
Abstract
Autism is an umbrella diagnosis with several different etiologies. Fragile X syndrome (FXS), one of the first identified and leading causes of autism, has been modeled in mice using molecular genetic manipulation. These Fmr1 knockout mice have recently been used to identify a new putative therapeutic target, the metabotropic glutamate receptor 5 (mGluR5), for the treatment of FXS. Moreover, mGluR5 signaling cascades interact with a number of synaptic proteins, many of which have been implicated in autism, raising the possibility that therapeutic targets identified for FXS may have efficacy in treating multiple other causes of autism.
Similar content being viewed by others

Introduction
Leo Kanner first described autism in 1943 [1]. It wasn’t until 1980 however, that autism was formally recognized in the Diagnostic and Statistical Manual of Mental disorders (DSM-III), and included as part of a new class, the Pervasive Developmental Disorders (PDD) [2]. At the same time, early psychodynamic theories of the etiology of autism [3] were being abandoned in favor of genetic ones. As early as 1975, case reports of monozygotic twins concordant for autism [4], followed by several systematic twin studies [5–10] substantiated the strong heritability of autism [11–13].
Standardization of diagnostic criteria [2], and improvements in our ability to reliably detect chromosomal abnormalities [14] allowed for the identification in the early 1980’s of the first genetic cause of autism—Fragile X syndrome (FXS) [15–17]. Subsequently, the Fragile X gene (FMR1) was discovered [18], and by 1994 the first animal model became available [19]. This genetically engineered Fmr1 knockout mouse (Fmr1 KO), has been validated for FXS, and is currently one of the leading animal models of autism [20].
Using this mutant mouse, we have been able to address the role of the FMR1 gene and the protein it encodes (fragile X mental retardation protein, FMRP) in brain development. Now, over 25 years since FXS was identified as a cause of autism, a new putative therapy has been proposed based on our understanding of the function of FMRP.
Modeling autism: a derailment of synaptic plasticity
Inherited mutations have the potential to disrupt brain development from the moment of fertilization onward; however, a genetic etiology does not preclude pathogenesis involving regulated processes later in development. Symptoms of autism typically present during the early postnatal period, usually between ages 1–3 years [20]. This epoch, the so-called ‘critical period’ [21], corresponds to a dynamic phase of brain development in which neurite outgrowth, maturation of inhibition and signaling, axon myelination, and synaptic plasticity are set in motion by the complex interplay of molecular genetic programs and experience [22]. Disruption of any of one of these processes could hypothetically lead to the characteristic symptoms of autism, which include abnormal social interaction and communication, stereotyped repetitive behaviors, often with co-morbid mental retardation, epilepsy, sleep disturbances, attention deficit and hyperactivity [23]. Thus, it has been tempting to speculate that the pathogenesis of autism involves a derailment of at least one of these developmental processes [24–26]. Given this framework, studies of synaptic plasticity in the Fmr1 KO mouse have been an obvious priority.
A potential breakthrough in understanding the pathogenesis of fragile X came from studies of group 1 metabotropic glutamate receptors (Gp1 mGluR) [27–31]. Gp1 mGluRs (which are further subdivided into mGluR1 and mGluR5 subtypes) couple to postsynaptic Gq-like G-proteins and phospholipase C (PLC) [32] as well as to extracellular signal-regulated kinase (ERK) transduction pathways [33, 34]. Their activation leads to the synthesis of new protein at the synapse [28, 35, 36], likely through the ERK signaling cascade [37, 38]. A functional consequence of Gp 1 mGluR-dependent protein synthesis in the hippocampus is long-term depression (LTD), a form of synaptic plasticity [29]. In the Fmr1 KO mouse, this mGluR-LTD is exaggerated and no longer protein synthesis-dependent [31, 39].
Meanwhile, studies of FMRP revealed that the expression of the protein is developmentally regulated [40, 41], such that in the post-natal brain it is largely cytoplasmic [42, 43], predominantly expressed in neurons [44, 45] and enriched postsynaptically at glutamatergic synapses [46]. Furthermore, FMRP is an RNA binding protein that co-localizes with polyribosomes [44, 47–55] which are found at the base of dendritic spines where they are thought to mediate local translational control of the synapse [56]. Indeed, both in vitro and in vivo metabolic labeling studies have now directly shown that FMRP functions as a repressor of protein synthesis [57–60].
