
Abstract
Gut inflammation has been correlated with cancerogenesis by disrupting gastrointestinal homeostasis. Numerous chronic inflammatory disorders of the tubular gastrointestinal tract (e.g., gastroesophageal reflux disease, Helicobacter pylori-induced and autoimmune chronic gastritis, celiac disease, and inflammatory bowel diseases) have been variably associated with an increased neoplastic risk. Gastrointestinal inflammation-induced neoplasms include epithelial tumors (esophageal squamous cell carcinoma and adenocarcinoma, gastric adenocarcinoma and neuroendocrine tumors, small bowel adenocarcinoma and neuroendocrine tumors, and colorectal cancer) and lymphomas (such as gastric marginal zone lymphomas and enteropathy-associated T cell lymphoma). In the last decades, numerous studies have investigated the pathogenetic mechanisms and the microenvironmental/microbiome changes that trigger genetic and/or epigenetic alterations eventually leading to tumorigenesis, often through a histologically recognizable inflammation-dysplasia-carcinoma cancerogenic sequence. In the present review, an overview of the current knowledge on the links between inflammatory diseases and neoplasms of the tubular GI tract, applying a site-by-site approach, is provided.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The link between inflammation and cancer was first postulated by the father of modern pathology, Rudolph Virchow, who, in 1863, described inflammatory cell infiltration within cancer growths. This led to the hypothesis that there was indeed a correlation between inflammation and carcinogenesis and that cancer could originate in sites of chronic inflammation and this has been proven by numerous research groups, and in various sites, over time [1].
Gut inflammation is known to disrupt gastrointestinal (GI) homeostasis and several chronic inflammatory disorders of the tubular GI tract (esophagitis, gastritis, enteritis and colitis) have been associated with an increased risk of developing solid and/or hematolymphoid neoplasms. Although there is evidence that some gut inflammatory disorders are caused by either infectious agents [e.g., Helicobacter pylori (HP)-related gastritis] or chemical injury (e.g., reflux esophagitis), many of them have an autoimmune or immune-mediated etiopathogenesis. In the last decades, numerous studies have investigated the biological mechanisms and microenvironmental/microbiome changes that trigger genetic and/or epigenetic alterations eventually leading to tumorigenesis in such conditions. In this review, we provide an overview of current knowledge on the links between inflammatory diseases and neoplasms of the tubular GI tract, applying a site-by-site approach.
Role of inflammation in the development of esophageal cancers
Two main types of epithelial cancers can affect the esophagus, namely squamous cell carcinoma (ESCC) and adenocarcinoma (EAC), which altogether cover more than 95% of esophageal malignancies. ESCC and EAC differ both for esophageal location (with involvement of middle and upper third of the esophagus by ESCC versus lower third and gastro-esophageal junction by EAC) and geographic distribution. EAC has in fact continued to increase in incidence in western countries in the last fifty years, thus overwhelming ESCC, which, however, still represents the predominant subtype globally.
Different causative agents have been correlated with cancer development in the esophagus, spanning from cigarette smoke, alcohol and/or hot beverage consumption, diet, infectious agents and gastroesophageal reflux disease (GERD). A major effort has been made by researchers to understand the molecular events which, following the exposition of esophageal epithelium to causative agents, lead to cancer development. In this context, a major role seems to be played by inflammation, both per se [2] and in combination with alterations of the microbiota [3].
Tobacco and alcohol consumption, which are the major risk factors for ESCC, promote cancer development via acetaldehyde which has a carcinogenic effect by forming DNA adducts and altering genes [4]. Acetaldehyde is a constituent of tobacco smoke and the first metabolite of ethanol; the combined use of tobacco and smoke act synergistically in the cascade of events driving progression from the normal squamous epithelium to pre-invasive and invasive neoplasia [5]. Moreover, tobacco and alcohol directly act on the inflammatory-immune system inducing production of several cytokines [i.e., interleukin (IL) 1–6–8] and forming free radicals (reactive oxygen and nitrogen species) which lead to oxidative stress and activation of the nuclear factor kappa B (NF-kB) family [6]. This creates a pro-inflammatory state responsible of inflammation and contributing to carcinogenesis [4].
The relationship between GERD and inflammation has been extensively studied in the last century. A major role is played both by gastric and bile acid in the gastro-esophageal refluxate. For several years the proposed model to explain the link between GERD and inflammation, was through direct damage of the superficial epithelial layer caused by acid refluxate, with necrosis and acute inflammation (neutrophils and eosinophils) permeating the squamous epithelium. This damage, in addition to the recruitment of inflammatory cells, is responsible for epithelial proliferation (manifesting as basal cell hyperplasia and papillary elongation) aimed at repairing, by substitution, the damaged surface epithelial cells. Furthermore, acid also damages the intercellular junctions, causing an increase in epithelial permeability (manifesting with dilatation of intercellular spaces) which enable the hydrogen ions to spread between epithelial cells [7]. In contrast with this hypothesis, in which damage starts from surface and proceeds to the deep portions of the mucosa, a rat model for reflux esophagitis suggests that inflammation starts in the submucosa, with recruitment of T-lymphocytes, mediated by release of pro-inflammatory cytokines (IL-8) by the squamous epithelium, and successively proceeds toward the surface [8]. This cytokine-mediated model with initial T-lymphocyte recruitment has also been confirmed in humans [9] and T-lymphocytes have been demonstrated to be significantly increased in biopsies of patients with both non-erosive and erosive reflux disease compared to healthy controls, while B-cells, Langerhans cells, Natural Killer cells and macrophages play a marginal role [10].
Similarly to exposure of the esophageal squamous epithelium to alcohol and smoke, the generation of reactive oxygen species, which stabilize and activate hypoxia-inducible factor (HIF)-2α, are linked to the pro-inflammatory IL cascade in GERD [11]. This cascade is responsible for the oxidative stress status which contributes to the development of Barrett’s esophagus (BE) and EAC [12].
Under the pressure of prolonged acid refluxate injury and inflammation, the esophageal squamous stratified epithelium is replaced by simple columnar epithelium thus starting the cascade of morphological and molecular events that from BE drive to EAC and Gastro-Esophageal Junction adenocarcinoma (GEJA) via low-grade dysplasia (LGD) and high-grade dysplasia (HGD) through an inflammation-dysplasia-carcinoma cancerogenic sequence. In each step of this cascade, inflammation plays an important role and an increase of T-cells, B cells, macrophages and dendritic cells, has been reported in BE and EAC/GEJA. Moons et al. have demonstrated that BE shows a predominant humoral immune response (Th2) while GERD shows a more pronounced cellular immune response (Th1) [13]. In this study, immunohistochemical analysis for the principal Th1 (macrophages and CD8+ T lymphocytes) and Th2 (plasma cells and mast cells) effector cells was performed, showing an increase in Th2 effector cells in BE with equal number of Th1 effector cells compared to GERD as well as a predominant expression of IgG and IgE by plasma cells. This shift toward a more humoral immune response in BE is associated with progressive depression of the cell-mediated immunity and this, on the one hand is correlated with angiogenesis while, on the other, causes a reduction of immune surveillance; these two sides of the coin are both involved in tumoral progression [13]. These observations have been confirmed and detailed in the study by Kavanagh et al. who, in addition to the Th1 vs Th2 profile shift from GERD to BE, demonstrated a significantly lower number of activated T-cells in EAC, with an increase in both pro and anti-inflammatory cytokines, probably leading to a mixed inflammatory profile in the final steps of the neoplastic cascade [14]. The recent studies by Lagisetty et al. and Sundaram et al. shed greater light on the dynamic changes in the immune landscape from normal esophagus, to BE, LGD- and HGD, and EAC using a sequential multiplex immunohistochemistry platform [15, 16]. Both studies have demonstrated a progressive increase of CD8+ T cells in the different steps of the neoplastic cascade, leading to decreased cytotoxic effector cells and an immunosuppressive microenvironment in EAC.
