic compounds have known immune-suppressive effects, effects on ICAM-1 expression in Caco-2 cells have—as far as we know—not been reported before. Therefore, after Caco-2 cells were fully differentiated, medium was replaced by medium containing the compound of interest in combination with an inflammation-inducing cocktail consisting of the cytokines IFNγ (100 U/mL) and IL-1β (50 U/mL). The compounds of interest were pre-incubated 30 min (sulfasalazine) or 2 h (dexamethasone, troglitazone or GW7647) before stimulation with the cytokine cocktail. After 16 h of cytokine stimulation, cells were used to determine cell surface ICAM-1 protein expression.
Fatty Acid Experiments
The effects of various fatty acids were evaluated using the same Caco-2 cell model. For this, various fatty acids were added at indicated concentrations 2 days after plating the cells and again for the following 22 days each time when the medium was refreshed. We used OA as a control n-9 MUFA because this is the most abundant fatty acid in the diet [9]. We further compared the effects of the n-6 PUFA ARA versus the n-3 PUFA EPA. We have explicitly chosen to use EPA instead of DHA since the proportion DHA in the colon mucosa of IBD patients was already higher as compared to control subjects [4, 5, 7]. In addition, EPA is most likely a more important eicosanoid precursor [8]. The fatty acids were dissolved in ethanol up to a final ethanol concentration in the medium of maximal 0.5% (v/v). To prevent cytotoxicity of the fatty acids the FA were bound to albumin, by pre-incubating the fatty acids dissolved in ethanol for 30 min at 37 °C in full culture medium together with 10% FCS, which also contained 0.1% BSA. Caco-2 cells were cultured with respectively 160 μM OA [C18:1(n-9)] versus 130 μM ARA [C20:4(n-6)] plus 30 μM OA (in total 160 μM fatty acids) or 6 μM EPA [C20:5(n-3)] plus 154 μM OA (in total also 160 μM fatty acids). By this approach the total molarity of fatty acids supplied was similar in all experiments, while supplying different amounts of the fatty acid of interest (i.e., 130 μM ARA or 6 μM EPA). These relatively low concentrations of OA (160 μM), ARA (130 μM) and EPA (6 μM) were chosen because they are four times higher than normally present in culture medium of Caco-2 cells containing 10% FCS. We have deliberately chosen for this low EPA concentration since the EPA concentration is very low in FCS. However, the EPA concentration used is—as for all fatty acids used in these experiments—already four times higher than normally present in culture medium. We cultured the cells for 22 days with these relatively low concentrations of fatty acids to simulate a realistic long-term in vivo change in dietary fatty acid intake. After 22 days culture with fatty acids, medium was replaced by medium enriched with the different fatty acids plus the cytokine cocktail [IFNγ (100 U/mL) and IL-1β (50 U/mL)]. After 16 h stimulation, ICAM-1 expression on living cells was measured and culture medium was collected to determine inflammatory protein expression profiles. To evaluate the effects of the fatty acids on NF-κB activity and the role of cyclooxygenase (COX)-enzymes, the experiments with the fatty acids ARA and EPA were repeated but now in our NF-κB reporter Caco-2 cell line with and without indomethacin (20 μM) added 2 h before and during cytokine stimulation. Prostaglandin PGE2 levels in the supernatant were quantitated using a PGE2 Biotrak enzyme-immunoassay (EIA) system (Amersham Biosciences Ltd, Buckinghamshire, UK) according to the high sensitivity enzyme immunoassay protocol 2.
Flow Cytometry Analysis of ICAM-1
In order to quantify cell surface ICAM-1 protein expression on living Caco-2 cells, we developed a flow cytometry assay. After 16 h of stimulation, the cells were washed three times with PBS and detached with trypsin–0.03% EDTA. Next, medium was added and cell suspensions were centrifuged for 5 min at 1,200 rpm at room temperature, followed by resuspending the pellets in 500 μL PBS-1% BSA. Cells were counted and diluted to 106 cells/mL in PBS-1% BSA. Recombinant-phycoerythrin (R-PE)-conjugated mouse-anti-human CD-54 monoclonal antibody (anti-ICAM-1) or isotype-matched control antibody (Becton Dickinson Biosciences, San Diego, CA; 20 μL/106 cells) was added and incubated for 30 min on ice in the dark. Next, cell suspensions were centrifuged for 5 min at 1,500 rpm and pellets were resuspended in 500 μL PBS-1% BSA. The amount of fluorescence of 10,000 living cells was counted and analyzed with the FACSort and CellQuest analysis software (Becton Dickinson, Franklin Lakes, NJ).
Stable Transfection of NF-κB in Caco-2 Cells
For evaluating the effects of the various interventions on transcriptional activity of NF-κB, a stable NF-κB reporter Caco-2 cell line was created. The 6κB-TK-luciferase (NF-κB reporter) plasmid and neomycin resistance plasmid were both kindly provided by Dr. R.C. Langen (Department of Pulmonology, Maastricht University, The Netherlands). Cells were transfected using Lipofectamine 2000 (Invitrogen Corporation, Paisley, UK) according the manufacturers’ instructions. Positive clones were selected by culturing with geneticin (1 mg/mL). To determine luciferase activity, non-stimulated and 3 h cytokine (100 U/mL IFNγ and 50 U/mL IL-1β) stimulated cells were lysed in luciferase lysis buffer (Promega, Madison, WI) and stored at −80 °C. Luciferase (Promega) activity was measured according to the manufacturers’ instructions and expressed relative to total protein (Bio-rad assay; Bio-rad, Hercules, CA).
