
Abstract
We present an open-source software able to automatically mutate any residue positions and find the best aminoacids in an arbitrary protein structure without requiring pairwise approximations. Our software, PROTDES, is based on CHARMM and it searches automatically for mutations optimizing a protein folding free energy. PROTDES allows the integration of molecular dynamics within the protein design. We have implemented an heuristic optimization algorithm that iteratively searches the best aminoacids and their conformations for an arbitrary set of positions within a structure. Our software allows CHARMM users to perform protein design calculations and to create their own procedures for protein design using their own energy functions. We show this by implementing three different energy functions based on different solvent treatments: surface area accessibility, generalized Born using molecular volume and an effective energy function. PROTDES, a tutorial, parameter sets, configuration tools and examples are freely available at http://soft.synth-bio.org/protdes.html.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
We can use molecular dynamics techniques to computationally design proteins if we employ the same ideas as in the inverse folding problem (Bowie et al. 1991), which consists on identifying sequences that will fold on a given protein structure. Physicochemically inspired protein design methods have so far proven successful (Bolon and Mayo 2001; Kuhlman and Baker 2000; Looger et al. 2003; López de la Paz and Serrano 2004; Röthlisberger et al. 2008). The first attempt for an automatic procedure (Hellinga and Richards 1994) optimized a semi-empirical energy function, simultaneously in sequence and structure space, using a Monte Carlo Simulated Annealing (MCSA) approach. Dead End Elimination (Dahiyat and Mayo 1996; Dahiyat et al. 1997) or Branch and Bound (Wernisch et al. 2000) techniques are exact methods for obtaining the optimal solutions for a given residue pair-wise energy function, but are restricted to smaller problems than with heuristic approaches such as MCSA.
All these methods usually relay on the precomputation of the different residue-residue energies for their subsequent use within an optimization tool (Archontis and Simonson 2005; Chowdry et al. 2007; Huang and Stultz 2007). This computation is usually done using a pair-wise approximation, which scores the arrangement of at most two different residue conformations at a time. To perform this computation, it is also usual to assume that the designed positions surrounding the considered residues are either devoid of their corresponding side chains (Wernisch et al. 2000) or occupied by some predefined aminoacid (e.g. alanine, Dahiyat and Mayo 1996). This approximation poses several problems, especially when treating the solvation energies, since the solvent environment plays a crucial role in determining the electrostatic energy of the protein, which has lead to the development of pair wise decomposable energy functions and approximations (continuum or empirical models, since the explicit inclusion of water molecules is currently intractable). Unfortunately, even the models with solvation energies decomposable in atom-atom pairs lead to unphysical precomputed residue-residue energy because the energy terms for each pair of interacting residues are context dependent: the solvation of one side-chain pair strongly depends on the shape of the protein, which depends on the conformation of all other side chains. This not only leads to a loss of accuracy when using combinatorial optimization, but in most cases it provides unrealistic results (Jaramillo and Wodak 2005).
The search for an appropriate protein design methodology that could incorporate an accurate estimation of the folding free energy is still far to be accomplished (Lippow and Tidor 2007), despite of the increasing number of successful examples of the use of molecular dynamics techniques in protein design (Dahiyat et al. 1997; Looger et al. 2003; Ogata et al. 2002). Work aiming to improve those energy functions for computational protein design includes understanding and validating their applicability, analyzing the different approximations that would improve their efficiency, checking new algorithms for their implementation as well as developing new potentials. The development of new energy functions requires testing various implicit solvents together with their corresponding models for the unfolded and folded state. For instance, solvations incorporating longer range energy terms are more sensible to the 3D structure of the unfolded model. In addition, the model for the folded state may also require a proper treatment of backbone flexibility, which may require to combine molecular dynamics with the protein design optimization. We propose a software that is able to combine both approaches, while allowing to easily explore improvements to current protein design methodologies. One of the best ways to achieve this is by extending a molecular dynamics package providing a set of implicit solvations. As the base simulation package we have considered CHARMM (Brooks et al. 1983; Schleif 2006), a widely used software for molecular dynamics simulation and analysis that, incorporates a panoply of implicit solvation models.