Another fundamental protagonist in the esophageal microenvironment, which can influence inflammation and is emerging as a potential driver of oncogenesis [17], is represented by the microbial flora. The microbial population changes in pathological conditions (GERD and BE) with respect to the normal healthy subject, with an increase in gram-negative bacteria. These gram-negative bacteria produce lipopolysaccharides (LPS) which cause high levels of pro-inflammatory cytokines via NF-κB activation with a simultaneous increase in IL-1β, IL-6, IL-8 and tumor necrosis factor (TNF) along the spectrum of GERD, BE and EAC. Altogether, the alteration in the microbiome of BE may lead to EAC by triggering chronic inflammation and propagating the inflammatory cascade [3].
Inflammation and gastric neoplasia
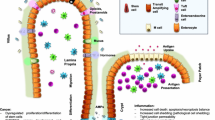
Longstanding mucosal inflammation is the main cause of the cancerogenic cascade leading to sporadic gastric neoplasia. The etiological agents responsible for gastritis, described in the Kyoto Classification, are environmental and host-related [18]. The environmental etiology is far more common, and includes both transmissible and non-transmissible agents. The epidemiological impact of the host-related category is significantly lower, and it includes different etiologies, most of which are immune-mediated disorders. Environmental and host-related etiologies may also overlap, as in the case of autoimmune gastritis triggered by HP infection. Both etiologic models result in the atrophic remodeling of the native gastric mucosa and chronic immune-system stimulation, promoting epithelial neoplastic lesions, following the inflammation-dysplasia-carcinoma sequence (Fig. 1), as well as gastritis-related lymphomas [19].

A histologic example of a gastric adenoma arising in the context of chronic atrophic gastritis of the oxyntic mucosa. Note in A the polypoid appearance of the adenoma and in B, at higher magnification, the inflammation with atrophy of the oxyntic mucosa and the high-grade cytoarchitectural features of the dysplastic adenomatous proliferation (A, B hematoxylin and eosin)
HP is both the most common and the best understood gastric carcinogen [20]. The prevalence of HP infection differs with age, geographic regions (up to 80% of middle-aged adults in the developing countries), socio-economic status, education level, living environment and occupation. Although the majority of HP infected patients remain asymptomatic, essentially all develop chronic inflammation, approximately 10% develop peptic ulcer disease, 1–3% progress to GC, and 0.1% develop mucosa-associated lymphoid tissue (MALT) lymphoma [21]. A positive association between consumption of salt-cured foods (fish, meat, vegetables), tobacco smoke, alcohol consumption, long-term use of proton pump inhibitors and increased risk of gastric cancer (GC) is described, which is stronger in subjects with HP infection [19].
HP produces a variety of virulence factors that may dysregulate host intracellular signaling pathways leading to neoplastic transformation. In particular, the HP-related toxin CagA (cytotoxin-associated gene A): (1) enhances proliferation by various molecular pathways; (2) it disrupts tight junctions leading to loss of polarity; (3) it interferes with oncogenes (such as p53 and Runt-related transcription factor3—RUNX3); (4) it activates the oncogenic RAS pathway by attenuating miRNA let-7 expression [21]. On the other hand, vacuolating cytotoxin A (VacA) is involved in epithelial tight junction disruption, apoptosis and suppressed T lymphocyte activation and proliferation by activation of Bax (Bcl 2 associated X protein) [21].
HP up-regulates pro-inflammatory cytokines such as IL-1, IL-6, IL-8, TNF-α, NF-κB, and it induces the generation of intracellular reactive oxygen species (ROS) and reactive nitrogen species (RNS) by host gastric epithelial cells and inflammatory cells. Moreover, HP may lead to hypermethylation of O6-methylguanine DNA methyltransferase (MGMT), with secondary silencing of many tumor suppressor genes such as Trefoil factor family 2, E-cadherin, p16, mismatch repair gene (hMLH1), fork head box, and RUNX3 [21].
Chronic inflammation leads to progressive accumulation of genetic alterations from the normal mucosa, intestinal metaplasia (IM), dysplasia to invasive carcinoma. In a long-term prospective Italian study, 69% of HGD progressed to GC, suggesting the presence of a molecular pathway developing in dysplastic lesions leading to GC onset [19]. IM is characterized by a higher mutation rate in DNA compared to normal gastric mucosa, it shows somatic copy number alterations (sCNAs) in 12.5% of cases and it shows higher levels of DNA methylation than normal mucosa [22]. With regards to dysplastic lesions, microsatellite instability (MSI)/defective mismatch repair (dMMR) profile, CDH1 inactivation (leading to loss of E-cadherin immunostaining), HER2 gene amplification and protein overexpression, aberrant p53 expression, increase from LGD to HGD, indicating that they are early drivers of carcinogenesis [23]. Interestingly, a relatively higher prevalence in PD-L1 positivity was observed among gastric dysplastic lesions compared to invasive carcinoma [24].
A less common environmental factor involved in gastric carcinogenesis is Epstein–Barr virus (EBV) infection. EBV infection is more common in men, in the Asian population, it is the highest in proximal tumors and those arising in gastric stump, and it is associated with lymphoepithelioma-like histotype [25]. EBV promotes carcinogenesis through DNA methylation of a series of tumor suppressor genes, resulting in uncontrolled cell growth and in promoting a pro-inflammatory environment. In particular, EBV infection is associated with PIK3CA mutation, inactivating mutations of ARID1A, and BCOR (encoding an anti-apoptotic protein) mutations. Recurrent JAK2 and ERBB2 amplification are observed whereas only rare TP53 mutations of cases are described. In all EBV-related cancers, CDKN2A (p16INK4A) promoter hypermethylation is described.