Taken together, these findings led to the hypothesis that Gp1 mGluRs and FMRP might work in functional opposition to regulate mRNA translation at the synapse, and that in the absence of FMRP, unchecked mGluR-dependent protein synthesis leads to the pathogenesis of the disease (Fig. 1) [61]. We have recently tested this so-called ‘mGluR theory’ and shown that increased levels of protein synthesis in the Fmr1 KO mouse [59, 60], are restored to wild type (WT) levels by selective reduction of mGluR5 signaling [60]. This manipulation also significantly decreases the magnitude of Gp1 mGluR-LTD in Fmr1 KO mice, confirming the role of mGluR5 in producing the exaggerated synaptic plasticity phenotype [60].

Opponent regulation of protein synthesis by FMRP and GpI mGluRs. FMRP is a negative regulator of translation at the synapse. Stimulation of GpI mGluRs with DHPG leads to the synthesis of proteins. Furthermore, many of the long-term consequences of Gp1 mGluR activation are protein synthesis dependent. The mGluR theory posits that in the absence of FMRP, as is the case in Fragile X syndrome, this balance between FMRP and Gp1 mGluRs is lost, and unchecked protein synthesis at the synapse leads to the characteristic features of the disease. Furthermore, this balance could be restored by reducing Gp1 mGluR activity at the synapse, by either knockdown or pharmacological blockade of the receptor. The therapeutic implication of the theory is that symptoms of FXS syndrome could be corrected by appropriate modulation of GpI mGluR signaling
The synapse is too small to be directly visualized by light microscopy. However, dendritic spines (the postsynaptic half of an excitatory synapse) can be visualized, and are used to estimate the number of excitatory synapses in the brain. Dendritic spines are highly modifiable structures, and changes in spine density and morphology have been correlated with synaptic plasticity [62]. Furthermore, abnormalities in dendritic spine morphology have long been associated with human mental retardation of unknown etiology [63], as well as with XLMR (x-linked mental retardation) [64], Down [65], Patau [65], Rett [66] and Fragile X syndromes [67, 68].
Dendritic spine structure is regulated by Gp1 mGluRs. Application of the selective mGluR5 agonist, DHPG, to cultured hippocampal neurons induces a protein synthesis dependent increase in the density of long thin spines [69]. Because DHPG application in cell culture also induces rapid protein synthesis dependent internalization of AMPA and NMDA receptors [70], receptor internalization may be the prelude to morphologic remodeling in response to plasticity inducing stimuli.
This response to stimulation with DHPG parallels spine changes seen in the Fmr1 KO mice, which lent support to the theory that exaggerated signaling through mGluR5 in the absence of FMRP could account for this morphologic correlate of synaptic plasticity [61]. Consistent with this idea, recent studies have shown that that AMPA receptor internalization is exaggerated in the absence of FMRP [71] and both this and the increased spine density phenotype seen in Fmr1 KO mice [60, 72–78] are rescued by selective reduction in mGluR5 signaling [60, 71].
Modeling autism: plasticity in vivo
While these in vitro and ex vivo demonstrations of opponent regulation by FMRP and mGluR5 provided the necessary foundation for identifying and correcting synaptic abnormalities, we also wanted to determine whether these interactions regulate circuit-level responses in the intact animal. Landmark studies of in vivo ocular dominance plasticity (ODP) in monkeys and cats [79–81] established a role for experience dependent plasticity in shaping the circuitry of the brain during the critical period. Moreover, because ODP occurs on the biologically relevant timescale, in response to perturbations of environmental stimuli using intrinsic patterns of neuronal activity, this paradigm is more readily translated to future studies in human patients (e.g. using visually evoked potentials [82] or transcranial magnetic stimulation [83]).
The development of transgenic technologies [19, 84, 85] and adaptation of the ODP paradigm to rodents [86–91] has allowed us to answer mechanistic questions about experience dependent plasticity in vivo. For example, ODP is in-part mGluR5 dependent [60], requires protein synthesis [92], and signals through ERK transduction [93]. In the Fmr1 KO mouse, this plasticity is exaggerated, such that bidirectional modifications that require 7 days of monocular deprivation (MD) in WT mice [91], occur after only 3 days in the absence of FMRP [60]. Significantly this hyper-plastic response is reminiscent of the exaggerated synaptic plasticity phenotype seen in the hippocampal slice [31], and is likewise restored to WT levels by 50% reduction of mGluR5 signaling [60].
Modeling autism: behavioral phenotypes
As mentioned above, epilepsy and mental retardation are both co-morbid features of autism [23]—an estimated 5–38% of autistic patients have seizure or subclinical epileptiform activity [94] while 70% have cognitive impairment [95, 96](but see, [97][98]). Thus, an important goal for modeling the disease is to establish behavioral tasks that recapitulate these symptoms in the Fmr1 KO mouse.