Peroxisome Proliferator-Activated Receptor (PPAR)γ and PPARα mRNA Expression of Differentiated Caco-2 Cells
Total RNA was extracted from differentiated Caco-2 cells with Trizol according to the manufacturers’ instructions (Gibco BRL, Gaithersburg, MD). Next, cDNA was made as described [31], and mRNA expression of PPARγ and PPARα was determined using commercially available Taqman gene expression assays (Applied Biosystems, Foster city, CA). Data were normalized against β-actin as housekeeping gene.
Fatty Acid Composition of Caco-2 Cells
Fatty acid incorporation into the Caco-2 cells was evaluated using extraction and analysis procedures as previously described [32]. Briefly, total lipids were extracted from 500 μL cell suspension in PBS-1% BSA according to the method of Bligh and Dyer [33]. Aminopropyl-bonded silica columns (Varian, Harbor City, CA) were used to separate phospholipids from the total lipid extract [34]. The phospholipids were then saponified, and the resultant fatty acids were methylated into their corresponding fatty acid methyl esters (FAMEs) [35]. Fatty acids were separated on an Autosystem (Perkin-Elmer, Norwalk, CT) gas chromatograph that was fitted with a silica-gel column (Cp-sil 88 for FAME, 50 m × 0.25 mm, 0.2-μm film thickness; Chrompack, Middelburg, The Netherlands) with helium gas (130 kPa) as the carrier gas. Both the injection and detection temperatures were set at 300 °C. The starting temperature of the column was 160 °C. Ten min after injection, the temperature was increased up to 190 °C at a rate of 2.5 °C/min. After 20 min at 190 °C, the temperature was increased up to 230 °C at a rate of 4 °C/min. The final temperature of 230 °C was maintained for 10 min. Data were analyzed by using CHROMCARD software (version 1.21; CE Instruments, Milan, Italy). The fatty acid compositions of the Caco-2 cells are expressed in relative amounts (% of total fatty acids identified; w/w).
Inflammatory Protein Expression Profiles Using an Antibody Array
Protein expression patterns of multiple cytokines, chemokines and growth factors, were detected simultaneously in Caco-2 cell culture media with the human cytokine antibody array III (Ray Biotech Inc., Norcross, GA) according to the manufacturers’ instructions. First, duplicates of cell culture media of Caco-2 cells cultured with ARA and EPA after cytokine stimulation were pooled. One millilitre of the pooled samples was added to the array membranes. After incubating and washing, the protein-bound membrane was incubated with a cocktail of biotin-labeled antibodies, followed by the addition of horseradish peroxidase-conjugated streptavidin. Array spot intensity was detected by using a LAS-3000 Lite Image reader (Raytest GmbH, Straubenhart, Germany) based on chemiluminecence imaging. Intensity of the spots was quantified in arbitrary units (a.u.) by densitometry using Aida software version 3.50 (Raytest GmbH), thereby correcting for background staining of the gel. Comparison of protein expression profiles was possible after normalization of each spot on an array using the positive controls, provided by the manufacturer. The sensitivity of the array is not the same for the various proteins. Differences in heights of bars from different proteins do therefore not necessarily represent differences in concentrations. The cytokines used for stimulation (IFNγ and IL-1β) were excluded from analysis.
Detection of ICAM-1 on Caco-2 Frozen Sections
To determine the localization of ICAM-1 in our in vitro Caco-2 cell model, Caco-2 cells were cultured and differentiated into small intestinal enterocyte on collagen-coated polyfluoroethylene transwell membrane inserts with a 0.4 μm membrane pore size (Corning Costar, Cambridge, MA). Differentiated Caco-2 cells were stimulated with IFNγ (100 U/mL) and IL-1β (50 U/mL) for 16 h, embedded in Tissue-Tek (Sakura Finetek, Zoeterwoude, The Netherlands) and rapidly frozen in 2-propanol (Fluka, Zwijndrecht, The Netherlands), dry-ice-cooled and stored at −80 °C. Serial cryosections (10 μm) were obtained using a Leica CM3050 cryostat (Leica Microsystems GmbH, Wetzlar, Germany) and thaw mounted on uncoated glass slides. Before processing or storage at −80 °C, the samples were air dried overnight. To detect ICAM-1 the sections were incubated 30 min in the dark at room temperature with recombinant-phycoerythrin (R-PE)-conjugated mouse–anti-human CD-54 monoclonal antibody or isotype-matched control antibody (Becton Dickinson Biosciences, San Diego, CA) 1:50 diluted in PBS-1% BSA. To detect cytokeratin (CK)-19 the sections were simultaneously incubated with a monoclonal antibody directed to CK-19, kindly obtained from Dr. E.B. Lane (University Dundee, Dundee, UK) 1:10 diluted. Then the sections were washed three times for 5 min in PBS. After that the secondary antibody goat anti-mouse IgG1 (ALEXA555) (Molecular Probes Europe, Leiden, The Netherlands) (1:500) against anti-CD54 (to evade fast quenching of the PE-label) and goat anti-mouse IgG2b (FITC) (Southern Biotech, Sanbio BV, Uden, The Netherlands) (1:50) against CK-19 diluted in PBS-1% BSA was added to the sections and incubated for 30 min. Again the sections were washed three times for 5 min with PBS. Finally, sections were mounted in Mowiol-TRIS pH 8.5 (Calbiochem, Omnilabo International, Etten-Leur, The Netherlands) containing 0.5 g/mL 4–6-diamino-2-phenylindole (DAPI; Molecular Probes Europe) to stain the nuclei. All sections were examined using a Nikon E800 fluorescence microscope (Uvikon, Bunnik, The Netherlands) coupled to a Basler A101C progressive scan colour CCD camera. By just a simple shift in filters, images were grabbed in fluorescence using the ALEXA excitation filter (540–580 nm), the FITC excitation filter (465–495 nm) and DAPI UV excitation filter (340–380 nm) in the red, green and blue channel, respectively. The images acquired were merged to examine the cellular localisation and level of expression of ICAM-1.