Autoimmune gastritis (AIG) is an autoimmune-mediated disease affecting the parietal cells in the body-fundus, which are the target of serum auto-anti-parietal cell (PCA) and anti-intrinsic factor antibodies [26]. AIG prevalence is significantly higher in middle-age females and may coexist with other autoimmune diseases, such as Hashimoto’s thyroiditis, insulin-dependent diabetes and vitiligo. An association with HP infection has been reported, due to molecular mimicry between HP and structural proteins of the parietal cells such as the gastric H+/K+ATPase [27]. Epidemiological studies providing data on AIG in the general population are lacking, due to the high rate of asymptomatic or pauci-symptomatic disease in early stages and frequent incomplete (lack of biopsies from the gastric body) mucosal sampling in patients undergoing gastroscopy. Prevalence of AIG has been estimated to be ~ 0.5–4.5% globally, increasing with age from 2.5% in the third decade to 12% in the eighth decade [28]. Interestingly, a few case series described AIG in pediatric patients affected by autoimmune disease [28]. With progressive loss of parietal cells and atrophy of oxyntic mucosa compartment, hydrochloric acid and intrinsic factor levels decrease, leading to stimulation of gastrin-producing cells and enterochromaffin-like (ECL) cells. ECL cell hyperplasia, through linear and micronodular phases, can advance to type 1 gastric neuroendocrine tumor (NET) [28].
Gastric mucosal atrophy may alter the gastric microbiota promoting a microenvironment (“cancerization field”) prone to the development of GC. However, a recent study found that the risk of GC, in naïve HP-negative AIG patients, is not increased [29]. The role of the atrophy-modulated gastric microbiota and its likely synergy with HP-induced inflammation in promoting the GC-prone microenvironment deserves further investigation.
Gastric MALT lymphoma is a low-grade lymphoma arising in the gastric mucosa driven by chronic HP infection. Gastric MALT lymphoma may regress with HP eradication while, in untreated patients, it can turn into extranodal-diffuse large B-cell lymphoma (eDLBCL), a high-grade lymphoma. Gastric MALT lymphoma can also be associated with hepatitis B virus, human immunodeficiency virus (HIV), EBV and human T-cell lymphotropic virus type 1 (HTLV-1) [30]. Inflammatory changes including production of a proliferation inducing ligand (APRIL), a member of the tumor-necrosis factor (TNF)-family, by macrophages, have been associated with lymphoma development [31]. In addition, HP can translocate the CagA protein directly into B-cells resulting in extracellular signal-regulated kinase activation and Bcl-2 expression up-regulation, leading to apoptosis inhibition. Normal B cells are transformed to malignant clone via three chromosomal translocations: t(11;18) (q21;q21), t(1;14)(p22;q32), and t(14;18)(q32;q21), which lead to the activation of NF-κB, which plays a role in immunity, inflammation, and cancerogenesis [32].
Small bowel inflammation and tumorigenesis
In the small bowel, the inflammation-dysplasia-carcinoma sequence is less well-characterized than in the other gut organs, due to the rarity of primary small intestinal adenocarcinomas and to the well-known technical-endoscopic issues in endoscopically exploring this intestinal tract. Nevertheless, a few immune-inflammatory disorders, including inflammatory bowel diseases (IBDs), celiac disease, as well as long-standing ileostomy, have been consistently associated with increased small bowel cancer risk [33].
In a recent population-based cohort study of patients with IBD diagnosed in Norway and Sweden from 1987 to 2016, the standardized incidence ratio of small bowel adenocarcinoma (SBA) was increased by more than eightfold in Crohn’s disease [34]. Importantly, in this investigation the first year of follow-up was excluded to reduce reverse causality, which may explain, at least in part, the excess risk estimates found during early follow-up. The highest SBA risks were found among patients with Crohn’s disease diagnosed before 40 years (often with a long disease duration before SBA diagnosis), those displaying stricturing behavior or those with inflammatory disease limited to the small bowel. Indeed, most SBAs associated with Crohn’s disease have been found in areas involved by active inflammation, which likely drives cancer development, and they have been found to be associated with metaplastic and/or dysplastic (conventional or non-conventional) mucosal changes, that often share with the adjacent cancer, the expression of gastro-pancreato-biliary markers [35,36,37]. On the other hand, small bowel resection and use of salicylates for more than two years seem to protect against SBA in patients with Crohn’s disease [38]. Some molecular alterations, such as IDH1 gene mutations, are enriched in SBAs associated with Crohn’s disease compared to sporadic cases, while APC mutations seem to be rarer in the former [39, 40].
In addition, Yu et al. reported that the risk of SBA is also increased (about twofold) in ulcerative colitis patients, where it was strongly associated with extensive disease. However, the relationship between ulcerative colitis and SBA is still uncertain, as a recent meta-analysis of 26 observational studies failed to find a significantly increased risk of SBA in ulcerative colitis [41]. Some subgroups of IBD patients may also have an increased risk of developing lymphoma; however, no association with disease severity was found [42]. Due to the low absolute risk of small bowel neoplasms, active surveillance of the small intestine is currently not recommended in IBD patients.
Interestingly, Yu et al. also found that the standardized incidence ratio of small intestinal NETs was increased (about twofold) both in Crohn’s disease and in ulcerative colitis patients, in the latter likely confined to patients with extensive colitis [34]. However, it should be noted that small intestinal NETs are usually not detected at sites of active inflammation, and they are often incidental findings in IBD surgical resection specimens. Therefore, the causal relationship between small intestinal mucosal inflammation and NET development remains to be elucidated. Interestingly, in IBD and NET patients, common patterns of microbiome composition (e.g., depletion of Faecalibacterium prausnitzii, which plays a role in modulating the immune system and to protect the gut barrier integrity by the production of butyrate) have been observed [43]; notwithstanding this observation, the role of intestinal microbiota in NET development requires further investigation. A recent investigation by Massironi et al. found that 13% of duodenal NETs were associated with duodenal gastric surface metaplasia, defined as the replacement of the normal duodenal epithelial cells with cells that resemble gastric foveolar epithelium [44]. Once again, this finding indirectly suggests that chronically inflamed microenvironment may play a role in the development of a subset of duodenal NETs, as duodenal gastric metaplasia is often related to chronic inflammation of the duodenal mucosa, due to abnormally high production of gastric acid triggered by HP infection or to drug-induced injury, celiac disease or Crohn’s disease. In addition, duodenal gastric metaplasia harboring KRAS or GNAS mutations may represent a precursor lesion of duodenal adenoma and adenocarcinoma [45].