We have developed a new bioinformatics tool in the form of a CHARMM script that the user could easily tailor to study any small system of interest in a reasonable amount of time. This approach is slower than software relying on combinatorial optimization, but is free of pairwise approximations. To exemplify the incorporation of new energy terms in PROTDES we have incorporated three protein design protocols based on different solvation models, one of them already tested experimentally for protein design (Jaramillo et al. 2002; Ogata et al. 2002) (the methodology based on accessible surface area). Another advantage of PROTDES is that it allows the consideration of DNA as template, allowing the study of DNA-protein interactions.
We remark that it is not the purpose of this paper to propose a new methodology for protein design, but to provide a tool allowing to implement already existing methodologies such as the DESIGNER (Wernisch et al. 2000) protocol. Additionally, two other solvation models, already implemented in CHARMM, are included in PROTDES. This shows the high versatility of our tool, that will allow the user to create novel protein design methodologies.
PROTDES uses scripting in all the design process, instead of dividing the calculation into energy precomputation and optimization. This contrasts with a recent attempt of using scripting in protein design to precompute a pair-wise energy matrix (Lopes et al. 2007). Several protein design problems are of small size and the speed of the optimization is not the critical step. In those problems (e.g. ligand binding) it is more important to incorporate a relaxation of the systems through molecular dynamics and a more careful treatment of the solvent. It is precisely the extensive use of scripting that makes PROTDES the only available tool allowing to integrate molecular dynamics within in silico mutagenesis in order to, for example, introduce a slight amount of backbone flexibility in the design.
Implementation
The stability of a protein can be described by its folding free energy: \(\Updelta G_{\rm folding} = G_{\rm folded} - G_{\rm ref}.\) To evaluate the fitness of various sequences for a fixed structure, we compare the folding free energy of the different sequences. At this point we make a strong assumption on the additivity and context independence effect of single mutations in the stability of the proteins, since we assume that the fixed backbone and fixed side chains will equally contribute to the score of each sequence. This way we can eliminate their contribution to the folding free energy. We are assuming here that a specific mutation will have an effect on the overall stability of the protein that is independent of its context, although this approximation can not be considered to hold in all cases (Cheng et al. 2008). Nevertheless this simplifying assumption can work in a specific setting, in fact this assumption is the same at the basis of classical QSAR analysis on congeneric series profitably used in pharmaceutical chemistry since more than 40 years (Hansch and Fujita 1964). We assume a fixed backbone, to neglect the change in the main chain entropy. The main contribution of entropy to ΔG comes from the difference in the side chain entropy between the reference and the folded state; this contribution is lower than 1kcal/(mol aminoacid) (Doig and Sternberg 1995) . The contribution of the side chains to the reference state energy is the sum of the contribution from each independent aminoacid. These contributions do not change with the aminoacid conformation or placement, are thus system independent, and can be pre-computed as has been previously done (Jaramillo et al. 2002). The reference energies provided with PROTDES involve no residue interactions in the reference state. The reference state has been modeled using a dipeptide for each residue. Finally, we obtain
The sum is over all aminoacids i in the sequence (rotamer r i ). We consider the following terms of the CHARMM22 force field: electrostatics, van der Waals and the solvation energy. For the solvation energy, PROTDES allows the user the election between three possible models.
Generalized Born using Molecular Volume (GBMV)
The solvation energy is given by:
where E screen ij is the electrostatic screening interaction energy between two charged groups in a dielectric medium, it is calculated with the generalized Born equation using the gbmv implementation (Lee et al. 2002) . ΔE self i is the solvation free energy of the group and ΔE nonp i is a surface area-dependent approximation to the hydrophobic solvation term
Accessible Surface Area (ASA)
This model for the solvation energy is based on the exposed surface area of each atom i; E sol = ∑ i σ i ASA i . The ASA is computed using the CHARMM22 van der Waals radii and the atomic solvation parameters, σ i , are measured in the vacuum to water transfer process (Ooi et al. 1987).