An estimated 20% of human patients with FXS have epileptiform activity or generalized seizure [99, 100]. Audiogenic seizure (AGS) is a robust paradigm for inducing seizure in the Fmr1 KO [60, 101–105] and recapitulates this neurologic feature of FXS and autism. Previous studies have not been able to account for increased epileptiform activity in Fmr1-KO mice by any of the anticipated mechanisms. For example, no differences have been observed between WT and Fmr1-KO mice in basal synaptic transmission, excitability, paired pulse facilitation, and long-term potentiation in the CA1 region of the hippocampus [106, 107].
Interestingly, it has been shown that agonists of group I mGluRs act as convulsants in rodents [32, 108] while selective Gp I mGluR antagonists block seizures in a range of rodent models of epilepsy [105, 109, 110]. Increases in epileptiform activity in response to mGluR5 stimulation are protein synthesis dependent [111, 112], suggesting that in addition to synapse specific changes, circuit level modulation of excitability is sensitive to the state of mGluR5 dependent protein synthesis [113]. Consistent with this idea, AGS seen in the Fmr1 KO is attenuated by 50% reduction of mGluR5 signaling [60].
Despite the moderate to severe mental retardation seen in human patients with FXS [114], cognitive phenotypes in the Fmr1 KO mice have been difficult to model [107, 115–117]. Inhibitory avoidance (IA) is a contextual (fear) conditioning paradigm used in animals to test hippocampus-based associative learning and memory [118]. IA extinction (IAE) is a paradigm that tests those conditioned responses in the face of contradictory contextual (safe) conditioning [119]. While IA learning is normal in Fmr1 KO mice on the C57-Bl6 background [19, 60], we have recently identified an IAE phenotype in the Fmr1 KO [60].
Although the synaptic mechanisms underlying IAE are not currently known, this behavior, like mGluR5-LTD, is protein synthesis dependent [119]. Furthermore, since both mGluR5-LTD and IAE are exaggerated in the Fmr1 KO mice and rescued by reduction of mGluR5 signaling [60], one interesting possibility is that mGluR5 LTD is the cellular mechanism subserving IAE learning. This mechanism is likely distinct from that which subserves IA, since IA training induces NMDA-LTP [120]and neither IA nor NMDA-LTP [106, 107] is disrupted in the Fmr1 KO on the C57-Bl6 background.
Therapeutic implications
In summary, we have discovered that FMRP is a protein that acts to regulate protein synthesis and synaptic plasticity triggered by Gp1 mGluRs. Understanding this balance between FMRP and mGluR-5 has allowed us to restore normal function in the Fmr1 KO model of autism—metabolic, morphologic, synaptic, circuit, and behavioral disruptions can all be corrected by reducing mGluR5 signaling by 50% [60]. Currently clinical trials based these and related findings are under way to determine safety and efficacy of mGluR modifying drugs in human patients with FXS and autism.
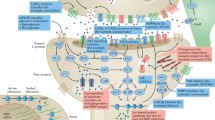
To put these findings in context, it is important to remember that mGluRs and FMRP do not exist in isolation at the synapse. As shown in Fig. 2, a number of other synaptic proteins that interact with the mGluRs either by direct physical contact or biochemical cascades, have also been identified as autism candidate genes [121–126] or single gene disorders associated with autism [127–134].

Autism as a synapsopathy. mGluR5 interacts with a number of postsynaptic proteins. Some of these have been identified as autism candidate genes (shown in purple; HOMER, SHANK, Neuroligin, Neurexin); others are proteins associated with single gene causes of autism (shown in red: FMRP/FXS, TSC/Tuberous Sclerosis, PTEN/ Hamartoma syndrome, MeCP2/Rett syndrome, E3A/Angelman’s syndrome)
For example, Gp1 mGluR signaling converges on transduction cascades also implicated in PTEN hamartoma syndrome and Tuberous sclerosis complex (TSC), which are other single gene causes of autism. PTEN inhibits PI3K-dependent signaling, which couples Gq signaling to the mTOR/S6K pathway for protein synthesis [128]. TSC 1/2 inhibits this same mTOR pathway, by acting as a GTPase-activating protein for the Ras-related small G protein Rheb [135].