Statistical Analysis
Data were expressed as means and standard deviations (SD) for the non-stimulated condition, the cytokine stimulated condition and cytokine stimulated–non-stimulated (called net stimulated) condition. To determine statistical significance unpaired t-tests (comparison between two interventions) or ANOVA (comparison between three interventions) with a Bonferonni post-hoc test when differences between interventions were significantly different, were performed. First non-stimulated values between interventions were tested to examine differences in basal values. Then, net stimulated values between interventions were tested to examine differences in cytokine-induced changes. Furthermore cytokines stimulated values were tested to examine differences in “end” values. All statistical analyses were performed using SPSS 11 for Mac Os X (SPSS, Chicago, IL). P-values of less than 0.05 were considered statistically significant.
Results
Model Validation
Stimulation with IL-1β and IFNγ increased cell-surface ICAM-1 protein expression on Caco-2 cells, which resulted in a net stimulated ICAM-1 expression of about 50 a.u. (Fig. 1, panels a and b). After pre-treatment of the cells for 2 h with dexamethasone, a corticosteroid with known therapeutic effects in IBD patients, the cytokine stimulated ICAM-1 expression was significantly decreased as compared to control (P = 0.044). Also the net stimulated ICAM-1 expression (25 a.u.) was significantly reduced as compared to control (P = 0.019). Next, we examined the effect of another frequently used therapeutic drug for IBD patients, sulfasalazine. Effects of sulfasalazine were comparable to those observed for dexamethasone. However, also ICAM-1 expression without cytokine stimulation (P = 0.004) (Fig. 1, panel a) was lowered. Altogether, these results show that in our cell model ICAM-1 expression is related to the clinical outcomes of drugs proven to treat IBD and could therefore be used as the main outcome parameter in the following experiments. To further validate characteristics of our in vitro model, we also localized the site of ICAM-1 expression on the Caco-2 cells by means of immunohistochemistry on frozen sections. As shown in Fig. 1 (panels c–f), ICAM-1 was expressed on the apical (lumen) side of the polarized Caco-2 cells. Besides a low constitutive expression (panel d), there was a clear increase after stimulation with the cytokine cocktail (panel e). This localisation is in line with the apical ICAM-1 expression, as found in intestinal biopsies from IBD patients [36].

Model validation. a ICAM-1 expression (in arbitrary units a.u.) on living control Caco-2 cells and after 2 h pre-treatment with 1 μM dexamethasone or 30 min 5 mM sulfasalazine with and without cytokine stimulation for 16 h (100 U/mL IFNγ and 50 U/mL IL-1β) and b net stimulated (stimulated–non-stimulated) ICAM-1 expression. Results represent means ± SD; n = 2. Representative representation of two independent experiments. a P < 0.05 versus control non-stimulated, b P < 0.05 versus control cytokine stimulated, c P < 0.05 versus control net stimulated. (c–f) Immunohistochemistry of ICAM-1 (red) on frozen sections of Caco-2 cells on a transwell showed increased apical expression after cytokine stimulation (100 U/mL IFNγ and 50 U/mL IL-1β). c Hematoxylin staining d Non-stimulated Caco-2 cells e Apical cytokine stimulated Caco-2 cells f Isotype control staining of ICAM-1. Red staining ICAM-1, green staining cytoskeleton (cytokeratin-19), blue staining nucleus. Magnification 40X
Effects of PPAR Agonists on ICAM-1 Expression and NF-κB Activation
Since PPARs are known modulators of inflammation and fatty acids are natural ligands for PPARs, we first examined PPAR expression in our differentiated Caco-2 cells. We found that PPARα and PPARγ mRNA are expressed in equal amounts (data not shown). Next we examined the effects of the PPARγ agonist troglitazone [a thiazolidinedione (TZD)] and the PPARα agonist GW7647 on ICAM-1 expression. Although both troglitazone and GW7647 significantly lowered the cytokine stimulated ICAM-1 expression as compared to control (P < 0.001 and P = 0.009, respectively), only troglitazone significantly (P = 0.008) reduced the net stimulated ICAM-1 expression (Fig. 2, panels a and b). Troglitazone and GW7647 were used at a concentration where they are selective for their receptor subtypes [37, 38]. It is known that PPARs suppress inflammation by inhibiting the transcription factor NF-κB, which is a key regulator of inflammation [39]. Therefore, we also examined effects of troglitazone on NF-κB transactivation in our Caco-2 NF-κB reporter cell line. These results showed that the cytokine cocktail induced after already 3 h a more than 200% increase in NF-κB transactivation. This increase of NF-κB transactivation was significantly (P = 0.008) lower after 2 h pre-treatment with the PPARγ agonist troglitazone (Fig. 2, panels c and d). Moreover, the inhibitory effect of pre-treatment with the PPARγ agonist troglitazone on cytokine induced NF-κB transactivation was significantly (P = 0.001) larger as compared to pre-treatment with the PPARα agonist GW7647 (data not shown).

