
Abstract
One of the outstanding questions in behavioral disorders is untangling the complex relationship between nurture and nature. Although epidemiological data provide evidence that there is an interaction between genetics (nature) and the social and physical environments (nurture) in a spectrum of behavioral disorders, the main open question remains the mechanism. Emerging data support the hypothesis that DNA methylation, a covalent modification of the DNA molecule that is a component of its chemical structure, serves as an interface between the dynamic environment and the fixed genome. We propose that modulation of DNA methylation in response to environmental cues early in life serves as a mechanism of life-long genome adaptation. Under certain contexts, this adaptation can turn maladaptive resulting in behavioral disorders. This hypothesis has important implications on understanding, predicting, preventing, and treating behavioral disorders including autism that will be discussed.
Similar content being viewed by others

Introduction
There has been a genocentric focus in our approach to human disease and specifically behavioral disorders such as autism in the last century. This has been driven by the high heritability of several common disorders such as autism. The underlying hypothesis has been that differences in gene sequence between individuals could result in alteration in genome function that would create phenotypic differences that manifest themselves in mental or physical disorders. A tremendous effort has been invested in genome-wide association studies of complex disease. This endeavor has resulted in delineation of an increasing list of associations between rare as well as common genetic variants and behavioral disorders such as autism. Several of these genes were also found to play an important role in the development of the nervous system and taught us important lessons on possible mechanisms of brain function (Grice and Buxbaum 2006; Kumar and Christian 2009; Levitt and Campbell 2009). It is believed that with the expansion in whole-genome technologies and by including copy number variations in addition to single-nucleotide polymorphisms in genomic studies, it will be possible to strengthen and broaden these associations (Weiss 2009; Weiss et al. 2009; Anney et al. 2010; Pinto et al. 2010). However, epidemiological data suggest an environmental component in most behavioral disorders including autism.
The DNA molecule bears two layers of information: the genetic information encoded in the sequence of bases in DNA and the pattern of distribution of methyl cytosines (Razin and Riggs 1980; Razin and Szyf 1984) and hydroxy-methyl cytosines (Kriaucionis and Heintz 2009). The methyl groups are added to the DNA molecule after replication by machinery that is distinct from the machinery that replicates the DNA sequence creating within the hard chemistry of the DNA molecule itself a potential for differential information. It has been known for several decades that the pattern of distribution of methyl cytosines is not equal in all tissues in a multicellular organism and that it exhibits tissue specificity (Razin and Szyf 1984). Thus, the DNA molecule has two identities: the ancestral identity encoded in the sequence and the cell-specific identity encoded in the pattern of DNA methylation. It has been almost an established dogma that DNA methylation patterns form during embryogenesis by innate organized developmental programs and that DNA methylation is mainly involved in cellular differentiation. It was therefore believed that DNA methylation patterns once formed remained fixed since cellular differentiation was believed to be terminal. Recent data suggest however that DNA methylation is more attentive to information from the environment including the social environment particularly early in life (Szyf et al. 2007). It is proposed here that the DNA methylation pattern bears not just the tissue-specific identity but also the individual identity formed as a response to interactions with the environment early in life and throughout life. The pattern of DNA methylation is proposed to play an important role in social adaptation of genome function and therefore in behavioral phenotypes. We will discuss the possible implications of this hypothesis on prediction, prevention, and treatment of neurodevelopmental behavioral disorders.
DNA methylation patterns
Vertebrate DNA is covalently modified by enzymatic addition of a methyl group to cytosines in DNA. The hallmark of DNA methylation in vertebrates and many other organisms is the fact that the distribution of the methyl groups in the DNA varies across different cell types in an organism. This creates a cell-type-specific pattern of DNA methylation. In vertebrates, the CG dinucleotide sequence is a principal target of DNA methylation (Gruenbaum et al. 1981) since it is preferentially recognized by vertebrate DNA methyltransferases (Gruenbaum et al. 1982). CG is the only dinucleotide sequence that contains a cytosine that is a palindrome and could be copied during cell division by a semiconservative DNA methyltransferase from the parental strand onto the daughter strand (Razin and Riggs 1980). Heritability of DNA methylation mark is critical for its role as an epigenetic signal that bears the specific cellular identity. This concept of automatic heritability was behind the prevailing dogma that once the innate programmed developmental processes form DNA methylation patterns during embryogenesis they remain fixed after birth to maintain cellular identity. Recent studies suggest that DNA methyation could occur in non-CG sequences at least in stem cells but it is unclear how prevalent non-CG methylation is in the brain (Lister et al. 2009; Fuso et al. 2010). In any case, the presence of non-CG methylated sites in our genomes suggests that mechanisms for inheritance of DNA methylation exist that are not defined automatically by the DNA methylation of the parental template strand of DNA. For example, when a new CC site is generated during DNA replication the complementary site on the parental strand of DNA is GG and it obviously cannot guide the methylation of the CC on the new strand of DNA. This has important implications on our understanding of the scope of biological functions that DNA methylation is involved in as will be discussed below (Szyf 2010). Many of the current assays of DNA methylation are biased to CG methylation. We might have missed important DNA methylation responses that involve non-CG methylation. For example, Fuso et al. 2010 have recently shown that during muscle differentiation, DNA demethylation of a regulatory region of the myogenin gene occurs mainly in methylated rich but CG poor sequence (Fuso et al. 2010). The possibility that DNA methylation could occur outside CG islands forces us to revisit this especially in the brain however it is as yet unclear whether non-CG methylation plays a role in postmitotic neurons.
Differential DNA methylation is an excellent candidate to serve as the “epigenetic” mechanism predicted by Waddington as an explanation for how one genome expresses multiple phenotypes in a multicellular organism (Waddington 1959; Razin and Riggs 1980). Since cellular phenotypes once generated during embryogenesis are extremely stable, this requires that DNA differential methylation patterns once generated would be inherited faithfully and accurately. Several lines of evidence supported this hypothesis. Cell-specific DNA methylation patterns were described almost two decades ago (Razin and Szyf 1984; Benvenisty et al. 1985) and were recently confirmed by whole-genome methylome mapping (Lister et al. 2009). Also, the prediction that a semiconservative enzyme copies the methyl groups from the palindromic CG on the template strand to the complementary CG on the daughter strand (Razin and Riggs 1980) was confirmed as well; providing a mechanism for faithful maintenance of the DNA methylation pattern in diverse cell lineages (Gruenbaum et al. 1982).