Another immune-mediated intestinal disorder, celiac disease, has also been found to be associated with an increased overall cancer risk (essentially confined to celiac individuals diagnosed after age 40) compared to the general population [46,47,48]. An increased risk of SBA, hemato-lymphoid (intestinal and non-intestinal) neoplasms, in particular enteropathy-associated T cell lymphoma (EATL), as well as other GI malignancies (e.g., pancreatic carcinoma) have also been described in celiac patients. Although previous studies reported a pooled odds ratio of 14.4 for SBA in celiac disease [49], a recent Swedish nationwide cohort of celiac individuals, accurately designed to reduce the risk of detection bias in the peri-diagnostic period, estimated the hazard risk of SBA and small bowel adenomas to be between 3.05 and 5.73 in celiac individuals in comparison with matched reference individuals, and the SBA risk was higher in the first 10 years of follow-up [50]. In the latter study, “mucosal healing” (defined as Marsh 0–2) after gluten-free diet was associated with a lower, albeit not statistically significant, risk of SBA in celiac individuals, suggesting the role of small bowel inflammation in the pathogenesis of SBA. It should be noted, however, that the absolute risk of SBA in celiac patients is low (0.06% in Emilsson’s study), thus not implying a need for surveillance. The hypothesis that SBAs arise from the classic “adenoma-to-carcinoma sequence” in celiac patients is still highly debated, due to the rarity of adenomatous dysplastic growths adjacent to the invasive adenocarcinoma [36]. Importantly, patients with celiac disease associated-SBA showed higher rates of MSI/dMMR and more favorable prognosis compared to patients with sporadic SBAs or SBAs associated with Crohn’s disease [51, 52]. On the contrary, EATL, a high-grade lymphoma typically associated with celiac disease, is a very aggressive disease, generally with an ominous outcome. While no risk factors for the development of SBA, have been identified, apart from the higher age at diagnosis of celiac disease, likely indicative of diagnostic delay, EATL may be preceded by type 2 refractory celiac disease. The latter is a rare form of complicated celiac disease characterized by villous atrophy and a monoclonal expansion of immunophenotypically abnormal intraepithelial T lymphocytes, which accumulate in the intraepithelial compartment driven by increased production of the potent anti-apoptotic and proliferative properties of IL-15 [53]. Moreover, rare cases of monomorphic epitheliotropic intestinal T cell lymphomas have been described in celiac patients [54]. Several studies provided evidence that strict gluten-free diet may decrease cancer risk and mortality, whereas non-adherence and/or non-responsiveness to a gluten-free diet may result in persistent mucosal chronic inflammation, which, eventually, might promote the development of lymphoma or carcinoma [47, 55].
Finally, intestinal T cell lymphomas have been described in patients with non-celiac enteropathies, such as autoimmune enteropathy [56], while patients with common variable immunodeficiency have been reported to be at increased risk for gastric adenocarcinoma and intestinal lymphomas, the latter usually arising in the setting of nodular lymphoid hyperplasia [57, 58].
Large bowel inflammation and tumorigenesis
Patients with IBD are at high risk for developing dysplasia and colorectal cancer (CRC) through an inflammation-dysplasia-carcinoma sequence [59]. IBD, encompassing Crohn’s disease and ulcerative colitis, is a chronic inflammatory disorder of the GI tract, caused by a dysregulated inflammatory and immune response in genetically susceptible individuals. An altered gut microbiome (dysbiosis), as well as other environmental factors, play an important role in triggering and perpetuating inflammation. Patients have a relapsing and remitting disease course, often with bloody diarrhea and abdominal pain in moments of active disease, interspersed with periods of remission. Individuals with IBD are at an increased risk of developing neoplasia, in particular CRC, but also SBA, intestinal lymphoma and anal cancer, as well as tumors in extraintestinal sites. Population-based studies have shown an estimated risk of CRC 2- to threefold that of the general population in ulcerative colitis patients [60], and Crohn’s disease patients appear to have a similar increased risk [60]. IBD-related CRCs often show peculiar histotypes (such as mucinous and signet ring carcinomas [61]), they are more often proximal in location and are high-grade malignancies with poorer overall survival compared to sporadic CRC.
Sporadic CRC follows the adenoma-carcinoma sequence, while IBD related CRC has been shown to follow the ‘inflammation–dysplasia–carcinoma’ sequence. In this context, inflammation plays a crucial role as the relapsing–remitting inflammatory nature of disease causes epithelial destruction and regeneration [62]. Chronic inflammation is involved in tumorigenesis through various mechanisms, including oxidative stress with DNA damage, abnormal immune response and involvement of the gut microbiota. In particular, epithelial proliferation induced by mucosal regeneration increases mutational burden and the selection of mutated clones. Mutagenesis is in part induced and driven by inflammation, by production of pro-inflammatory cytokines (IL-1, IL-6, TNF-α) and chemokines and the generation of reactive oxygen species and lipid peroxidation leading to increased inflammation-induced oxidative DNA damage (with accumulation of mutations). The inflammation-induced activation of nuclear transcription factors (NF-kB and STAT3) which perpetuate inflammation and promote carcinogenesis via the loss of the p53 tumor suppressor gene leads to unchecked cell growth and inhibition of apoptosis with increase of cytokine-mediated DNA damage. Indeed, TP53 mutations have been observed in non-dysplastic epithelial cells in inflamed mucosa underlining how inflammation plays an initial and pivotal role in the development of IBD-related CRC.
Inflammation, not only drives the initiation of cancer but it is also involved in disease progression and this can be observed from a morphologic point of view also. The cancerogenic sequence therefore starts with intestinal mucosa which has been genetically modified by chronic active inflammation and on this basis, the sequence from LGD to HGD to IBD-related CRC is initiated [63].
The standardized classification system of IBD-related dysplasia was introduced by Riddell et al. in 1983 dividing dysplasia into categories, including (indefinite for dysplasia) LGD, HGD and invasive carcinoma [64]. Conventional (or intestinal type) dysplasia is the most well-recognized form of dysplasia, and the identification and grading of dysplasia in IBD (according to Riddell) is the cornerstone of management of these patients. Recently, SCENIC (Surveillance for Colorectal Endoscopic Neoplasia Detection and Management in Inflammatory Bowel Disease Patients) guidelines [65] have stressed another important feature of dysplasia, specifically whether it is endoscopically visible or invisible. These endoscopic features guide patient management, as polypoid/visible dysplasia (even HGD) can be treated endoscopically while colectomy is the treatment of choice for flat/invisible dysplasia (especially in HGD or multifocal LGD). Endoscopic surveillance of IBD patients is therefore fundamental for recognizing early lesions which can be treated conservatively and which reduce neoplastic risk in these patients.
While conventional (intestinal type) dysplasia has garnered, up till now, most interest, new non-conventional patterns have been collected and described in recent years. Seven morphologic categories have been described including hypermucinous dysplasia (the most common), goblet cell-deficient, crypt cell dysplasia, increased Paneth cell differentiation and serrated lesions. Recognition of these non-conventional dysplastic (NCD) lesions is important as they are common in IBD patients with dysplasia (up to 33% of dysplastic lesions are non-conventional) and IBD patients harboring CRC (45% of IBD-associated CRC had associated NCD lesions in one series) [66, 67]. NCD lesions may be seen either adjacent to CRC or within the same segment, they may be found associated with conventional dysplasia and, despite their low-grade appearance, they are associated with high grade (poorly differentiated) CRC. Furthermore, new studies have shown that NCD lesions (especially hypermucinous, goblet cell-deficient, and crypt cell dysplasia), often graded as LGD, have a higher rate of aneuploidy, KRAS mutations and appear to have a higher risk of progression to HGD/CRC compared to conventional dysplasia. These NCD lesions are more frequently flat/invisible (40% in NCD lesions compared to 18% for conventional dysplastic lesions) making endoscopic surveillance and treatment ever more important. An extremely recent contribution has shown that increased histologic inflammation is an independent risk factor for NCD, showing an increased cumulative inflammation burden compared to non-dysplastic UC patients [68].