a, b ICAM-1 expression (in arbitrary units a.u.) on living control Caco-2 cells and after pre-treatment with PPAR agonists (2 h; PPARγ troglitazone 100 μM and PPARα GW7647 1 μM) and c, d NF-κB transactivation measured by luciferase activity (in RLU/mg) in a NF-κB reporter Caco-2 cell-line of control cells and after troglitazone pre-treatment with and without cytokine stimulation for 3 h (100 U/mL IFNγ and 50 U/mL IL-1β) and net stimulated (stimulated–non-stimulated). Results represent means ± SD; n = 2. Representative representation of two independent experiments. bP < 0.05 versus control cytokine stimulated, cP < 0.05 versus control net stimulated
Fatty Acid Incorporation in Phospholipids
Table 1 shows that the fatty acids kept for 22 days in the culture medium were incorporated into the phospholipid fraction (% of total fatty acids) of the Caco-2 cells. Fatty acid incorporation into phospholipids showed the same pattern as the fatty acid composition of total lipids (data not shown), although changes in the total lipids were more pronounced. Moreover, the fatty acid profiles of phospholipids (Table 1) and total lipids (data not shown) were similar in non-stimulated and stimulated Caco-2 cells. Cells cultured with ARA showed in particular an increase in the proportion of long chain n-6 PUFAs (mainly ARA) and a decrease in n-9 MUFAs (mainly OA) as compared with cells cultured with only OA. The cells cultured with EPA showed an increase in the proportion of n-3 fatty acids (mainly EPA) whereas the proportions of n-9 MUFAs and n-6 PUFAs did not change much as compared with cells cultured with only OA.
Effects of Fatty Acids on ICAM-1 Expression and NF-κB Activation
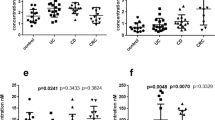
ICAM-1 expression was analyzed on living cells. FACS analysis did not show significant changes in cell populations cultured with the various fatty acids, suggesting no significant cell death. As shown in Fig. 3 (panel a), ARA significantly elevated ICAM-1 expression as compared to OA or EPA (non-stimulated P < 0.001 ARA versus OA and ARA versus EPA, and stimulated P = 0.010 ARA versus OA, and P = 0.013 ARA versus EPA). Interestingly, stimulated and non-stimulated ICAM-1 expression on cells cultured with EPA did not significantly differ from those cultured with OA (Fig. 3, panel a). Despite these clear differences in non-stimulated and stimulated ICAM-1 expression between ARA and EPA or OA, the net stimulated ICAM-1 expression did not differ between the groups (ANOVA P = 0.134). Since dietary interventions aiming at lowering mucosal ARA content are most likely more successful by dietary EPA enrichments than by OA enrichment–because OA is the most abundant fatty acid present in our diet [9], making higher intakes not realistic–in further experiments we have focused on effects of ARA versus EPA. First, we examined the effects on NF-κB activation in the stable NF-κB reporter Caco-2 cell line (Fig. 4). NF-κB transactivation in stimulated Caco-2 cells cultured with ARA was significantly increased as compared to that in stimulated cells cultured with EPA (Fig. 4, panels a and b) (stimulated P = 0.017 and net stimulated P = 0.001). To evaluate whether the effects were related to the production of different families of eicosanoids we measured PGE2 production and examined the effects of the different fatty acids in the presence of the cyclooxygenase (COX)-inhibitor indomethacin. As expected, PGE2 production by Caco-2 cells cultured with ARA was significantly higher than by cells cultured with EPA (data not shown). Stimulation of the cells with the cytokine cocktail did however, not influence PGE2 production. Since cytokine stimulation enhanced NF-κB activation, this suggests that PGE2 is not directly involved in NF-κB activation. Indomethacin decreased PGE2 production of Caco-2 cells by approximately 70% (data not shown), but had no effect on NF-κB transactivation (Fig. 4, panels c and d) as compared to untreated Caco-2 cells. Also ICAM-1 expression was not affected (data not shown). However, as shown in Fig. 4, the effects of ARA versus EPA in the presence of indomethacin treatment (panel e) showed an identical pattern as observed without indomethacin (panel a). Also in the presence of indomethacin the net cytokine stimulated NF-κB transactivation was significantly higher after ARA as compared to EPA (P = 0.047) (Fig. 4, panel f). This indicates that the increased basal PGE2 production by ARA cannot explain the different effects of ARA and EPA on NF-κB activation.

a–b ICAM-1 expression (in arbitrary units a.u.) on living Caco-2 cells cultured for 22 days with 160 μM oleic acid (OA) or 130 μM arachidonic acid (ARA) plus 30 μM OA or 6 μM eicosapentaenoic acid (EPA) plus 154 μM OA with and without cytokine stimulation for 16 h (100 U/mL IFNγ and 50 U/mL IL-1β) and net stimulated (stimulated–non-stimulated). Results represent means ± SD; n = 2. Representative representation of two independent experiments. ANOVA between groups, non-stimulated P < 0.001, stimulated P = 0.006 and net stimulated P = 0.134. aBonferroni P < 0.05 versus OA and EPA non-stimulated, bBonferroni P < 0.05 versus OA and EPA cytokine stimulated

NF-κB transactivation with and without cytokine stimulation for 3 h (100 U/mL IFNγ and 50 U/mL IL-1β) (a, c, e) and net stimulated (stimulated–non-stimulated) (b, d, f) in a NF-κB reporter Caco-2 cell line measured by luciferase activity (in RLU/mg). a–b Reporter Caco-2 cells cultured for 22 days with 130 μM arachidonic acid (ARA) plus 30 μM oleic acid (OA) or 6 μM eicosapentaenoic acid (EPA) plus 154 μM OA. c–d Control reporter Caco-2 cells and after 2 h pre-treatment with the COX-inhibitor indomethacin (20 μM). e–f Reporter Caco-2 cells cultured for 22 days with 130 μM arachidonic acid (ARA) plus 30 μM OA or 6 μM eicosapentaenoic acid (EPA) plus 154 μM OA and 2 h pre-treatment with the COX-inhibitor indomethacin (20 μM). Results represent means ± SD; n = 2. b P < 0.05 versus EPA cytokine stimulated, c P < 0.05 versus EPA or EPA + indomethacin net stimulated
Effects of Fatty Acids on Inflammatory Proteins Expression Profiles
Finally, to explore the effects of ARA and EPA on a broader scale, we evaluated protein expression profiles consisting of various inflammatory mediators by using antibody arrays. Figure 5 shows that ARA treatment particularly increased expression of monocyte chemotactic protein (MCP)-1 and angiogenin, while EPA treatment increased IL-10, IL-6, macrophage inflammatory protein (MIP)-1δ, and growth regulated protein (GRO) expression.