The involvement of DNA methylation in cellular differentiation and in the “canalization” leading to terminal differentiation might imply that the DNA methylation pattern in differentiated cells is also terminal. However, there is considerable epidemiological data suggesting that experience after birth has a profound effect on the phenotype later in life. How could a static and relatively fixed genome adapt to different environmental contexts? This question is especially pertinent in the nervous system where nurture after birth plays an important role in brain development. One of the important new concepts in DNA methylation has been the idea that DNA methylation might be diversifying genome function not only in response to innate developmental programs but also in response to environmental exposures enabling the same genome to express different phenotypes (Szyf 2009b). DNA methylation is therefore proposed to be a general mechanism of genome adaptation in addition to its role in conferring cell type identity to genomes during differentiation.
Differential DNA methylation is a strong candidate to regulate differential gene expression. There is an overall inverse correlation between DNA methylation in several regulatory regions of genes and gene expression which was discovered in the early eighties (Razin and Szyf 1984) (Razin and Riggs 1980) and was confirmed by whole-genome approaches (Rauch et al. 2009). However, exceptions to this rule exist and unmethylated promoters of genes that are silenced are frequent in the genome. It is possible that methylation dependent regulatory regions that were not examined in the published studies control these genes. Alternatively, it is important to note that DNA methylation and gene expression studies describe different levels of gene expression regulation. DNA methylation patterns program gene expression potential and condition genes to respond to appropriate environmental and physiological signals whereas expression studies reveal a transient state of expression.
One of the early landmark discoveries in the field was the inverse correlation between active chromatin configuration and DNA methylation. Highly methylated DNA regions are packaged in the nucleus in inactive chromatin configuration (Razin and Cedar 1977). This data points to a relationship between two critical elements of the epigenome: chromatin modification and DNA methylation. Several studies support a bilateral relationship between DNA methylation and chromatin structure; DNA methylation directs the formation of inactive chromatin while inactive chromatin targets DNA methylation. The converse is also true; loss of methylation targets activating histone modification marks and active histone methylation marks such as histone acetylation facilitate DNA demethylation (D’Alessio and Szyf 2006).
Regulation of promoter activity by DNA methylation
Although global changes in DNA methylation that cover wide regions of the genome occur in pathological conditions such as cancer (Feinberg and Vogelstein 1983) and lupus (Cornacchia et al. 1988; Yung and Richardson 1994), the most established role of DNA methylation is in regulation of promoter activity and gene regulatory regions (Stein et al. 1982). These account for a minor fraction of the changes in DNA methylation documented in literature. At least two mechanisms were demonstrated for inhibition of gene activity by DNA methylation. A methyl group positioned in a recognition element for a transcriptional factor can block binding of the transcription factor to the promoter (Comb and Goodman 1990; Inamdar et al. 1991). Alternatively, methylated DNA attracts methylated DNA binding proteins such as the Rett syndrome protein methyl-CpG binding protein 2 (MeCP2), which in turn recruit histone modification enzymes such as histone deacetylases (HDAC)s to the gene precipitating an inactive gene-silencing chromatin configuration (Nan et al. 1997).
Although the main focus of most studies has been on promoter DNA methylation, recent whole-genome methylation analyses revealed an interesting positive relationship between DNA methylation in bodies of genes and gene expression (Hellman and Chess 2007; Lister et al. 2009; Rauch et al. 2009). A revealing example is X inactivation in females; although promoters in the active X are unmethylated relative to the inactive X the reverse is true for the bodies of genes which are hypermethylated on the active X (Hellman and Chess 2007). A large fraction of the methylation in DNA however occurs in inter-genic regions and in repetitive sequences, and this methylation might play yet unknown roles. Methylation of retroviral elements in the genome was proposed to silence ectopic expression from these parasitic elements that could disrupt normal genome function (Yoder et al. 1997).
DNA methylation enzymes
To understand how DNA methylation patterns are formed it is important to decipher the machinery that lays down the DNA methylation pattern as well as the machinery responsible for its maintenance (see Fig. 1 for review of DNA methylation reactions). Since brain maturation doesn’t stop at birth and is responsive not only to innate signals (nature) but also to the environment (nurture) it is critical to determine whether the DNA methylation pattern is fixed at birth as had been the accepted dogma or whether it continues to change during early life. A related question is whether the DNA methylation machinery is responsive to environmental signals specifically the social environment, or whether it is solely defined by an innate program. If indeed environmental signals could affect DNA methylation patterns, this could set life-long gene expression programs that could result in behavioral pathologies such as autism spectrum disorders.

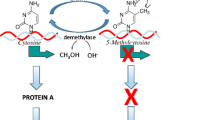
DNA methylation reactions. DNA methyltransferases transfer methyl groups (CH 3 ) from the methyl donor S-adenosyl-l-methionine to cytosines in DNA. Two kinds of DNA methylation reactions are shown; de novo methyltransferases (DNMT3a and DNMT3b) add new methyl groups to cytosines in DNA, maintenance DNA methyltransferase (DNMT1) copies the DNA methylation pattern from the template strand following DNA replication. Passive demethylation: if DNMT1 is absent during DNA replication, the DNA will be copied without being methylated and will be thus “passively” demethylated. DNA could be actively demethylated by demethylases that remove the methyl groups from DNA in the absence of DNA replication (see Fig. 2)
DNA methyltransferases
The DNA methylation reaction is catalyzed by DNA methyltransferase(s) (DNMT) (Razin and Cedar 1977). Methylation of DNA occurs immediately after replication by a transfer of a methyl moiety from the donor S-adenosyl-l-methionine (SAM, AdoMet) in a reaction catalyzed by DNMT (Turnbull and Adams 1976). Three distinct phylogenic DNA methyltransferases were identified in mammals. DNMT1 shows preference for hemimethylated DNA in vitro, which is consistent with its role as a maintenance DNMT, whereas DNMT3A and DNMT3B methylate unmethylated and methylated DNA at an equal rate which is consistent with a de novo DNMT role (Okano et al. 1998) (Fig. 1). Two additional DNMT homologs were found; DNMT2, whose substrate and DNA methylation activity is unclear (Vilain et al. 1998) but was shown to methylate tRNA (Goll et al. 2006; Rai et al. 2007) and DNMT3L which is essential for the establishment of maternal genomic imprints but lacks key methyltransferase motifs, and is possibly a regulator of methylation rather than an enzyme that methylates DNA (Bourc’his et al. 2001; Bourc’his and Bestor 2004). Knockout mouse data indicate that DNMT1 is responsible for a majority of DNA methylation marks in the mouse genome (Li et al. 1992) as well as the human genome (Chen et al. 2007), whereas DNMT3A and DNMT3B are responsible for some but not all de novo methylation during development (Okano et al. 1999).