Risk factors for malignancy have been identified in IBD patients, and the most important are correlated with inflammation. In particular long-standing IBD has been shown to be correlated with increased cancer risk. Older series reported CRC risk as high as 15% in patients with 30 years of active disease [69], while more recent estimates, based on large population-based studies and meta-analyses, identify lower (though absolutely not negligible) percentage risks [70]. Furthermore, active disease and severity of inflammation increase the risk of dysplasia and CRC as well as disease extent [71]. Other culprits of increased CRC risk in IBD patients include primary sclerosing cholangitis (threefold increase), family history of CRC (twofold increase) and younger onset-IBD (likely attributable to longer disease duration) [72].
Recent contributions have also investigated the effect of gut microbiome on IBD-related (and sporadic) CRC and are gaining an increasing level of interest [73]. Various theories concerning bacterial involvement in IBD-related CRC have been proposed, where dysbiosis is probably an active participant in the inflammation-dysplasia-cancer sequence. The passage of gut bacteria (such as E. coli and enterotoxigenic Bacteroides fragilis) from the lumen into the subepithelial tissue, through mucosa barrier disruption, sustains inflammation, with an increase in pro-inflammatory and pro-carcinogenic mediators increasing the risk of developing CRC.
Conclusions
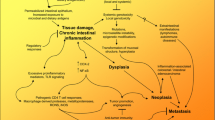
The etiologically heterogeneous inflammatory disorders affecting the diverse organs of the tubular GI tract predispose to diverse epithelial and non-epithelial neoplasms, as summarized in Fig. 2. An inflammation-dysplasia-carcinoma sequence has been well characterized in some conditions, such as in the BE-associated EAC, HP-gastritis-related GC or in IBD-related CRCs, whereas the tumorigenic processes are poorly known in other sites, such as the small bowel. The identification and modulation of cancer-inducing molecular mechanisms and gut dysbiosis may open the door for treatment and prevention of GI neoplasms (e.g., IBD-related CRC) in the future.

A schematic summary of the principal inflammatory conditions which predispose to cancer development in various sites along the gastrointestinal tract
Data Availability
Not applicable.
References
Wen Y, Zhu Y, Zhang C, Yang X, Gao Y, Li M, Yang H, Liu T, Tang H (2022) Chronic inflammation, cancer development and immunotherapy. Front Pharmacol 13:1040163. https://doi.org/10.3389/fphar.2022.1040163
Tambunting L, Kelleher D, Duggan SP (2022) The Immune Underpinnings of Barrett’s-associated adenocarcinogenesis: a retrial of nefarious immunologic co-conspirators. Cell Mol Gastroenterol Hepatol 13:1297–1315. https://doi.org/10.1016/j.jcmgh.2022.01.023
D’Souza SM, Houston K, Keenan L, Yoo BS, Parekh PJ, Johnson DA (2021) Role of microbial dysbiosis in the pathogenesis of esophageal mucosal disease: a paradigm shift from acid to bacteria? World J Gastroenterol 27:2054–2072. https://doi.org/10.3748/wjg.v27.i18.2054
Chela HK, Gangu K, Ertugrul H, Juboori AA, Daglilar E, Tahan V (2022) The 8th Wonder of the Cancer World: esophageal cancer and inflammation. Diseases 10:44. https://doi.org/10.3390/diseases10030044
Prabhu A, Obi KO, Rubenstein JH (2014) The synergistic effects of alcohol and tobacco consumption on the risk of esophageal squamous cell carcinoma: a meta-analysis. Am J Gastroenterol 109:822–827. https://doi.org/10.1038/ajg.2014.71
Gonçalves RB, Coletta RD, Silvério KG, Benevides L, Casati MZ, da Silva JS, Nociti FH Jr (2011) Impact of smoking on inflammation: overview of molecular mechanisms. Inflamm Res 60:409–424. https://doi.org/10.1007/s00011-011-0308-7
Fiocca R, Mastracci L, Milione M, Parente P, Savarino V; Gruppo Italiano Patologi Apparato Digerente (GIPAD), Società Italiana di Anatomia Patologica e Citopatologia Diagnostica/International Academy of Pathology, Italian division (SIAPEC/IAP) (2011) Microscopic esophagitis and Barrett's esophagus: the histology report. Dig Liver Dis 43(Suppl 4):S319–S330. https://doi.org/10.1016/S1590-8658(11)60588-4
Souza RF, Huo X, Mittal V, Schuler CM, Carmack SW, Zhang HY, Zhang X, Yu C, Hormi-Carver K, Genta RM, Spechler SJ (2009) Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology 137:1776–1784. https://doi.org/10.1053/j.gastro.2009.07.055
Dunbar KB, Agoston AT, Odze RD, Huo X, Pham TH, Cipher DJ, Castell DO, Genta RM, Souza RF, Spechler SJ (2016) Association of acute gastroesophageal reflux disease with esophageal histologic changes. JAMA 315:2104–2112. https://doi.org/10.1001/jama.2016.5657
Mastracci L, Bruzzone M, Pacella E, Tinelli C, Zentilin P, Savarino E, De Silvestri A, Fiocca R, Grillo F (2016) The contribution of intraepithelial inflammatory cells to the histological diagnosis of microscopic esophagitis. Esophagus 13:80–87. https://doi.org/10.1007/s10388-015-0501-9
Ustaoglu A, Nguyen A, Spechler S, Sifrim D, Souza R, Woodland P (2020) Mucosal pathogenesis in gastro-esophageal reflux disease. Neurogastroenterol Motil 32:e14022. https://doi.org/10.1111/nmo.14022
Abdel-Latif MM, Duggan S, Reynolds JV, Kelleher D (2009) Inflammation and esophageal carcinogenesis. Curr Opin Pharmacol 9:396–404. https://doi.org/10.1016/j.coph.2009.06.010
Moons LM, Kusters JG, Bultman E, Kuipers EJ, van Dekken H, Tra WM, Kleinjan A, Kwekkeboom J, van Vliet AH, Siersema PD (2005) Barrett’s oesophagus is characterized by a predominantly humoral inflammatory response. J Pathol 207:269–276. https://doi.org/10.1002/path.1847
Kavanagh ME, Conroy MJ, Clarke NE, Gilmartin NT, O’Sullivan KE, Feighery R, MacCarthy F, O’Toole D, Ravi N, Reynolds JV, O’Sullivan J, Lysaght JV (2016) Impact of the inflammatory microenvironment on T-cell phenotype in the progression from reflux oesophagitis to Barrett oesophagus and oesophageal adenocarcinoma. Cancer Lett 370:117–124. https://doi.org/10.1016/j.canlet.2015.10.019