Protein expression profile in the culture medium of Caco-2 cells cultured for 22 days with 130 μM arachidonic acid (ARA) plus 30 μM oleic acid (OA) or 6 μM eicosapentaenoic acid (EPA) plus 154 μM OA after 16 h cytokine stimulation (100 U/mL IFNγ and 50 U/mL IL-1β) measured with an antibody array. Results are represented as mean ± SD; n = 2. CSF colony stimulating factor, ENA epithelial-derived neutrophil activating protein, MCP monocyte chemotactic protein, MDC, macrophage derived chemokine, MIG monokine induced by gamma interferon, MIP macrophage inflammatory protein, SDF stromal cell-derived factor, GRO growth regulated protein, MCSF Macrophage colony stimulating factor, Tpo thrombopoietin, EGF epidermal growth factor, OSM oncostatin M, VEGF Vascular endothelial growth factor, SCF stem cell factor, IL interleukin, TNF tumor necrosis factor, Ang angiogenin
Discussion
ICAM-1 seems important in the pathology of IBD [15]. Whether fish oils, which have been shown to protect against relapses in IBD patients on remission [13], have effects on ICAM-1 expression, is however, unknown. In addition, a direct side-by-side comparison of fish oils with ARA, which is postulated to have pro-inflammatory effects [40] and is elevated in the colon mucosa of IBD patients [4, 7], has never been made. The transcription factor NF-κB is important in regulating intestinal inflammation and is elevated in IBD patients [21, 22, 26]. We have now shown that the n-3 PUFA EPA, as compared to the n-6 PUFA ARA, clearly reduced cytokine stimulated NF-κB activation and ICAM-1 expression in enterocytes in vitro. Moreover, effects of OA on ICAM-1 expression were comparable to those of EPA. Because EPA and OA resulted in comparable changes in the proportions of ARA in the phospholipids of the enterocytes, the reported effects may be ascribed to the increased ARA proportion in the ARA cultured cells. Thus, decreasing cellular ARA levels seems to be a crucial step. Since OA is already the most abundant fatty acid present in our diet [9] and in the colon mucosa [4], decreasing mucosal ARA levels may be easier by increasing fish oil intake than by increasing OA intake. Moreover, EPA does compete with ARA for incorporation into tissue phospholipids [10, 11].
Our data showed that replacing ARA for EPA or OA decreased ICAM-1 expression and NF-κB activation in Caco-2 enterocytes. In line with our observations in enterocytes, n-6 PUFAs also increased NF-κB activation as compared to n-3 PUFAs in monocytes [41] and macrophages [42]. Also, earlier in vitro studies have demonstrated that fish oils reduced cytokine stimulated ICAM-1 expression in endothelial cells [43] and monocytes [44] as compared to conditions without addition of fatty acids. Moreover, in vivo ICAM-1 expression (surface and mRNA) on peritoneal macrophages was reduced in mice fed fish oils compared to that in mice fed coconut oil [45]. In humans, dietary fish oil supplementation lowered expression of ICAM-1 on ex vivo stimulated monocytes as compared to no supplementation [46]. However, our study is the first that examined effects of EPA versus ARA on ICAM-1 expression and NF-κB activation in enterocytes. We however, realize that, although enterocytes play an important role in intestinal inflammation, immune modulating effects of fatty acids in CD patients will be influenced not only by enterocytes, but also the interaction with other intestinal immune and non-immune cells is important. Therefore, in future experiments effects on other cell types, e.g. isolated from mucosal biopsies of CD patients should be evaluated. Moreover, to validate if the effects of EPA versus ARA are also applicable in the pathogenesis or even the treatment of CD patients, these effects should be confirmed in appropriate animal models of IBD.
We used an approach of supplying different amounts of the fatty acids of interest (i.e., 130 μM ARA or 6 μM EPA), while the total molarity of fatty acids supplied was similar in all experiments by adding OA. These different concentrations of ARA and EPA were deliberately chosen because they were both four times the amount, in which the cells grow normally (i.e., the fatty acid composition of culture medium with 10% FCS). Since the EPA concentration is very low in FCS, we have deliberately chosen for a low EPA concentration. Higher concentrations of EPA would be interesting to examine, however, are difficult to be achieved with dietary interventions in vivo. Using iso-molaric total concentrations of fatty acids is essential, because an increase in total fat can be immune suppressive [47]. Therefore, we used OA as a reference fatty acid to make total fatty acid concentrations between experimental fatty acid conditions iso-molaric, i.e. OA was exchanged for ARA or EPA. In addition, we evaluated the condition of OA only. The results of this latter condition showed that decreasing ARA levels in the mucosa seems to be more important than increasing EPA levels.