Effective energy function (EEF1)
An excluded volume implicit solvation model combined with an empirical screening (Lazaridis and Karplus 1999). The sum over atomic contributions is performed:
E ref is computed in a reference state where the atom is completely accessible to the solvent and f, the solvation free energy density is a Gaussian function.
Backbone-flexibility
Along the previous lines, we have assumed that the backbone of the protein stayed fixed. Nevertheless, there has been some successful attempts to introduce backbone flexibility within computational protein design (Desjarlais and Handel 1999; Sood and Baker 2006; Su and Mayo 1997). One of the most powerful tools available in CHARMM is its capacity to perform molecular dynamics simulations. This advantage is inherited by PROTDES, where backbone flexibility can be introduced together with aminoacid sequence selection, using the molecular dynamics simulations tools implemented in CHARMM, to allow for local adjustments of the backbone.
The implementation in PROTDES is uses the following default parameters and modifies the region inside a 9 Å sphere surrounding the C α of the designed position. The meaning of the different parameter can be found in the CHARMM documentation. dynamics verlet timestep 0.0005 nstep 500 nprint 500 iprfrq 100 - firstt 240 finalt 300 twindh 10.0 ieqfrq 200 ichecw 1 - iasors 0 iasvel 1 inbfrq 20
Software and interface
PROTDES has been developed as a CHARMM script (1200 code lines) and can run in any machine with an installed version of CHARMM. PROTDES is a free, open-source software package licensed under a free Creative Commons Attribution Noncommercial 3.0 (http://creativecommons.org/licenses/by-nc/3.0), that can be freely downloaded from our web site: (http://soft.synth-bio.org/protdes.html). The program’s distribution includes the source code, documentation, usage examples (phage λ cI repressor protein core redesign, MHC-I binding peptides design and phage λ cI repressor protein specificity redesign with and without backbone flexibility) and a configuration utility to guide the user through the definition of the protein structure, the positions to mutate, the position dependent libraries, the specification of the reference energies and the number of iterations of the heuristic algorithm.
Input Data PROTDES needs a minimum of initial data to star the designing process
-
Protein structure, in the form of a PDB file
-
List of positions to mutate
-
Solvation model to be used: GBMV, ASA or EEF1
-
Rotamer library for each variable position. The rotamer library is a text file containing the different rotamers to be used at each position. For each rotamer, there is a line in this file containing the name of the aminoacid and the values of its dihedral angles An initial library (Tuffery et al. 1991) is provided with PROTDES, but the user may choose generate his proper libraries to include local backbone configuration and side chain preferences (Dunbrack and Karplus 1994)
-
The reference energies to model the unfolded state, which depend on the chosen solvation model. The reference energies provided with PROTDES have been computed without residue interactions and the unfolded (or reference state) has been modeled using a dipeptide. An additional utility of PROTDES is that can be switched to a “compute reference energy” mode that allows analysing the effect of different methodologies to model the unfolded state.
Initially, PROTDES starts with a sequence in an initial conformation; the different input data are read and the designed positions are mutated to glycine. Then, the optimization procedure starts, and the following is repeated N times
-
(i)
Randomly choose a position, p.
-
(i′)
The following step would be to remove the side chains from p to compute the solvation energy of the surrounding region in the absence of any rotamer. This solvation energy would then be subtracted in the computation of the energy corresponding to any rotamer. Fortunately, the different energies we will compare share this common term, rendering futile its computation.
-
(i′)
-
(ii)
For each of the allowed aminoacids in position p, randomly choose a conformation (rotamer), patch it, and perform a local minimization (keeping the backbone fixed) to allow slight conformational variations. In case that p correspond to C or N terminus, special patches are used.
-
(iii)
Among the chosen rotamers find the one, r p improving most the total folding free energy and patch it: equation \(r_p | E(r_p) = \hbox{min} \left\{ E_{(r_i,\rm prot)}-E_{r_i}^{\rm ref} \right \}\) equation
-
(iv)
If desired, perform a molecular dynamics to introduce backbone flexibility and slightly vary the backbone to adapt it to the introduced mutation.