Lagisetty KH, McEwen DP, Nancarrow DJ, Schiebel JG, Ferrer-Torres D, Ray D, Frankel TL, Lin J, Chang AC, Kresty LA, Beer DG (2021) Immune determinants of Barrett's progression to esophageal adenocarcinoma. JCI Insight 6:e143888. https://doi.org/10.1172/jci.insight.143888
Sundaram S, Kim EN, Jones GM, Sivagnanam S, Tripathi M, Miremadi A, Di Pietro M, Coussens LM, Fitzgerald RC, Chang YH, Zhuang L (2022) Deciphering the immune complexity in esophageal adenocarcinoma and pre-cancerous lesions with sequential multiplex immunohistochemistry and sparse subspace clustering approach. Front Immunol 13:874255. https://doi.org/10.3389/fimmu.2022.874255
Sharma T, Gupta A, Chauhan R, Bhat AA, Nisar S, Hashem S, Akhtar S, Ahmad A, Haris M, Singh M, Uddin S (2022) Cross-talk between the microbiome and chronic inflammation in esophageal cancer: potential driver of oncogenesis. Cancer Metastasis Rev 41:281–299. https://doi.org/10.1007/s10555-022-10026-6
Sugano K, Tack J, Kuipers EJ, Graham DY, El-Omar EM, Miura S, Haruma K, Asaka M, Uemura N, Malfertheiner P, Faculty members of Kyoto Global Consensus Conference (2015) Kyoto global consensus report on Helicobacter pylori gastritis. Gut 64:1353–1367. https://doi.org/10.1136/gutjnl-2015-309252
Rugge M, Genta RM, Di Mario F, El-Omar EM, El-Serag HB, Fassan M, Hunt RH, Kuipers EJ, Malfertheiner P, Sugano K, Graham DY (2017) Gastric cancer as preventable disease. Clin Gastroenterol Hepatol 15:1833–1843. https://doi.org/10.1016/j.cgh.2017.05.023
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans Personal habits and indoor combustions: volume 100 E—a review of human carcinogens (2012) IARC monographs on the evaluation of carcinogenic risks to humans/World Health Organization. Int Agency Res Cancer 100:1–538
Wang F, Meng W, Wang B, Qiao L (2014) Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett 34:196–202. https://doi.org/10.1016/j.canlet.2013.08.016
Businello G, Angerilli V, Parente P, Realdon S, Savarino E, Farinati F, Grillo F, Vanoli A, Galuppini F, Paccagnella S, Pennelli G, Mastracci L, Saragoni L, Fassan M (2021) Molecular landscapes of gastric pre-neoplastic and pre-invasive lesions. Int J Mol Sci 22:9950. https://doi.org/10.3390/ijms22189950
Angerilli V, Pennelli G, Galuppini F, Realdon S, Fantin A, Savarino E, Farinati F, Mastracci L, Luchini C, Fassan M (2022) Molecular subtyping of gastroesophageal dysplasia heterogeneity according to TCGA/ACRG classes. Virchows Arch 481:545–552. https://doi.org/10.1007/s00428-022-03392-7
Fassan M, Brignola S, Pennelli G, Alberti G, Angerilli V, Bressan A, Pellino A, Lanza C, Salmaso R, Lonardi S, Pucciarelli S, Spolverato G, Scarpa M, Realdon S, Farinati F, Luchini C, Rugge M, Loupakis F (2020) PD-L1 expression in gastroesophageal dysplastic lesions. Virchows Arch 477:151–156. https://doi.org/10.1007/s00428-019-02693-8
Chen XZ, Chen H, Castro FA, Hu JK, Brenner H (2015) Epstein–Barr virus infection and gastric cancer: a systematic review. Medicine (Baltimore) 94:e792. https://doi.org/10.1097/MD.0000000000000792
Pennelli G, Grillo F, Galuppini F, Ingravallo G, Pilozzi E, Rugge M, Fiocca R, Fassan M, Mastracci L (2020) Gastritis: update on etiological features and histological practical approach. Pathologica 112:153–165. https://doi.org/10.32074/1591-951X-163
Bergman MP, D’Elios MM (2010). Cytotoxic T cells in H. pylori-related gastric autoimmunity and gastric lymphoma. J Biomed Biotechnol 2010:104918. https://doi.org/10.1155/2010/104918
Lenti MV, Rugge M, Lahner E, Miceli E, Toh BH, Genta RM, De Block C, Hershko C, Di Sabatino A (2020) Autoimmune gastritis. Nat Rev Dis Primers 6:56. https://doi.org/10.1038/s41572-020-0187-8
Rugge M, Bricca L, Guzzinati S, Sacchi D, Pizzi M, Savarino E, Farinati F, Zorzi M, Fassan M, Dei Tos AP, Malfertheiner P, Genta RM, Graham DY (2022) Autoimmune gastritis: long-term natural history in naïve Helicobacter pylori-negative patients. Gut. https://doi.org/10.1136/gutjnl-2022-327827 (epub ahead of print)
Pizzi M, Sabattini E, Parente P, Bellan A, Doglioni C, Lazzi S (2020) Gastrointestinal lymphoproliferative lesions: a practical diagnostic approach. Pathologica 112:227–247. https://doi.org/10.32074/1591-951X-161
Raderer M, Kiesewetter B (2021) What you always wanted to know about gastric MALT- lymphoma: a focus on recent developments. Ther Adv Med Oncol 13:17588359211033824. https://doi.org/10.1177/17588359211033825
Violeta Filip P, Cuciureanu D, Sorina Diaconu L, Maria Vladareanu A, Silvia Pop C (2018) MALT lymphoma: epidemiology, clinical diagnosis and treatment. J Med Life 11:187–193. https://doi.org/10.25122/jml-2018-0035
Giuffrida P, Vanoli A, Arpa G, Bonometti A, Luinetti O, Solcia E, Corazza GR, Paulli M, Di Sabatino A (2018) Small bowel carcinomas associated with immune-mediated intestinal disorders: the current knowledge. Cancers (Basel) 11:31. https://doi.org/10.3390/cancers11010031
Yu J, Refsum E, Perrin V, Helsingen LM, Wieszczy P, Løberg M, Bretthauer M, Adami HO, Ye W, Blom J, Kalager M (2022) Inflammatory bowel disease and risk of adenocarcinoma and neuroendocrine tumors in the small bowel. Ann Oncol 33:649–656. https://doi.org/10.1016/j.annonc.2022.02.226
Svrcek M, Piton G, Cosnes J, Beaugerie L, Vermeire S, Geboes K, Lemoine A, Cervera P, El-Murr N, Dumont S, Scriva A, Lascols O, Ardizzone S, Fociani P, Savoye G, Le Pessot F, Novacek G, Wrba F, Colombel JF, Leteurtre E, Bouhnik Y, Cazals-Hatem D, Cadiot G, Diebold MD, Rahier JF, Delos M, Fléjou JF, Carbonnel F (2014) Small bowel adenocarcinomas complicating Crohn’s disease are associated with dysplasia: a pathological and molecular study. Inflamm Bowel Dis 20:1584–1592. https://doi.org/10.1097/MIB.0000000000000112