Regarding the pathways underlying the anti-inflammatory effect of fish oils, several suggestions have been made. As compared to n-6 PUFAs, n-3 PUFAs may have different effects on (I) signal transduction pathways, and (II) the types and levels of eicosanoids synthesized [40]. Regarding the first mechanism, our finding that EPA lowered NF-κB activation and ICAM-1 expression as compared to ARA indeed showed that EPA and ARA differently affected the NF-κB signal transduction pathway. In this respect, two peroxisome proliferators-activated receptors (PPARs), PPARα and PPARγ, seem relevant, since fatty acids are natural ligands for these PPARs. Both PPARs seem to have anti-inflammatory effects by inhibiting NF-κB activation [48, 49]. In our in vitro model, the PPARγ agonist troglitazone inhibited NF-κB activation, and cytokine and net stimulated ICAM-1 expression, while the PPARα agonist GW7647 only inhibited cytokine-stimulated ICAM-1 expression. This suggests that both PPARγ and PPARα have anti-inflammatory effects on enterocytes, in which effects of PPARγ seem more pronounced. This is in agreement with the finding that PPARγ agonists but not a PPARα agonist inhibited IL-8 expression of Caco-2 cells and HT-29 cells [50]. Differences between fatty acids in activation of PPARs might explain the activation of NF-κB by ARA and not by EPA in our Caco-2 cells. However, our findings are not supported by the PPARγ binding affinities of EPA and ARA, which are about the same [51], while we used much lower EPA concentrations than ARA concentrations. Secondly, not only EPA but also OA lowered intestinal ICAM-1 expression while OA is a poor PPARγ ligand [51]. Therefore, although PPARγ activation certainly protects against inflammation, our results do not suggest that the protective effects of EPA and OA as compared to ARA on intestinal inflammation are PPARγ-mediated. Therefore, there should be another explanation why effects of OA and EPA were comparable but different from those of ARA.
The mechanism underlying a second possible explanation relates to the incorporation of fatty acids into cell membrane phospholipids and eicosanoids synthesis. Eicosanoids can modulate the intensity and duration of the inflammatory response. [8, 40] Replacement of ARA by EPA in the culture medium of the cells resulted in a decrease in the proportion of ARA in cell membranes and an increase in the EPA proportion and as expected the PGE2 production of Caco-2 cells cultured with EPA was also lower than PGE2 production of cells cultured with ARA. However, PGE2 production was not different between non-stimulated and stimulated cells, while stimulation resulted in an abundant elevation of NF-κB activation. To our opinion this rules out the role of PGE2 in the NF-κB activation of ARA and EPA. Moreover, although in our experiments indomethacin–a blocker of COX–indeed inhibited PGE2 production, it did not lower NF-κB activation. This is in line with findings of De Catherina et al. [43] showing that in endothelial cells the effects of DHA on VCAM-1 expression could not be inhibited by indomethacin, although prostacyclin production was completely suppressed. Therefore, we fully agree that in an in vivo situation pro-inflammatory effects of ARA are probably mediated by eicosanoids synthesized from ARA, however, these effects are not derived from a direct eicosanoid mediated activation of NF-κB as shown in our model.
Finally, we evaluated inflammatory protein signatures of Caco-2 cell culture medium after 22 days treatment with EPA versus ARA. Since these signatures were analyzed in pooled material we cannot draw conclusions based on statistical analysis. However, this array was intended to generate hypotheses about differences in immune modulating effect of ARA versus EPA based on the protein expression profiles. These profiles indicate, that both fatty acids induced specific changes in various inflammation mediators. Most of these proteins, but not all, are regulated by NF-κB. For example, IL-6 protein expression was higher after EPA as compared to ARA, whereas NF-κB activation was higher after ARA. It should however, be considered that although highly important, NF-κB is not the only transcription factor involved in the regulation of IL-6 and other inflammatory proteins. Thus, expression of individual proteins cannot be predicted by the level of NF-κB activation solely. In addition, we found clear differences between EPA and ARA, i.e., that some proteins were expressed higher after EPA (IL-6, IL-10, MIP-1δ and GRO), while others were higher expressed after ARA (MCP-1, and angiogenin). The precise role of these individual proteins in the process of intestinal inflammation is not clear. However, the increased MCP-1 expression after ARA treatment is in line with the finding that MCP-1 expression is upregulated in the mucosa of IBD patients and correlates with disease activity [52]. The increased expression of the Th2 cytokine IL-10 after EPA treatment fits with the anti-inflammatory role for IL-10 in IBD pathology. In this respect, IL-10 knockout mice develop chronic intestinal inflammation and delivery of recombinant IL-10 to the intestinal mucosa by the bacterium L. lactis attenuated mucosal inflammation in two mouse models [53]. In humans, a pilot study using these bacteria showed also promising effects [54].
In conclusion, we have shown that ARA but not EPA and OA activates NF-κB and elevates ICAM-1 expression in Caco-2 enterocytes and we hypothesize that the effects are not related to PPARγ activation or eicosanoid formation.