Razin and Riggs proposed that the DNA methylation pattern was accurately and automatically inherited during replication since maintenance DNMT could only methylate hemimethylated sites generated on the nascent DNA strand during replication of a methylated CG dinucleotide in the template strand (Fig. 1). DNA methylation was therefore proposed to be truly heritable by an automatic semiconservative mechanism (Razin and Riggs 1980).
It is becoming clear now, however, that DNMTs are targeted to specific sequences and that the targeting factors are required not only for generating the patterns of methylation but also for maintaining the pattern of DNA methylation. For example, ubiquitin-like, containing PHD and RING finger domains 1, also known as NP95 in mouse and ICBP90 in human is required for targeting DNMT1 to newly replicating hemimethylated DNA, (Bostick et al. 2007). DNMTs are found in complexes with other proteins that include other chromatin modifying proteins such as HDAC1 and HDAC2 (Fuks et al. 2000, 2001). The discovery that DNMT1 and other DNMTs are targeted to specific sites by chromatin modifying enzymes suggests that DNA methylation is not exclusively automatic and provides a mechanism for a targeted change in DNA methylation in response to activation of signaling pathways
DNA demethylases
For DNA methylation to act as a responsive signal after birth in nondividing neurons, it needs to be reversible in postmitotic cells like other biological signals (Ramchandani et al. 1999). It was originally believed that demethylation could occur only through a “passive” process of replication in absence of DNMT1 (Fig. 1). The most controversial issue in the DNA methylation field is the question of whether the DNA methylation reaction is reversible (Ramchandani et al. 1999). Several authors have provided evidence for replication-independent demethylation (Wilks et al. 1984; Szyf et al. 1995; Oswald et al. 2000; Lucarelli et al. 2001; Bruniquel and Schwartz 2003), and it has been shown that brain extracts are capable of demethylating “naked” DNA substrate in vitro (Mastronardi et al. 2007; Dong et al. 2008). The strongest evidence for dynamic methylation–demethylation comes from several studies showing active demethylation in postmitotic neurons (Weaver et al. 2004; Levenson et al. 2006; Miller and Sweatt 2007; Feng et al. 2010). Conditional knockout of DNMT1 in postmitotic neurons results in DNA demethylation suggesting the presence of demethylase activity in nondividing neurons (Feng et al. 2010).
The main open question is what is the mechanism of DNA demethylation (see Fig. 2 for review of DNA demethylation reactions). The most widely accepted mechanisms for active DNA demethylation involve DNA repair and replacement of the methylated cytosine nucleotide by an unmethylated cytosine nucleotide by either base or nucleotide excision repair. One proposed mechanism is removal of the methylated cytosine by a glycosylase activity; the abasic site is then repaired and replaced with an unmethylated cytosine (Razin et al. 1986; Jost 1993). A study proposed that DNMTs participate in demethylation by deamination of the methyl cytosine to thymidine creating a C/T mismatch, which is then corrected by a mismatch-repair mechanism (Kangaspeska et al. 2008). DNMTs were previously shown to deaminate 5-methylcytosines (Shen et al. 1992; Zingg et al. 1998) under conditions of low SAM. Growth arrest and DNA-damage-inducible, alpha (GADD45A), a DNA repair protein was proposed to participate in catalysis of active DNA demethylation by an unknown DNA repair-based mechanism (Barreto et al. 2007). However, this was disputed (Jin et al. 2008). A very recent study suggested that demethylation in zebra fish embryos involves a complex sequence of coupled enzymatic reactions; activation-induced cytidine deaminase (AID; which converts 5-meC to thymine), a G/T mismatch-specific thymine glycosylase methyl-CpG binding domain protein 4 and repair promoted by GADD45A (Rai et al. 2008). AID has been implicated in the global demethylation in mouse primordial germ cells as well (Popp et al. 2010).

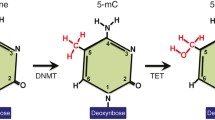
Mechanisms of active DNA demethylation. Several demethylation reactions were suggested. Direct demethylation by a demethylase enzyme (dMTase; MBD2 is a putative candidate) could release a methyl moiety (CH 3 ) in the form of either methanol or formaldehyde. Alternatively, the methyl cytosine ring could be modified by either deamination catalyzed for example by AID or by the DNA methyltransferases (DNMT) which were shown to catalyze deamination of 5-methylcytidine in the absence of SAM or hydroxylation of the methyl moiety catalyzed by TET1. The modified base is then excised and repaired. Alternatively, the bond between the sugar and the base is cleaved (by glycosylases such as MBD4 or 5-methylcytosine glycosylase 5-MCDG) followed by repair. Repair proteins shown to be associated with demethylation were GADD45(a and b)
In contrast to these repair-based mechanisms, we have previously proposed that demethylation is truly a reversible reaction that involves removal of the methyl moiety rather than breaking the DNA and fixing it with an unmethylated cytosine (Ramchandani et al. 1999). We proposed that the methylated DNA binding protein (MBD2) was a bona fide demethylase that removed methyl groups from DNA and truly reversed the DNA methylation reaction. This is to date the only described bona fide demethylase. MBD2 has been implicated in the activation of both methylated and unmethylated genes (Fujita et al. 2003; Angrisano et al. 2006). Several groups (Ng et al. 1999; Wade et al. 1999) have contested the demethylase and transcriptional activating properties of MBD2. Studies by Detich et al. have demonstrated MBD2 demethylase activity in vitro (Detich et al. 2002). Hamm et al. (Hamm et al. 2008) have proposed an oxidative mechanism of 5-cytosine DNA demethylation by MBD2. According to this mechanism, oxidation of the methyl moiety generates 5-hydroxymethylcytosine by oxidation, which is followed by release of the methyl residue in formaldehyde. Interestingly, 5-hydroxymethylcytosine was recently discovered in mammalian DNA (Pelizzola et al. 2008). A recent study has shown that TET1, an enzyme that converts methylcytosine to hydroxymethylcytosine, is required for maintaining the demethylated state of nanog in mouse ES cells supporting a possible role for TET1 and 5-hydroxymethylcysoine as an intermediary in the demethylation reaction (Ito et al. 2010). In summary, although there is no agreement as of yet on the mechanism of DNA demethylation, the presence of active demethylation in somatic cells is widely acknowledged.