Vanoli A, Di Sabatino A, Martino M, Klersy C, Grillo F, Mescoli C, Nesi G, Volta U, Fornino D, Luinetti O, Fociani P, Villanacci V, D’Armiento FP, Cannizzaro R, Latella G, Ciacci C, Biancone L, Paulli M, Sessa F, Rugge M, Fiocca R, Corazza GR, Solcia E (2017) Small bowel carcinomas in celiac or Crohn’s disease: distinctive histophenotypic, molecular and histogenetic patterns. Mod Pathol 30:1453–1466. https://doi.org/10.1038/modpathol.2017.40
Arpa G, Vanoli A, Grillo F, Fiocca R, Klersy C, Furlan D, Sessa F, Ardizzone S, Sampietro G, Macciomei MC, Nesi G, Tonelli F, Capella C, Latella G, Ciardi A, Caronna R, Lenti MV, Ciccocioppo R, Barresi V, Malvi D, D’Errico A, Rizzello F, Poggioli G, Mescoli C, Rugge M, Luinetti O, Paulli M, Di Sabatino A, Solcia E (2021) Prognostic relevance and putative histogenetic role of cytokeratin 7 and MUC5AC expression in Crohn’s disease-associated small bowel carcinoma. Virchows Arch 479:667–678. https://doi.org/10.1007/s00428-021-03109-2
Piton G, Cosnes J, Monnet E, Beaugerie L, Seksik P, Savoye G, Cadiot G, Flourie B, Capelle P, Marteau P, Lemann M, Colombel JF, Khouri E, Bonaz B, Carbonnel F (2008) Risk factors associated with small bowel adenocarcinoma in Crohn’s disease: a case-control study. Am J Gastroenterol 103:1730–1736. https://doi.org/10.1111/j.1572-0241.2008.01847.x
Liao X, Li G, McBride R, Houldsworth J, Harpaz N, Polydorides AD (2020) Clinicopathological and molecular characterisation of Crohn’s Disease-associated small bowel adenocarcinomas. J Crohns Colitis 14:287–294. https://doi.org/10.1093/ecco-jcc/jjz135
Aparicio T, Svrcek M, Henriques J, Afchain P, Lièvre A, Tougeron D, Gagniere J, Terrebonne E, Piessen G, Legoux JL, Lecaille C, Pocard M, Gornet JM, Zaanan A, Lavau-Denes S, Lecomte T, Deutsch D, Vernerey D, Puig PL (2021) Panel gene profiling of small bowel adenocarcinoma: Results from the NADEGE prospective cohort. Int J Cancer 148:1731–1742. https://doi.org/10.1002/ijc.33392
Wan Q, Zhao R, Xia L, Wu Y, Zhou Y, Wang Y, Cui Y, Shen X, Wu XT (2021) Inflammatory bowel disease and risk of gastric, small bowel and colorectal cancer: a meta-analysis of 26 observational studies. J Cancer Res Clin Oncol 147:1077–1087. https://doi.org/10.1007/s00432-020-03496-0
Afif W, Sandborn WJ, Faubion WA, Rahman M, Harmsen SW, Zinsmeister AR, Loftus EV Jr (2013) Risk factors for lymphoma in patients with inflammatory bowel disease: a case-control study. Inflamm Bowel Dis 19:1384–1389. https://doi.org/10.1097/MIB.0b013e318281325e
Vitale G, Dicitore A, Barrea L, Sbardella E, Razzore P, Campione S, Faggiano A, Colao A; NIKE, Albertelli M, Altieri B, Bottiglieri F, De Cicco F, Di Molfetta S, Fanciulli G, Feola T, Ferone D, Ferraù F, Gallo M, Giannetta E, Grillo F, Grossrubatscher E, Guadagno E, Guarnotta V, Isidori AM, Lania A, Lenzi A, Calzo FL, Malandrino P, Messina E, Modica R, Muscogiuri G, Pes L, Pizza G, Pofi R, Puliani G, Rainone C, Rizza L, Rubino M, Ruggieri RM, Sesti F, Venneri MA, Zatelli MC (2021) From microbiota toward gastro-enteropancreatic neuroendocrine neoplasms: are we on the highway to hell? Rev Endocr Metab Disord 22:511–525. https://doi.org/10.1007/s11154-020-09589-y
Massironi S, Rossi RE, Milanetto AC, Andreasi V, Campana D, Nappo G, Partelli S, Gallo C, Scaravaglio M, Zerbi A, Panzuto F, Pasquali C, Falconi M, Invernizzi P, On Behalf Of ItaNet Italian Association For Neuroendocrine Tumours Study Group (2022) Duodenal gastric metaplasia and duodenal neuroendocrine neoplasms: more than a simple coincidence? J Clin Med 11:2658. https://doi.org/10.3390/jcm11092658
Matsubara A, Ogawa R, Suzuki H, Oda I, Taniguchi H, Kanai Y, Kushima R, Sekine S (2015) Activating GNAS and KRAS mutations in gastric foveolar metaplasia, gastric heterotopia, and adenocarcinoma of the duodenum. Br J Cancer 112:1398–1404. https://doi.org/10.1038/bjc.2015.104
Elfström P, Granath F, Ekström Smedby K, Montgomery SM, Askling J, Ekbom A, Ludvigsson JF (2011) Risk of lymphoproliferative malignancy in relation to small intestinal histopathology among patients with celiac disease. J Natl Cancer Inst 103:436–444. https://doi.org/10.1093/jnci/djq564
Marafini I, Monteleone G, Stolfi C (2020) Association between Celiac Disease and cancer. Int J Mol Sci 21:4155. https://doi.org/10.3390/ijms21114155
Lebwohl B, Green PHR, Emilsson L, Mårild K, Söderling J, Roelstraete B, Ludvigsson JF (2022) Cancer risk in 47,241 individuals with celiac disease: a Nationwide Cohort Study. Clin Gastroenterol Hepatol 20:e111–e131. https://doi.org/10.1016/j.cgh.2021.05.034
Han Y, Chen W, Li P, Ye J (2015) Association between Coeliac Disease and risk of any malignancy and gastrointestinal malignancy: a meta-analysis. Medicine (Baltimore) 94:e1612. https://doi.org/10.1097/MD.0000000000001612
Emilsson L, Semrad C, Lebwohl B, Green PHR, Ludvigsson JF (2020) Risk of small bowel adenocarcinoma, adenomas, and carcinoids in a Nationwide Cohort of Individuals with Celiac Disease. Gastroenterology 159:1686-1694.e2. https://doi.org/10.1053/j.gastro.2020.07.007
Potter DD, Murray JA, Donohue JH, Burgart LJ, Nagorney DM, van Heerden JA, Plevak MF, Zinsmeister AR, Thibodeau SN (2004) The role of defective mismatch repair in small bowel adenocarcinoma in celiac disease. Cancer Res 64(19):7073–7077. https://doi.org/10.1158/0008-5472.CAN-04-1096
Vanoli A, Di Sabatino A, Furlan D, Klersy C, Grillo F, Fiocca R, Mescoli C, Rugge M, Nesi G, Fociani P, Sampietro G, Ardizzone S, Luinetti O, Calabrò A, Tonelli F, Volta U, Santini D, Caio G, Giuffrida P, Elli L, Ferrero S, Latella G, Ciardi A, Caronna R, Solina G, Rizzo A, Ciacci C, D’Armiento FP, Salemme M, Villanacci V, Cannizzaro R, Canzonieri V, Reggiani Bonetti L, Biancone L, Monteleone G, Orlandi A, Santeusanio G, Macciomei MC, D’Incà R, Perfetti V, Sandri G, Silano M, Florena AM, Giannone AG, Papi C, Coppola L, Usai P, Maccioni A, Astegiano M, Migliora P, Manca R, Martino M, Trapani D, Cerutti R, Alberizzi P, Riboni R, Sessa F, Paulli M, Solcia E, Corazza GR (2017) Small bowel carcinomas in coeliac or Crohn’s Disease: clinico-pathological, molecular, and prognostic features. A study from the Small Bowel Cancer Italian Consortium. J Crohns Colitis 11:942–953. https://doi.org/10.1093/ecco-jcc/jjx031