Additional molecular dynamics simulations could be performed each time an aminoacid is introduced in a given position, unfortunately the comparison between the obtained values for the folding free energy considering each aminoacid (Eq. 1, would be less clear. Nevertheless we have added this capability to PROTDES to allow the user to explore new computational design methodologies.
The previously stated algorithm can be modify and PROTDES can be forced to follow an exhaustive approach, where point ii) is modified and for each aminoacid all its possible rotamers are considered. This is extremely convenient if we have a very low number of designed positions. The different options within PROTDES are modified by flags in the configuration file.
Finally PROTDES collects the best sequence and its energy. A scheme of PROTDES’ algorithm is presented in Fig. 1.

PROTDES algorithm for computational protein design. The initial configuration is loaded into CHARMM; the designed positions are mutated to glycine. The heuristic algorithm proceeds iteratively (i) choose a position (ii) for each allowed aminoacid randomly choose a rotamer (iii). The rotamer providing the best folding energy (fixing the aminoacid content of the other positions) for that position is patched, until the total number of iterations N is reached
Results
We have used PROTDES software to perform the examples that can be found in the documentation included in the software. There, we provide a detailed analysis of the different configuration files used in each of the four designs we made: phage λ cI repressor protein core redesign, MHC-I binding peptides design and phage λ cI repressor protein specificity redesign with and without backbone flexibility. Here we report the results obtained in two of the cases.
MHC-I binding peptides design
Major histocompatibility complex (MHC) class I molecules are peptide binding proteins that play a key role in the mammalian immune system, allowing the immune system to both recognize foreign antigens and to remain tolerant to self-derived peptides.
There is a huge variability in the peptides proteins from the MHC-I bind. In this work, following previous results (Ogata et al. 2002), we designed peptides likely to bind the HLA-A2 member of this family. MCH-I proteins have a binding site formed by two α-helices and a β-sheet. Peptides formed by 8–10 aminoacids are able to enter the binding pocket. The HLA-A2 proteins have additional specific binding pockets that allows them to preferentially bind 9 aminoacid peptides containing the so called “anchor residues”: L, I, M or V in the second position of the peptide (P2) and V, I, A or M at C-terminus (Madden 1995).
Our departure point was the 2.5 Å resolution structure for the HLA-A2-peptide complex (Madden et al. 1993), [PDB:1HHK]. We allowed to mutate 8 out of the 9 positions in the peptide and we left P6 fixed. No additional constraint on the aminoacid contain was introduced and the whole rotamer library (Tuffery et al. 1991) (214 rotamers) was considered at each position.
EEF1 solvation
We run PROTDES 100 times with 200 and 500 heuristic steps. We obtained similar results in both cases: (8 out of the 10 sequences with lower energy were identical in both cases.) With 500 heuristic steps we obtained the sequences in Table 1, where only the 4th sequence fulfills the requirements regarding the anchor residues.
ASA solvation
Once again we run PROTDES 100 with 200 and 500 steps in each iteration and the results with 200 and 500 iterations were very similar. The list of the top 10 sequences with the highest predicted binding energy for the 200 iterations case is shown in Table 2. We can see that out of the 10 predicted sequences with a higher affinity, all but the last one have the appropriate anchor residues.
GBMV solvation
This resulted the most time consuming procedure. Fortunately it proved to be accurate enough to converge we a small number of heuristic iterations. As a result we needed only to perform 50 runnings using 200 heuristic steps, since, even with this limitations the results were highly satisfactory: between the 10 highest ranking solution 6 of them had the required anchor residues, as can be seen in Table 3.
If we compare the three different solvation models, we realize that the EEF1 model yields not very accurate results. On the other hand, both ASA and GBMV models yield fairly appropriate solutions, as far as the anchor residues are concerned. In addition, if we consider that the ASA solvation model is much faster that the GBMV, we will have to conclude that, for this problem ASA is the most adequate solvation to use.