DNA methylation as a genome adaptation mechanism and as a source of mental health disorders
Our new understanding of the DNA methylation and demethylating enzymes sets the stage for a life-long role for DNA methylation in modulation and fine tuning of DNA methylation patterns. It is also consistent with the idea that DNA methylation could be responsive to environmental signals and allow the genome to adapt its programs to signals from the social environment. It is interesting to note that there is evolutionary evidence for involvement of DNA methylation in sociality especially in the caste structure in the honeybee (Kucharski et al. 2008; Maleszka 2008; Lyko et al. 2010). It is possible that DNA methylation evolved to translate social information to genomic programs of social behavior, which is especially pertinent for autism. Epidemiological data points to the importance of early-life experience in setting life-long health and mental health trajectories in humans (Power et al. 2006). It is therefore hypothesized that early life is an especially sensitive time for DNA methylation adaptation in the brain and system wide. It is proposed that a DNA methylation genome-wide adaptation can turn “maladaptive” under specific contexts later in life and result in mental health pathology. It is predicted that an “adaptation” mechanism leading to pathology will involve several gene circuits and a system-wide adaptation of the DNA methylation pattern to an anticipated environment. This is consistent with the complex gene expression manifestation of many mental health disorders including autism. This mechanism suggests that mental health disabilities would emerge as a consequence of an organized fine tuning of DNA methylation patterns that is a response for cues coming from the early-life social environment. However, this hypothesis remains to be tested as it is unknown whether large changes in DNA methylation occur in the brains of individuals with autism.
An alternative mechanism is that stochastic DNA methylation errors in the germ line similar to single-nucleotide polymorphisms or DNA methylation errors limited to certain tissues would result in disruption of gene function leading to mental health pathology. The existence of stochastic differences in DNA methylation in the germ line and existence of such differences in DNA methylation even between identical twins were recently demonstrated (Kaminsky et al. 2006; Petronis 2006; Mill et al. 2008; Kaminsky et al. 2009; Flanagan et al. 2006). There are fundamental differences in the possible implications of an adaptive versus a stochastic mechanism of emergence of DNA methylation differences on prevention, prediction and treatment of behavioral pathologies. We will present several lines of evidence suggesting that the early social environment plays an active role in adaptations of the DNA methylation pattern in the brain and the immune system.
Epigenetic programming by maternal care in rodents
There are several models that measure the impact of early-life social environment on behavior and other health phenotypes later in life. Animal models could be used to test whether the impact of early-life social environment on the phenotype is mediated by “genetic” or “epigenetic” mechanisms. Maternal behavior plays a cardinal role in the behavioral development of mammals. Models of maternal deprivation in primates and rodents and natural variation in maternal care in rodents were used to demonstrate the profound impact of maternal care and “nurture” on a panel of phenotypes in the offspring that last into adulthood (Ruppenthal et al. 1976; Suomi et al. 1976).
Hippocampal Glucocorticoid receptor (GR) controls the negative feedback of the HPA axis by glucocorticoids. In the rat, the adult offspring of mothers that exhibit increased levels of pup licking/grooming (i.e., High LG mothers) over the first week of life show increased hippocampal (GR) expression, enhanced glucocorticoid feedback sensitivity, decreased hypothalamic corticotrophin releasing factor (CRF) expression and more modest HPA stress responses compared to animals reared by Low LG mothers (Liu et al. 1997; Francis et al. 1999). The GR/NR3C1 gene encoding the glucocorticoid receptor (GR exon 17 promoter) exhibits differences in DNA methylation and histone acetylation in the hippocampus of the offspring of high and low LG mothers. Differences in epigenetic programming in response to differences in maternal LG emerged early in life and remained stable into adulthood illustrating how epigenetic programming early in life could set up life-long behavioral trajectories (Weaver et al. 2004).
The basic concepts of this study were repeated more recently in several other models of early-life social adversity. Exposure of infant rats to stressed caretakers that displayed abusive behavior produced persisting changes in methylation of BDNF gene promoter in the adult prefrontal cortex (Roth et al. 2009). Early-life stress in mice caused sustained DNA hypomethylation of an important regulatory region of the arginine vasopressin (AVP) gene (Murgatroyd et al. 2009).
Evidence for DNA methylation variation associated with early-life adversity in humans
An extremely important question is whether the results in rodents could be translated to humans? The state of methylation of ribosomal RNA (rRNA) gene promoters and GR were examined a cohort of suicide victims in Quebec who were abused as children and their control group. rRNA forms the skeleton of the ribosome, the protein synthesis machinery. Protein synthesis is essential for building new memories and creating new synapses in the brain. Our genome contains around 400 copies of the genes encoding rRNA. One possible way to control the protein synthesis capacity of a cell is through changing the fraction of active rRNA alleles in a cell (Brown and Szyf 2007). We have previously shown that the fraction of rRNA genes that is active and is associated with the RNA Pol1 transcription machinery is unmethylated while the fraction that is inactive is methylated (Brown and Szyf 2007). Our results showed that the suicide victims who experienced childhood abuse had higher overall methylation in their rRNA genes and expressed less rRNA. This difference in methylation was region specific: it was present in the hippocampus and was not observed in the cerebellum. Moreover, although significant methylation differences were observed between the controls and the suicide victims, no sequence differences were observed. The fact that the difference in methylation was brain-region specific and that no sequence differences were observed further strengthens the conclusion that this difference in methylation was driven by environmental rather than genetic variation (McGowan et al. 2008). These data point to the possibility that the effects of early-life adversity might not be limited to the usual suspects of highly brain-specific genes but that ubiquitously expressed genes could be involved as well. Modulation of expression of ubiquitous genes might be important in modulating brain function.