Soderquist CR, Bhagat G (2021) Cellular and molecular bases of refractory celiac disease. Int Rev Cell Mol Biol 358:207–240. https://doi.org/10.1016/bs.ircmb.2020.12.001
Lenti MV, Biagi F, Lucioni M, Di Sabatino A, Paulli M, Corazza GR (2019) Two cases of monomorphic epitheliotropic intestinal T-cell lymphoma associated with coeliac disease. Scand J Gastroenterol 54:965–968. https://doi.org/10.1080/00365521.2019.1647455
Pelizzaro F, Marsilio I, Fassan M, Piazza F, Barberio B, D’Odorico A, Savarino EV, Farinati F, Zingone F (2021) The risk of malignancies in celiac disease—a literature review. Cancers (Basel) 13:5288. https://doi.org/10.3390/cancers13215288
Ciccocioppo R, Croci GA, Biagi F, Vanoli A, Alvisi C, Cavenaghi G, Riboni R, Arra M, Gobbi PG, Paulli M, Corazza GR (2018) Intestinal T-cell lymphoma with enteropathy-associated T-cell lymphoma-like features arising in the setting of adult autoimmune enteropathy. Hematol Oncol 36:481–488. https://doi.org/10.1002/hon.2494
Washington K, Stenzel TT, Buckley RH, Gottfried MR (1996) Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia. Am J Surg Pathol 20:1240–1252. https://doi.org/10.1097/00000478-199610000-00010
Leone P, Vacca A, Dammacco F, Racanelli V (2018) Common variable immunodeficiency and gastric malignancies. Int J Mol Sci 19:451. https://doi.org/10.3390/ijms19020451
Jess T, Rungoe C, Peyrin-Biroulet L (2012) Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin Gastroenterol Hepatol 10:639–645. https://doi.org/10.1016/j.cgh.2012.01.010
Olén O, Erichsen R, Sachs MC, Pedersen L, Halfvarson J, Askling J, Ekbom A, Sørensen HT, Ludvigsson JF (2020) Colorectal cancer in Crohn’s disease: a Scandinavian population-based cohort study. Lancet Gastroenterol Hepatol 5:475–484. https://doi.org/10.1016/S2468-1253(20)30005-4
Remo A, Fassan M, Vanoli A, Bonetti LR, Barresi V, Tatangelo F, Gafà R, Giordano G, Pancione M, Grillo F, Mastracci L (2019) Morphology and molecular features of rare colorectal carcinoma histotypes. Cancers (Basel) 11:1036. https://doi.org/10.3390/cancers11071036
Nagao-Kitamoto H, Kitamoto S, Kamada N (2022) Inflammatory bowel disease and carcinogenesis. Cancer Metastasis Rev 41:301–316. https://doi.org/10.1007/s10555-022-10028-4
Porter RJ, Arends MJ, Churchhouse AMD, Din S (2021) Inflammatory bowel disease-associated colorectal cancer: translational risks from mechanisms to medicines. J Crohns Colitis 15:2131–2141. https://doi.org/10.1093/ecco-jcc/jjab102
Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, Ahren C, Correa P, Hamilton SR, Morson BC et al (1983) Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum Pathol 14:931–968. https://doi.org/10.1016/s0046-8177(83)80175-0
Laine L, Kaltenbach T, Barkun A, McQuaid KR, Subramanian V, Soetikno R, SCENIC Guideline Development Panel (2015) SCENIC international consensus statement on surveillance and management of dysplasia in inflammatory bowel disease. Gastrointest Endosc 81:489–501.e26. https://doi.org/10.1016/j.gie.2014.12.009
Lee H, Rabinovitch PS, Mattis AN, Lauwers GY, Choi WT (2021) Non-conventional dysplasia in inflammatory bowel disease is more frequently associated with advanced neoplasia and aneuploidy than conventional dysplasia. Histopathology 78:814–830. https://doi.org/10.1111/his.14298
Choi WT, Yozu M, Miller GC, Shih AR, Kumarasinghe P, Misdraji J, Harpaz N, Lauwers GY (2020) Nonconventional dysplasia in patients with inflammatory bowel disease and colorectal carcinoma: a multicenter clinicopathologic study. Mod Pathol 33:933–943. https://doi.org/10.1038/s41379-019-0419-1
Nguyen ED, Wang D, Lauwers GY, Choi WT (2022) Increased histologic inflammation is an independent risk factor for nonconventional dysplasia in ulcerative colitis. Histopathology 81:644–652. https://doi.org/10.1111/his.14765
Eaden JA, Abrams KR, Mayberry JF (2001) The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 48:526–535. https://doi.org/10.1136/gut.48.4.526
Jess T, Simonsen J, Jørgensen KT, Pedersen BV, Nielsen NM, Frisch M (2012) Decreasing risk of colorectal cancer in patients with inflammatory bowel disease over 30 years. Gastroenterology 143:375–81.e1. https://doi.org/10.1053/j.gastro.2012.04.016 (quiz e13–4)
Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A (2004) Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 126:45145–45149. https://doi.org/10.1053/j.gastro.2003.11.010
Faye AS, Holmer AK, Axelrad JE (2022) Cancer in inflammatory bowel disease. Gastroenterol Clin North Am 51:649–666. https://doi.org/10.1016/j.gtc.2022.05.003
Quaglio AEV, Grillo TG, De Oliveira ECS, Di Stasi LC, Sassaki L (2022) Gut microbiota, inflammatory bowel disease and colorectal cancer. World J Gastroenterol 28:4053–4060. https://doi.org/10.3748/wjg.v28.i30.4053
Funding
Open access funding provided by Università degli Studi di Pavia within the CRUI-CARE Agreement. None.
Author information
Authors and Affiliations
Contributions
Conception/design of the work: AV, FG; literature search and analysis: all authors; drafting the work: all authors. All authors commented on previous versions of the manuscript and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights statement
This review article did not involve research on human participants and/or animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vanoli, A., Parente, P., Fassan, M. et al. Gut inflammation and tumorigenesis: every site has a different tale to tell. Intern Emerg Med 18, 2169–2179 (2023). https://doi.org/10.1007/s11739-023-03320-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-023-03320-w