Figure 2 is a graphical representation of the results obtained with PROTDES using the ASA model with 200 iterations. The calculation was performed in a 2.13GHz CPU and each complete run of PROTDES took around 40 min.

Structure of the highest binding predicted peptide, using the ASA solvation, in the HLA-A2 binding pocket. The peptide is represented by a stick model and the HLA-A2 molecule is represented by its molecular surface. Figure generated using the Pymol (DeLano 2002) viewer software
The presence of the correct anchor residues is a necessary condition for binding (Madden 1995), so although we cannot claim that our designed peptides will actually bind to the MHC-I we can affirm that they fulfill all the known requisites, since the needed anchor residues appear in our designs. In fact, our results show similar anchor residues as the calculations reported with DESIGNER (Ogata et al. 2002). Nevertheless, sequence based algorithms specially designed to deal with the prediction of peptides able to bind a given complex provide accurate predictions (Selz et al. 2007), nevertheless a combination of both sequence-based and structural methods will likely yield the best results.
PROTDES has shown its performance and versatility in the design of peptides able to bind to the MHC-I, The prediction of protein binding sites and together with it the prediction of the possible binding peptides is still an open question. PROTDES can allow for a certain amount of backbone flexibility within the design and this will clearly help to refine the predictions. In fact the recent work by (Haliloglu et al. 2008) approaches specifically this same problem from a dynamical point of view.
Specificity change of the λ cI repressor protein
We have focused on the DNA binding region of the phage λ cI repressor protein. This is an extensively studied protein with well known transcriptional regulatory functions, since it acts as a transcription factor (TF) regulating the transcription of the cI protein and the Cro protein.. In order to introduce networks performing well defined functions in an organism, independence between the new functional biological components and the pre-existing cellular networks has to be attained to free the regulation of the introduced networks of interference with the chosen chassis.
To obtain TFs with a weak interaction with already existing genetic circuits within the considered organism, protein design can be a powerful tool. Our goal will be to redesign the λ cI binding region and simultaneously mutate its binding site in the DNA so that the new TF has a weaker interaction with the wild type DNA and the wild type TF has a low binding to the mutated DNA, this would assure independence between the introduced networks regulated by the designed TF and DNA and the ones present in the organism. We have considered the 1.8 Å resolution structure [PDB:1LMB] (Beamer and Pabo 1992) and analyzed the DNA-protein binding region, shown in Fig. 3. We used CHARMM to introduced the following DNA mutations: A4C (chain LMBA) and T38G (chain LMBB). This process is detailed documented in the examples file distributed with PROTDES, together with the CHARMM scripts used to rebuild the final DNA structure shown in Fig. 4.

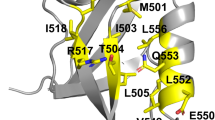
Structure of the DNA-protein interaction region of cI protein: the original structure, [PDB:1LMB] that shows the two H-bonds formed between Q44 and A4

Structure of mutated DNA interacting with the wild type λ cI protein: we have mutated A4C, (chain LMBA) and T38G (chain LMBB) and as a consequence the interaction between Q44 and DNA has weakened
Nowadays the consideration of DNA as a template within PROTDES, restricts the available solvation models to use to only the ASA model. The analysis of the interaction between cI and the mutated DNA, (Fig. 4), led us to consider positions 44 and 33 (chain LMBC) in the designing procedure. Furthermore, we allowed PROTDES to choose between all possible aminoacids in position 44 so we used a 214 rotamer library (Tuffery et al. 1991), on the other hand, in position 33 we only allowed for configurational changes, so we used a 19 rotamer library containing different configurations for Gln.
When introducing the DNA mutations, we assumed that DNA structure would not be greatly altered, this is perhaps on of the strongest assumptions in this designing process, so we allowed a certain amount of backbone flexibility. We introduced a molecular dynamics and we allowed the atoms inside a 9Å sphere surrounding the designed positions to vary their positions, including the atoms belonging to the protein’s or DNA backbone.