Individuals with treatment-resistant forms of major depression show decreased GR expression and increased HPA activity. Site-specific differences in DNA methylation in the GR exon 1f promoter between suicide completers who had reported social adversity early in life and suicide completers who did not experience social adversity early in life were detected in this study. These differences in DNA methylation were associated with reduced expression of the GR gene (McGowan et al. 2009). Differences in DNA methylation of the GR promoter promoter were observed also in peripheral blood cells; the GR promoter was more methylated in lymphocytes in newborns exposed prenatally to maternal depression than control newborns (Oberlander et al. 2008). This lends support to the hypothesis that DNA methylation differences in response to social adversity are system wide and are not limited to brain-specific regions.
Epigenetic modulation of other candidate genes was implicated in suicide; the gamma-aminobutyric acid A receptor alpha 1 subunit promoter (Linthorst et al. 1995) within the frontopolar cortex (Poulter et al. 2008) and tropomyosin-related kinase B in the frontal cortex of suicide completers (Ernst et al. 2009). It is unknown yet whether these changes in DNA are also associated with early-life adversity.
Genome- and system-wide effects of early-life adversity
Genetics of complex disease has taught us that several genes must be involved. However, most of our studies were biased toward the candidate gene approach assuming that phenotype associated with early-life adversity would involve a few critical brain-specific genes. In addition, unsurprisingly the first line of studies examining the impact of early-life adversity on DNA methylation focused on specific brain regions. However, it is becoming clear that genes work in networks and that the total output of a network could be significantly affected by a combination of subtle changes in several nodes of a network. The fact that methylation differences in suicide victims were found in rRNA genes (McGowan et al. 2008) supports the idea that genes outside the usual brain-specific suspects are involved. Also it is clear, by looking at the multiple phenotypes associated with early-life adversity (Power et al. 2006) that the response to early-life adversity is not limited to the brain and that it is a system-wide response.
We recently performed a detailed mapping of five megabases of DNA spanning the locus of the GR gene from both directions and identified numerous differences in DNA methylation between the suicide and the control groups. Recent high-density epigenome mapping of chromosome 18 in the adult rat offspring of high and low maternal care reveals broad differences in DNA methylation and histone acetylation that cover wide regions of chromosome 18. High maternal care results in hypomethylation of some regions and hypermethylation of others and an inverse picture is observed with histone acetylation (McGowan et al. 2011). This can explain why the adult offspring of high and low LG mother exhibit wide spread differences in gene expression (Weaver et al. 2006).
We examined association between early-life adversity and differential DNA methylation in white blood cells. Our unpublished data indicates associations between DNA methylation patterns in 45-year-old adults and social adversity early in life (Borghol et al., unpublished). The emerging picture supports the hypothesis that early-life adversity results in modulation of DNA methylation of many functional gene circuitries resulting in a change in the total output of these circuitries. This could result in adaptation to environments later in life or maladaptation. The fact that DNA methylation changes in blood were associated with social adversity support the hypothesis that the response to early-life adversity is system wide and that it will be possible to follow DNA methylation changes that are relevant to behavioral pathologies in peripheral blood cells. However, it is unclear how the specific changes detected in blood correlate with specific changes in DNA methylation in the brain.
Signaling pathways as conduits between social exposures and DNA methylation changes
If indeed these reported changes in DNA methylation are adaptive mechanisms that respond to social cues there must be signaling pathways linking social exposure and DNA methylation and histone acetylation changes. A reasonable hypothesis is that exposure to social cues results in firing of a signaling cascade that activates transacting factors that deliver DNA and chromatin modifying enzymes to specific regulatory sequences of genes (Fig. 3). Evidence for such a mechanism comes from the rat maternal care model. Maternal behavior triggers a signaling pathway that involves the serotonin receptor, increase in cAMP, recruitment of the transcription factor nerve growth factor-induced protein A, which in turn recruits the histone acetyltransferase CREB binding protein (CBP), and the methylated DNA binding protein and candidate DNA demethylase MBD2 (Weaver et al. 2007) to the GR promoter. Our hypothesis is that the increased histone acetylation triggered by CBP or by other recruited histone acetyltransferases facilitates the demethylation of the gene by MBD2 or other DNA demethylases (Weaver et al., unpublished data). These data chart a possible conduit through which exposure to a social behavior such as maternal behavior results in epigenetic modification of a specific gene in the brain. Although it is certain that there are other molecular pathways that link social exposure and changes in DNA methylation in particular positions in the genome, this example provides evidence for the feasibility of transducing a social signal into a DNA methylation mark.

The early-life environment modulates the DNA methylation equilibrium; reversibility of DNA methylation. The DNA methylation equilibrium is laid down during embryogenesis by innate developmental programs. Increased methylation of promoters is associated with a hypoacetylated and poorly transcribed gene (right) and hypomethylated promoter is hyperacetylated and highly transcribed (left). A balance of DNA methylation and demethylation activities dynamically maintains this pattern and is attuned to signals from the early environment that can modulate the pattern through activation of signaling pathways that facilitate either increased demethylation or increased methylation. Two examples were previously reported; maternal licking and grooming increases serotonin firing increasing cAMP and activation of protein kinase A, which in turn activates NGFIA facilitating demethylation and histone acetylation of the GR exon 17 promoter. Early-life stress triggers activation of CAMKII kinase resulting in phosphorylation of MeCP2 (P). Phosphorylated MeCP2 facilitates demethylation and increased transcription of genes such as AVP. The DNA methylation balance set by early-life environment could be reversed during adulthood using epigenetic modifiers such as the histone deacetylase inhibitor TSA or methionine the precursor of the methyl donor SAM resulting in an epigenetic and behavioral reversal of the phenotype defined by maternal care. (AC histone acetylation, CH 3 DNA methylation)
A different signaling cascade linking social exposure to DNA demethylation provides a mechanism for how early-life stress results in persistent life-long hypomethylation of the AVP gene. The AVP promoter is methylated and bound by MECP2. Depolarization of hypothalamic neurons triggers phosphorylation of MeCp2 at Ser438 by calcium-dependent calmodulin kinase II (Murgatroyd et al. 2009). This phosphorylation converts MeCp2 from a transcriptional silencer with high affinity to methylated DNA into a transcriptional activator with low affinity to methylated DNA (Zhou et al. 2006). This facilitates demethylation of the AVP gene. Phosphorylation of MeCP2 in response to neuronal activation was shown before to facilitate demethylation of the BDNF promoter (Chen et al. 2003). This signaling pathway delineates a direct link between neuronal activation and the phosphorylation state of a protein interacting with methylated genes in the brain. Neuronal activation resulting in signaling through phosphorylation of proteins interacting with methylated DNA might be a general pathway that links social exposure and the activation of neurons. Future studies are required to map the signaling pathways that link early-life adversity to the DNA methylation/demethylation of gene circuitries in brain and T cells. MeCP2 is especially interesting in autism since deficiencies in MeCP2 were shown in ASD (Nagarajan et al. 2006). It will be interesting to map the downstream consequences of reduced MeCP2 on the methylome in ASD.