Abbreviations
- ARA:
-
Arachidonic acid
- CD:
-
Crohn’s disease
- COX:
-
Cyclooxygenase
- EPA:
-
Eicosapentaenoic acid
- GRO:
-
Growth regulated protein
- IBD :
-
Inflammatory bowel disease
- ICAM:
-
Intercellular adhesion molecule
- IFN :
-
Interferon
- IL:
-
Interleukin
- MCP:
-
Monocyte chemotactic protein
- MIP :
-
Macrophage inflammatory protein
- MUFA:
-
Monounsaturated fatty acid
- NF-κB:
-
Nuclear factor-kappa B
- OA:
-
Oleic acid
- PGE2 :
-
Prostaglandin E2
- PPAR:
-
Peroxisome proliferator-activated receptor
- PUFA:
-
Polyunsaturated fatty acid
- UC:
-
Ulcerative colitis
References
Kromann N, Green A (1980) Epidemiological studies in the Upernavik district, Greenland. Incidence of some chronic diseases 1950–1974. Acta Med Scand 208:401–406
Shoda R, Matsueda K, Yamato S, Umeda N (1996) Epidemiologic analysis of Crohn disease in Japan: increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan. Am J Clin Nutr 63:741–745
Belluzzi A (2002) n-3 Fatty acids for the treatment of inflammatory bowel diseases. Proc Nutr Soc 61:391–395
Nishida T, Miwa H, Shigematsu A, Yamamoto M, Iida M, Fujishima M (1987) Increased arachidonic acid composition of phospholipids in colonic mucosa from patients with active ulcerative colitis. Gut 28:1002–1007
Buhner S, Nagel E, Korber J, Vogelsang H, Linn T, Pichlmayr R (1994) Ileal and colonic fatty acid profiles in patients with active Crohn’s disease. Gut 35:1424–1428
Pacheco S, Hillier K, Smith C (1987) Increased arachidonic acid levels in phospholipids of human colonic mucosa in inflammatory bowel disease. Clin Sci (Lond) 73:361–364
Fernandez-Banares F, Esteve-Comas M, Mane J, Navarro E, Bertran X, Cabre E, Bartoli R, Boix J, Pastor C, Gassull MA (1997) Changes in mucosal fatty acid profile in inflammatory bowel disease and in experimental colitis: a common response to bowel inflammation. Clin Nutr 16:177–183
Calder PC (2006) n-3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr 83:1505S–1519S
Hulshof KFAM, Jansen-van der Vliet M, Westenbrink S, Doest tD (2004) De inneming van vetzuren en vetzuurclusters (voedselconsumptiepeiling 1997–1998). TNO Voeding, Zeist
Li B, Birdwell C, Whelan J (1994) Antithetic relationship of dietary arachidonic acid and eicosapentaenoic acid on eicosanoid production in vivo. J Lipid Res 35:1869–1877
Zhou L, Nilsson A (2001) Sources of eicosanoid precursor fatty acid pools in tissues. J Lipid Res 42:1521–1542
Hawthorne AB, Daneshmend TK, Hawkey CJ, Belluzzi A, Everitt SJ, Holmes GK, Malkinson C, Shaheen MZ, Willars JE (1992) Treatment of ulcerative colitis with fish oil supplementation: a prospective 12 month randomised controlled trial. Gut 33:922–928
Belluzzi A, Brignola C, Campieri M, Pera A, Boschi S, Miglioli M (1996) Effect of an enteric-coated fish-oil preparation on relapses in Crohn’s disease. N Engl J Med 334:1557–1560
MacLean CH, Mojica WA, Newberry SJ, Pencharz J, Garland RH, Tu W, Hilton LG, Gralnek IM, Rhodes S, Khanna P et al. (2005) Systematic review of the effects of n-3 fatty acids in inflammatory bowel disease. Am J Clin Nutr 82:611–619
Bernstein CN, Sargent M, Gallatin WM (1998) Beta2 integrin/ICAM expression in Crohn’s disease. Clin Immunol Immunopathol 86:147–160
Goke M, Hoffmann JC, Evers J, Kruger H, Manns MP (1997) Elevated serum concentrations of soluble selectin and immunoglobulin type adhesion molecules in patients with inflammatory bowel disease. J Gastroenterol 32:480–486
Matsuzawa J, Sugimura K, Matsuda Y, Takazoe M, Ishizuka K, Mochizuki T, Seki SS, Yoneyama O, Bannnai H, Suzuki K, et al. (2003) Association between K469E allele of intercellular adhesion molecule 1 gene and inflammatory bowel disease in a Japanese population. Gut 52:75–78
Hamamoto N, Maemura K, Hirata I, Murano M, Sasaki S, Katsu K (1999) Inhibition of dextran sulphate sodium (DSS)-induced colitis in mice by intracolonically administered antibodies against adhesion molecules (endothelial leucocyte adhesion molecule-1 (ELAM-1) or intercellular adhesion molecule-1 (ICAM-1)). Clin Exp Immunol 117:462–468
Rijcken E, Krieglstein CF, Anthoni C, Laukoetter MG, Mennigen R, Spiegel HU, Senninger N, Bennett CF, Schuermann G (2002) ICAM-1 and VCAM-1 antisense oligonucleotides attenuate in vivo leucocyte adherence and inflammation in rat inflammatory bowel disease. Gut 51:529–535
van Deventer SJ, Tami JA, Wedel MK (2004) A randomised, controlled, double blind, escalating dose study of alicaforsen enema in active ulcerative colitis. Gut 53:1646–1651
Schreiber S (2005) The complicated path to true causes of disease: role of nuclear factor kappaB in inflammatory bowel disease. Gut 54:444–445
Neurath MF, Becker C, Barbulescu K (1998) Role of NF-kappaB in immune and inflammatory responses in the gut. Gut 43:856–860
Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M (1995) Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 270:286–290
Gan HT, Chen YQ, Ouyang Q (2005) Sulfasalazine inhibits activation of nuclear factor-kappaB in patients with ulcerative colitis. J Gastroenterol Hepatol 20:1016–1024
Bantel H, Berg C, Vieth M, Stolte M, Kruis W, Schulze-Osthoff K (2000) Mesalazine inhibits activation of transcription factor NF-kappaB in inflamed mucosa of patients with ulcerative colitis. Am J Gastroenterol 95:3452–3457
Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, Knuechel R, Baeuerle PA, Scholmerich J, Gross V (1998) Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology 115:357–369