Due to the low number of positions to design we have utilized the exhaustive procedure in the optimization so that all the rotamers in library will be patched in each iteration, we found that even with 5 heuristic iteration convergence was achieved, the results of 5 out of 10 runnings, corresponding to the minimal energy found was identical. Increasing the number of steps provided the same results. PROTDES proposed the Q44I mutation, that together with the side chain modeling of Q33 managed to lower the energy of the complex by 3.2 kcal/mol. The interactions with DNA have also changed, now its backbone is involved as can be seen in Fig. 5.

Structure of the mutated DNA interacting with the mutated cI repressor protein: the DNA mutations A4C, (chain LMBA) and T38G (chain LMBB) and the cI protein mutation Q44I have change the protein DNA interaction
In Fig. 6 the original backbone and the structure resulting from the molecular mechanics procedure are compared The root mean square deviation between the backbone atoms is 0.210 Å (Pymol).

Structure comparision: Comparision between the structures of the original structure of the cI DNA complex and the mutated ones
Conclusions
In summary we have developed a new bioinformatics tool for protein design based on CHARMM, a widely used set of force fields for molecular dynamics and molecular dynamics simulation and analysis package. PROTDES aims to exploit CHARMM’s power. As such, PROTDES has been written in the form of a script that the user can easily modify to study any small system of interest in a reasonable amount of time.
This scripting approach where no energy matrix are precomputed before the optimization process has the advantage of having no pair wise energy approximations, on the other hand is slower than software approaching the combinatorial optimization using other methods like MCSA. In addition, already existing software to generate CHARMM scripts (Sunhwan et al. 2008) can help the user to modify PROTDES it self. The versatility of PROTDES will allow the user to create and test new protein design methodologies.
References
Archontis G, Simonson T (2005) A residue-pairwise generalized born scheme, suitable for protein design simulations. J Phys Chem B 109:22667–22673
Beamer L, Pabo C (1992) Refined 1.8 angstrom crystal structure of the lambda repressor operator complex. J Mol Biol 227:177–196
Bolon D, Mayo S (2001) Enzyme-like proteins by computational design. Proc Natl Acad Sci USA 98:14274–14279
Bowie J, Lathy R, Heisenberg D (1991) A method to identify protein sequences folding into a known three-dimensional structure. Science 253:164–170
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) Charmm: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem 4(2):187–217
Cheng TMK, Lu Y-E, Vendruscolo M, Pietro L, Blundell TL (2008) Prediction by graph theoretic measures of structural effects in proteins arising from non-synonymous single nucleotide polymorphisms. PLoS Comput Biol 4(7):e1000135
Chowdry AB, Reynolds KA, Hanes MS, Vorhies M, Pokala N, Handel TM (2007) An object-oriented library for computational protein design. J Comp Chem 28(14):2378–2388
Dahiyat B, Mayo S (1996) Protein design automation. Protein Sci 5:895–903
Dahiyat C, Sarisky BI, Mayo S (1997) De-novo protein design: towards fully automated sequence selection. J Mol Biol 273(4):789–796
DeLano WL (2002) The pymol molecular graphics system
Desjarlais JR, Handel TM (1999) Hydrophobic core design and structure prediction with backbone flexibility. J Mol Biol 189:305–318
Doig AJ, Sternberg MJE (1995) Side-chain conformational entropy in protein folding. Protein Sci 4:2247–2251
Dunbrack RL, Karplus M (1994) Conformational analysis of the backbone-dependent rotamer preferences of protein sidechains. Nat Struct Biol 1(5):334–340
Haliloglu T, Seyrek E, Erman B (2008) Prediction of binding sites in receptor-ligand complexes with the gaussian network model. Phys Rev Lett 100(22):228102–228106