Reversibility of epigenetic programming
One of the most exciting implications of the epigenetic mechanism of behavioral disorders is the possibility of reversing maladaptive DNA methylation marks by either pharmacological agents (Szyf 1994, 2001) or social interventions (Szyf 2009b). Our line of reasoning presented in this review implies that social intervention could potentially be as effective as pharmacological intervention. If DNA methylation reaction is reversible as proposed here, then the DNA methylation pattern could be changed even in postmitotic tissues in either direction by manipulation of either DNA methylation/demethylation enzymes or chromatin modification enzymes (Fig. 3).
Our previous studies have shown that increasing histone acetylation using the HDAC inhibitor trichostatin A (TSA) facilitates replication-independent demethylation of non-replicating plasmid DNA in human cells (Cervoni and Szyf 2001; Cervoni et al. 2002). This experiment demonstrated that human somatic cells contain the enzymatic machinery required to demethylate DNA in the absence of DNA replication and that it is possible to alter the DNA methylation pattern using pharmacological agents that change histone acetylation (Cervoni and Szyf 2001; Cervoni et al. 2002). We tested whether a similar strategy would reverse epigenetic states established through maternal care and whether they would result in a change in the phenotype. Injection of the HDAC inhibitor TSA into the brains of adult offspring of low LG maternal care reversed the epigenetic programming of the GR exon 17 promoter and reestablished stress responsivity and open-field behavior that was indistinguishable from the offspring of high LG maternal care (Weaver et al. 2004).
We have previously shown that it is possible to alter the state of methylation of a non-replicating plasmid by treating cells with the methyl donor SAM which inhibited the DNA demethylation reaction (Detich et al. 2003.). Injection of the amino acid methionine, the precursor of SAM, into the brains of adult offspring of high maternal LG resulted in increased DNA methylation and downregulation of GR as well as heightened stress responsivity and an open-field behavior that was indistinguishable from the adult offspring of low maternal LG (Weaver et al. 2004, 2005). These data suggest that both the methylating and demethylating enzymes are present in the adult neuron and are amenable to pharmacological modulation. Although the number of DNA methylation and demethylation inhibitors is limited and they are extremely nonspecific, such agents have shown promise in cancer therapy (for review, see (Szyf 2009a)). Development of highly specific inhibitors of proteins from the DNA methylation machinery might revolutionize the pharmacological approach to mental health.
Summary and prospective
Autism demonstrates robust heritability suggesting a strong genetic component. However, our calculations of genetic heritability are based to a large extent on monozygotic twin studies. Monozygotic twins share not only genes but also environments and a common germ line DNA methylation pattern (Kaminsky et al. 2009). It is therefore possible that the genetic contribution to autism has been overestimated and the epigenetic component underestimated. The possibility that autism has an epigenetic basis that is driven by either stochastic drifts in DNA methylation, the perinatal environment or the early-life social environment must be investigated. Several studies point to an epigenetic basis for autism. RETT syndrome a mental retardation disorder that is similar in certain aspects with autistic spectrum disorders is caused by a deficiency in MeCP2 an epigenetic protein that binds methylated DNA and has several roles in interpretation of the DNA methylation pattern as well in controlling DNA methylation states (Amir et al. 1999). Alterations in DNA methylation in the promoter of MeCP2 were shown in autistic brains linking autism, DNA methylation and MeCP2 (Nagarajan et al. 2006). An attractive prospect is that an epigenetic molecular link will be unraveled between certain early-life social environments and autism.
One difficulty in exploring the epigenetic contribution to autism is that currently it is not possible to monitor in the brain of subjects with autism the tissue- and cell-type-specific changes that can occur in the epigenome. This is not a factor for de novo or inherited genetic mutations, which are identical in all cells and therefore can be monitored in any peripheral cell source. DNA methylation signatures are potential diagnostics and predictors of autism but if the changes in DNA methylation are limited to the brain they will have no diagnostic utility. Obviously, analysis of postmortem autistic brains will provide valuable information on the mechanisms involved but will have no diagnostic value. However, as discussed above there is evidence to suggest that some of the responses to early-life adversity are not limited to the brain. Therefore, it is tempting to speculate that it might be possible to delineate DNA methylation signatures of autism in peripheral blood cells. Although it is stands to reason that the DNA methylation signatures in T cells will not inform us about methylation changes in brain-specific genes, it is plausible that they will inform us about a systemic component of the epigenetic processes that contributed to the disorder. This possibility could only be tested in future studies that would directly examine whether there is a DNA methylation signature in T cells or other immune cell type that associates with autism. If indeed there is a DNA methylation signature of autism, a critical question is whether changes in immune cells DNA methylation is mechanistically related to the behavioral effects or whether these changes are parallel consequences of a system wide epigenetic response. Bilateral crosstalk between the immune system and the brain is well documented but any role in autism is speculative at this stage.
If indeed there is a systemic epigenetic defect that is associated with the disease, epigenetic intervention through changes in dietary methyl donor components, epigenetic drugs, or behavioral interventions might be effective. Peripheral DNA methylation marks in immune cells would allow monitoring of the response of the methylome to the therapy.
We raised in this review the possibility that disorders such as autism are caused by organized adaptive responses of the epigenome to early-life environment rather than a stochastic DNA methylation drift. Understanding the nature of these responses could provide guidance as to the link between the early environment and autism and direct possible preventative strategies.
In difference from genetic polymorphisms that are fixed and terminal, epigenetic changes are potentially reversible. One unexplored but nevertheless provocative possibility is that epigenetic or behavioral interventions that have epigenetic effects will serve as therapeutic alternatives for treating and reversing autism. Epigenetic markers might provide in the future guidance as well for non-pharmacological interventions. Although the evidence for the involvement of DNA methylation and autism is still sparse, the tremendous potential of such a mechanism calls for an intensive effort to test this hypothesis.