Schreiber S, Nikolaus S, Hampe J (1998) Activation of nuclear factor kappa B in inflammatory bowel disease. Gut 42:477–484
De Vry CG, Prasad S, Komuves L, Lorenzana C, Parham C, Le T, Adda S, Hoffman J, Kahoud N, Garlapati R, et al. (2007) Non-viral delivery of nuclear factor-kappaB decoy ameliorates murine inflammatory bowel disease and restores tissue homeostasis. Gut 56:524–533
Neurath MF, Pettersson S, Meyer zum Buschenfelde KH, Strober W (1996) Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med 2:998–1004
Hidalgo IJ, Raub TJ, Borchardt RT (1989) Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 96:736–749
Thijssen MA, Malpuech-Brugere C, Gregoire S, Chardigny JM, Sebedio JL, Mensink RP (2005) Effects of specific CLA isomers on plasma fatty acid profile and expression of desaturases in humans. Lipids 40:137–145
Wensing AG, Mensink RP, Hornstra G (1999) Effects of dietary n-3 polyunsaturated fatty acids from plant and marine origin on platelet aggregation in healthy elderly subjects. Br J Nutr 82:183–191
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Kaluzny MA, Duncan LA, Merritt MV, Epps DE (1985) Rapid separation of lipid classes in high yield and purity using bonded phase columns. J Lipid Res 26:135–140
Morrison WR, Smith LM (1964) Preparation Of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J Lipid Res 5:600–608
Parkos CA, Colgan SP, Diamond MS, Nusrat A, Liang TW, Springer TA, Madara JL (1996) Expression and polarization of intercellular adhesion molecule-1 on human intestinal epithelia: consequences for CD11b/CD18-mediated interactions with neutrophils. Mol Med 2:489–505
Willson TM, Brown PJ, Sternbach DD, Henke BR (2000) The PPARs: from orphan receptors to drug discovery. J Med Chem 43:527–550
Brown PJ, Stuart LW, Hurley KP, Lewis MC, Winegar DA, Wilson JG, Wilkison WO, Ittoop OR, Willson TM (2001) Identification of a subtype selective human PPARalpha agonist through parallel-array synthesis. Bioorg Med Chem Lett 11:1225–1227
Delerive P, Fruchart JC, Staels B (2001) Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol 169:453–459
Teitelbaum J, Walker W (2001) Review: the role of omega 3 fatty acids in intestinal inflammation. J Nutr Biochem 12:21–32
Camandola S, Leonarduzzi G, Musso T, Varesio L, Carini R, Scavazza A, Chiarpotto E, Baeuerle PA, Poli G (1996) Nuclear factor kB is activated by arachidonic acid but not by eicosapentaenoic acid. Biochem Biophys Res Commun 229:643–647
Novak TE, Babcock TA, Jho DH, Helton WS, Espat NJ (2003) NF-kappa B inhibition by omega -3 fatty acids modulates LPS-stimulated macrophage TNF-alpha transcription. Am J Physiol Lung Cell Mol Physiol 284:L84–89
De Caterina R, Cybulsky MI, Clinton SK, Gimbrone MA Jr. Libby P (1994) The omega-3 fatty acid docosahexaenoate reduces cytokine-induced expression of proatherogenic and proinflammatory proteins in human endothelial cells. Arterioscler Thromb 14:1829–1836
Hughes DA, Southon S, Pinder AC (1996) (n-3) Polyunsaturated fatty acids modulate the expression of functionally associated molecules on human monocytes in vitro. J Nutr 126:603–610
Miles EA, Wallace FA, Calder PC (2000) Dietary fish oil reduces intercellular adhesion molecule 1 and scavenger receptor expression on murine macrophages. Atherosclerosis 152:43–50
Hughes DA, Pinder AC, Piper Z, Johnson IT, Lund EK (1996) Fish oil supplementation inhibits the expression of major histocompatibility complex class II molecules and adhesion molecules on human monocytes. Am J Clin Nutr 63:267–272
Luyer MD, Buurman WA, Hadfoune M, Jacobs JA, Konstantinov SR, Dejong CH, Greve JW (2004) Pretreatment with high-fat enteral nutrition reduces endotoxin and tumor necrosis factor-alpha and preserves gut barrier function early after hemorrhagic shock. Shock 21:65–71
Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391:79–82
Poynter ME, Daynes RA (1998) Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem 273:32833–32841
Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD (1999) A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest 104:383–389
Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, et al. (1999) Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell 3:397–403
Herfarth H, Goke M, Hellerbrand C, Muhlbauer M, Vogl D, Scholmerich J, Rogler G (2003) Polymorphism of monocyte chemoattractant protein 1 in Crohn’s disease. Int J Colorectal Dis 18:401–405
Stokkers PC, Hommes DW (2004) New cytokine therapeutics for inflammatory bowel disease. Cytokine 28:167–173
Braat H, Rottiers P, Huyghebaert N, Zelinkova Z, Remaut E, Remon JP, Deventer vS, Neirynck S, Peppelenbosch M, Steidler L et al. (2005) Interleukin-10 producing Lactococcus lactis for the treatment of Crohn’s disease. Gastroenterology 128:A104
Acknowledgments
The authors thank Hasibe Aydeniz (Department of Human Biology, Maastricht University, The Netherlands) for analyzing the fatty acid composition of the Caco-2 cells, Maurice Konings (Department of Human Biology) for analyzing PPARγ and PPARα mRNA expression in Caco-2 cells and Martine Hulsbosch (Department of Human Biology) for PGE2 measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ramakers, J.D., Mensink, R.P., Schaart, G. et al. Arachidonic Acid but not Eicosapentaenoic Acid (EPA) and Oleic Acid Activates NF-κB and Elevates ICAM-1 Expression in Caco-2 Cells . Lipids 42, 687–698 (2007). https://doi.org/10.1007/s11745-007-3071-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11745-007-3071-3