Hansch C, Fujita T (1964) p-σ−π analysis. a method for the correlation of biological activity and chemical structurecomputational design of a biologically active enzyme. J Am Chem Soc 86:1616–1626
Hellinga HW, Richards FM (1994) Optimal sequence selection in proteins of known structure by simulated evolution. Proc Nat Acad Sci USA 91(13):5803–5807
Huang C, Stultz A (2007) Conformational sampling with implicit solvent models: application to the phf6 peptide in tau protein. Biophys J 92:34–45
Jaramillo A, Wodak SJ (2005) Computational protein design is a challenge for implicit solvation models. Biophys J 88(1):156–171
Jaramillo A, Wernisch L, Hery S, Wodak S (2002) Folding free energy function selects native-like protein sequences in the core but not on the surface. Proc Nat Acad Sci USA 99(21):13554–13559
Kuhlman B, Baker D (2000) Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci USA 97(19):10383–10388
Lazaridis T, Karplus M (1999) Effective energy function for proteins in solution. Proteins 35(3):133–152
Lee MS, Salsbury FR, Brooks CL (2002) Novel generalized born methods. J Comp Chem 116:10606–10614
Lippow SM, Tidor B (2007) Progress in computational protein design. Current Opin Biotechnol 18(4):305–311
Looger LL, Dwyer MA, Smith JJ, Hellinga HW (2003) Computational design of receptor and sensor proteins with novel functions. Nature 423(6936):185–190
Lopes A, Alexandrov A, Bathelt C, Archontis G, Simonson T (2007) Computational sidechain placement and protein mutagenesis with implicit solvent models. Proteins 67(4):853–867
López de la Paz M, Serrano L (2004) Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci USA 101(1):87–92
Madden DR (1995) The 3-dimensional structure of peptide-mhc complexes. Annu Rev Immunol 13:587–622
Madden DR, Garboczi DN, Wiley D (1993) The antigenic identity of peptide-mhc complexes: a comparison of the conformations of five viral peptides presented by hla-. Cell 75:693–708
Ogata K, Jaramillo A, Cohen W, Briand J-P, Connan F, Choppin J, Muller S, Wodak SJ (2002) Automatic sequence design of mhc class-i binding peptides impairing cd8+ t cell recognition. J Biol Chem 278(2):1281–1290
Ooi T, Oobatake M, Nemethy G, Scheraga H (1987) Accesible surface areas as a measure of the thermodynamics parameters of hydration of peptides. Proc Natl Acad Sci USA 84:3086–3090
Röthlisberger D, Khersonsky O, Wollacott AM, Jiang L, Dechancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D (2008) Kemp elimination catalysts by computational enzyme design. Nature 453:190–195
Schleif R (2006) Analysis of protein structure and function: a Beginner’s guide to CHARMM. http://gene.bio.jhu.edu
Selz KA, Samoylova TI, Samoylov AM, Vodyanoy VI, Mandell AJ (2007) Designing allosteric peptide ligands targeting a globular protein. Biopolymers 85(1):38–59
Sood V, Baker D (2006) Recapitulation and design of protein binding peptide structures and sequences. J Mol Biol 357:917–927
Su A, Mayo SL (1997) Coupling backbone flexibility and amino acid sequence selection in protein design. Protein Sci 6:1701–1707
Sunhwan J, Taehoon K, Vidyashankara GI, Wonpil I (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29(11):1859–1865
Tuffery P, Etchebest C, Hazout S, Lavery R (1991) A new approach to the rapid determination of sidechains. J Biomol Struct Dyn 8:1267–1289
Wernisch L, Hery S, Wodak S (2000) Automatic protein design with all atom force-fields by exact and heuristic optimization. J Mol Biol 301(3):713–736
Acknowledgements
P.T. acknowledges financial support from the EMBO long-term fellowship program. We acknowledge financial support from the grant BIOMODULARH2 FP6-NEST-Pathfinder 043340. and grant FP6-NEST-PATH-SYN-043338 Emergence.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Suárez, M., Tortosa, P. & Jaramillo, A. PROTDES: CHARMM toolbox for computational protein design. Syst Synth Biol 2, 105–113 (2008). https://doi.org/10.1007/s11693-009-9026-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11693-009-9026-7