References
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2 [see comments]. Nat Genet. 1999;23:185–8.
Angrisano T, Lembo F, Pero R, Natale F, Fusco A, Avvedimento VE, et al. TACC3 mediates the association of MBD2 with histone acetyltransferases and relieves transcriptional repression of methylated promoters. Nucleic Acids Res. 2006;34:364–72.
Anney R et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19:4072–82.
Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–5.
Benvenisty N, Szyf M, Mencher D, Razin A, Reshef L. Tissue-specific hypomethylation and expression of rat phosphoenolpyruvate carboxykinase gene induced by in vivo treatment of fetuses and neonates with 5-azacytidine. Biochemistry. 1985;24:5015–9.
Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–4.
Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–9.
Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–9.
Brown SE, Szyf M. Epigenetic programming of the rRNA promoter by MBD3. Mol Cell Biol. 2007;27:4938–52.
Bruniquel D, Schwartz RH. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol. 2003;4:235–40.
Cervoni N, Szyf M. Demethylase activity is directed by histone acetylation. J Biol Chem. 2001;276:40778–87.
Cervoni N, Detich N, Seo SB, Chakravarti D, Szyf M. The oncoprotein Set/TAF-1beta, an inhibitor of histone acetyltransferase, inhibits active demethylation of DNA, integrating DNA methylation and transcriptional silencing. J Biol Chem. 2002;277:25026–31.
Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–9.
Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–6.
Comb M, Goodman HM. CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP−2. Nucleic Acids Res. 1990;18:3975–82.
Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol. 1988;140:2197–200.
D’Alessio AC, Szyf M. Epigenetic tete-a-tete: the bilateral relationship between chromatin modifications and DNA methylation. Biochem Cell Biol. 2006;84:463–76.
Detich N, Theberge J, Szyf M. Promoter-specific activation and demethylation by MBD2/demethylase. J Biol Chem. 2002;277:35791–4.
Detich N, Hamm S, Just G, Knox JD, Szyf M. The Methyl Donor S-Adenosylmethionine Inhibits Active Demethylation of DNA: A CANDIDATE NOVEL MECHANISM FOR THE PHARMACOLOGICAL EFFECTS OF S-ADENOSYLMETHIONINE. J Biol Chem. 2003;278:20812–20.
Dong E, Nelson M, Grayson DR, Costa E, Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci USA. 2008;105:13614–9.
Ernst C, Deleva V, Deng X, Sequeira A, Pomarenski A, Klempan T, et al. Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Arch Gen Psychiatry. 2009;66:22–32.
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92.
Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–30.
Flanagan JM, Popendikyte V, Pozdniakovaite N, Sobolev M, Assadzadeh A, Schumacher A, et al. Intra- and interindividual epigenetic variation in human germ cells. Am J Hum Genet. 2006;79:67–84.
Francis D, Diorio J, Liu D, Meaney MJ. Nongenomic transmission across generations of maternal behavior and stress responses in the rat. Science. 1999;286:1155–8.
Fujita H, Fujii R, Aratani S, Amano T, Fukamizu A, Nakajima T. Antithetic effects of MBD2a on gene regulation. Mol Cell Biol. 2003;23:2645–57.
Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24:88–91.
Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001;20:2536–44.
Fuso A, Ferraguti G, Grandoni F, Ruggeri R, Scarpa S, Strom R, et al. Early demethylation of non-CpG, CpC-rich, elements in the myogenin 5′-flanking region: a priming effect on the spreading of active demethylation. Cell Cycle. 2010;9:3965–76.
Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311:395–8.
Grice DE, Buxbaum JD. The genetics of autism spectrum disorders. Neuromolecular Med. 2006;8:451–60.
Gruenbaum Y, Stein R, Cedar H, Razin A. Methylation of CpG sequences in eukaryotic DNA. FEBS Lett. 1981;124:67–71.
Gruenbaum Y, Cedar H, Razin A. Substrate and sequence specificity of a eukaryotic DNA methylase. Nature. 1982;295:620–2.
Hamm S, Just G, Lacoste N, Moitessier N, Szyf M, Mamer O. On the mechanism of demethylation of 5-methylcytosine in DNA. Bioorg Med Chem Lett. 2008;18:1046–9.
Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–3.
Inamdar NM, Ehrlich KC, Ehrlich M. CpG methylation inhibits binding of several sequence-specific DNA- binding proteins from pea, wheat, soybean and cauliflower. Plant Mol Biol. 1991;17:111–23.
Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33.
Jin SG, Guo C, Pfeifer GP. GADD45A does not promote DNA demethylation. PLoS Genet. 2008;4:e1000013.
Jost JP. Nuclear extracts of chicken embryos promote an active demethylation of DNA by excision repair of 5-methyldeoxycytidine. Proc Natl Acad Sci USA. 1993;90:4684–8.
Kaminsky Z, Wang SC, Petronis A. Complex disease, gender and epigenetics. Ann Med. 2006;38:530–44.
Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–5.
Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–5.
Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30.
Kucharski R, Maleszka J, Foret S, Maleszka R. Nutritional control of reproductive status in honeybees via DNA methylation. Science. 2008;319:1827–30.
Kumar RA, Christian SL. Genetics of autism spectrum disorders. Curr Neurol Neurosci Rep. 2009;9:188–97.
Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–73.
Levitt P, Campbell DB. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest. 2009;119:747–54.
Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26.
Linthorst AC, Flachskamm C, Muller-Preuss P, Holsboer F, Reul JM. Effect of bacterial endotoxin and interleukin-1 beta on hippocampal serotonergic neurotransmission, behavioral activity, and free corticosterone levels: an in vivo microdialysis study. J Neurosci. 1995;15:2920–34.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22.
Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, et al. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997;277:1659–62.
Lucarelli M, Fuso A, Strom R, Scarpa S. The dynamics of myogenin site-specific demethylation is strongly correlated with its expression and with muscle differentiation. J Biol Chem. 2001;276:7500–6.
Lyko F, Foret S, Kucharski R, Wolf S, Falckenhayn C, Maleszka R. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 2010;8:e1000506.
Maleszka R. Epigenetic integration of environmental and genomic signals in honey bees: the critical interplay of nutritional, brain and reproductive networks. Epigenetics. 2008;3:188–92.
Mastronardi FG, Noor A, Wood DD, Paton T, Moscarello MA. Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J Neurosci Res. 2007;85:2006–16.
McGowan PO, Sasaki A, Huang TC, Unterberger A, Suderman M, Ernst C, et al. Promoter-wide hypermethylation of the ribosomal RNA gene promoter in the suicide brain. PLoS ONE. 2008;3:e2085.
McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–8.
McGowan PO, Suderman M, Sasaki A, Huang TCT, Hallett M, et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS ONE. 2011;6(2):e14739. doi:10.1371/journal.pone.0014739.
Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711.
Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–69.
Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmuhl Y, Fischer D, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12:1559–66.
Nagarajan RP, Hogart AR, Gwye Y, Martin MR, Lasalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:172–82.
Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–81.
Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex [see comments]. Nat Genet. 1999;23:58–61.
Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106.
Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases [letter]. Nat Genet. 1998;19:219–20.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57.
Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–8.
Pelizzola M, Koga Y, Urban AE, Krauthammer M, Weissman S, Halaban R, et al. MEDME: an experimental and analytical methodology for the estimation of DNA methylation levels based on microarray derived MeDIP-enrichment. Genome Res. 2008;18:1652–9.
Petronis A. Epigenetics and twins: three variations on the theme. Trends Genet. 2006;22:347–50.
Pinto D et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72.
Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–5.
Poulter MO, Du L, Weaver IC, Palkovits M, Faludi G, Merali Z, et al. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biol Psychiatry. 2008;64:645–52.
Power C, Jefferis BJ, Manor O, Hertzman C. The influence of birth weight and socioeconomic position on cognitive development: does the early home and learning environment modify their effects? J Pediatr. 2006;148:54–61.
Rai K, Chidester S, Zavala CV, Manos EJ, James SR, Karpf AR, et al. Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Genes Dev. 2007;21:261–6.
Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–12.
Ramchandani S, Bhattacharya SK, Cervoni N, Szyf M. DNA methylation is a reversible biological signal. Proc Natl Acad Sci USA. 1999;96:6107–12.
Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci USA. 2009;106:671–8.
Razin A, Cedar H. Distribution of 5-methylcytosine in chromatin. Proc Natl Acad Sci USA. 1977;74:2725–8.
Razin A, Riggs AD. DNA methylation and gene function. Science. 1980;210:604–10.
Razin A, Szyf M. DNA methylation patterns. Formation and function. Biochim Biophys Acta. 1984;782:331–42.
Razin A, Szyf M, Kafri T, Roll M, Giloh H, Scarpa S, et al. Replacement of 5-methylcytosine by cytosine: a possible mechanism for transient DNA demethylation during differentiation. Proc Natl Acad Sci USA. 1986;83:2827–31.
Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. 2009;65:760–9.
Ruppenthal GC, Arling GL, Harlow HF, Sackett GP, Suomi SJ. A 10-year perspective of motherless-mother monkey behavior. J Abnorm Psychol. 1976;85:341–9.
Shen JC, Rideout WMd, Jones PA. High frequency mutagenesis by a DNA methyltransferase. Cell. 1992;71:1073–80.
Stein R, Razin A, Cedar H. In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells. Proc Natl Acad Sci USA. 1982;79:3418–22.
Suomi SJ, Collins ML, Harlow HF, Ruppenthal GC. Effects of maternal and peer separations on young monkeys. J Child Psychol Psychiatry. 1976;17:101–12.
Szyf M. DNA methylation properties: consequences for pharmacology. Trends Pharmacol Sci. 1994;15:233–8.
Szyf M. Towards a pharmacology of DNA methylation. Trends Pharmacol Sci. 2001;22:350–4.
Szyf M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol. 2009a;49:243–63.
Szyf M. The early life environment and the epigenome. Biochim Biophys Acta. 2009b;1790:878–85.
Szyf M. A dynamic methylome; implications of non-CG methylation/demethylation. Cell Cycle. 2010;9:3846–7.
Szyf M, Theberge J, Bozovic V. Ras induces a general DNA demethylation activity in mouse embryonal P19 cells. J Biol Chem. 1995;270:12690–6.
Szyf M, McGowan P, Meaney MJ. The social environment and the epigenome. Environ Mol Mutagen. 2007;49:46–60.
Turnbull JF, Adams RL. DNA methylase: purification from ascites cells and the effect of various DNA substrates on its activity. Nucleic Acids Res. 1976;3:677–95.
Vilain A, Apiou F, Dutrillaux B, Malfoy B. Assignment of candidate DNA methyltransferase gene (DNMT2) to human chromosome band 10p15.1 by in situ hybridization. Cytogenet Cell Genet. 1998;82:120.
Waddington CH. Canalization of development and genetic assimilation of acquired characters. Nature. 1959;183:1654–5.
Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation [see comments]. Nat Genet. 1999;23:62–6.
Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–54.
Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, et al. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci. 2005;25:11045–54.
Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci USA. 2006;103:3480–5.
Weaver IC, D’Alessio AC, Brown SE, Hellstrom IC, Dymov S, Sharma S, et al. The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J Neurosci. 2007;27:1756–68.
Weiss LA. Autism genetics: emerging data from genome-wide copy-number and single nucleotide polymorphism scans. Expert Rev Mol Diagn. 2009;9:795–803.
Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–8.
Wilks A, Seldran M, Jost JP. An estrogen-dependent demethylation at the 5′ end of the chicken vitellogenin gene is independent of DNA synthesis. Nucleic Acids Res. 1984;12:1163–77.
Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites [see comments]. Trends Genet. 1997;13:335–40.
Yung RL, Richardson BC. Role of T cell DNA methylation in lupus syndromes. Lupus. 1994;3:487–91.
Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–69.
Zingg JM, Shen JC, Jones PA. Enzyme-mediated cytosine deamination by the bacterial methyltransferase M.MspI. Biochem J. 1998;332:223–30.
Acknowledgments
Studies from MS lab discussed here were supported by grants from the Canadian Institutes of Health Research the National Cancer Institute of Canada, The Sackler program in psychobiology and epigenetics at McGill University, the National Institute of Child Health and Disease (USA). MS is a fellow of the Canadian Institute for Advanced Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Szyf, M. DNA methylation, the early-life social environment and behavioral disorders. J Neurodevelop Disord 3, 238–249 (2011). https://doi.org/10.1007/s11689-011-9079-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11689-011-9079-2