Structural proteins within the synapse also interconnect Gp1 mGluRs to various autism candidate genes. For example, both Shank and Homer proteins crosslink mGluR5 to the postsynaptic density [136], and misregulated Homer1b and PSD-95 have been implicated in the pathogenesis of FXS [137, 138]. The Neuroligin/Neurexin complex, important for synapse formation and implicated in autism, is in turn tethered to the synapse via its interaction with PSD-95 [125]. AlphaCaMKII, a major regulatory protein in synaptic plasticity [139] is also tethered to the synapse by PSD-95; absence of inhibitory phosphorylation of alphaCaMKII by UBE3a, has been implicated in Angelman syndrome [132]. Interestingly, mGluR5 stimulated protein synthesis of alphaCaMKII and PSD-95 are impaired in synaptoneurosomes from Fmr1 KO mice [140]. Furthermore, CAMKII dependent phosphorylation of MeCP2 links these synaptic proteins to Rett syndrome, another single gene disorder associated with autism, and transcriptional regulation of brain derived nerve growth factor (BDNF) [141]. In turn, TrkB mediated BDNF signals through ERK, regulates dendritic spine formation [142], and has also been implicated in the pathogenesis of FXS [143].
Together, these results suggest it may be useful to think of autism as a synapsopathy [144]—a disease where disruption of the synapse during development produces a common clinical picture, despite a heterogeneity of interconnected causes. It also raises the interesting possibility that treatments for one cause, such as fragile X, may have efficacy in treating other causes of autism.
References
Kanner L. Autistic disturbances of affective contact. Nerv Child. 1943;2:217–50.
Lotspeich L. Autism and pervasive developmental disorders. In: Bloom F, Kupfer D, editors. Psychopharmacology: The fourth generation of progress, Vol. 4. Nashville: American College of Neuropsychopharmacology; 2000.
Bettelheim B. The empty fortress: Infantile autism and the birth of the self. New York: Free; 1967.
McQuaid PE. Infantile autism in twins. Br J Psychiatry. 1975;127:530–4.
Folstein S, Rutter M. Genetic influences and infantile autism. Nature. 1977;265(5596):726–8.
Steffenburg S, et al. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30(3):405–16.
Bailey A, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25(1):63–77.
Barton M, Volkmar F. How commonly are known medical conditions associated with autism? J Autism Dev Disord. 1998;28(4):273–8.
Trottier G, Srivastava L, Walker CD. Etiology of infantile autism: a review of recent advances in genetic and neurobiological research. J Psychiatry Neurosci. 1999;24(2):103–15.
McCauley JL, et al. Genome-wide and Ordered-Subset linkage analyses provide support for autism loci on 17q and 19p with evidence of phenotypic and interlocus genetic correlates. BMC Med Genet. 2005;6:1.
For comparison, autism has an estimated heritability of 0.9while the heritability of breast cancer is only 0.27—where 1 equals population variance exclusively due to genetics and 0 equals population variance exclusively due to environmental factors [12].
Freitag CM. The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007;12(1):2–22.
Lichtenstein P, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85.
Sutherland GR. Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science. 1977;197(4300):265–6.
Brown WT, et al. Association of fragile X syndrome with autism. Lancet. 1982;1(8263):100.
August GJ. A genetic marker associated with infantile autism. Am J Psychiatry. 1983;140(6):813.
Brown WT, et al. Fragile X and autism: a multicenter survey. Am J Med Genet. 1986;23(1–2):341–52.
Verkerk AJ, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–14.
Consortium TD-BFX. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78(1):23–33.
Bernardet M, Crusio WE. Fmr1 KO mice as a possible model of autistic features. ScientificWorldJournal. 2006;6:1164–76.
Armstrong VL. What is so critical?: a commentary on the reexamination of critical periods. Dev Psychobiol. 2006;48(4):326–31. discussion 332-6.
Rice D, Barone S Jr. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–33.
Association AP. Diagnostic and statistical manual of mental disorders IV-TR. 4th ed. Washington, DC: American Psychiatric Association; 2000.
Hughes JR. Autism: the first firm finding = underconnectivity? Epilepsy Behav. 2007;11(1):20–4.
Polleux F, Lauder JM. Toward a developmental neurobiology of autism. Ment Retard Dev Disabil Res Rev. 2004;10(4):303–17.
Courchesne E. Brain development in autism: early overgrowth followed by premature arrest of growth. Ment Retard Dev Disabil Res Rev. 2004;10(2):106–11.
Dudek SM, Bear MF. A biochemical correlate of the critical period for synaptic modification in the visual cortex. Science. 1989;246(4930):673–5.
Weiler IJ, Greenough WT. Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci U S A. 1993;90(15):7168–71.
Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288(5469):1254–7.
Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis–dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86(1):321–5.
Huber KM, et al. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99(11):7746–50.
Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–37.
Thandi S, Blank JL, Challiss RA. Group-I metabotropic glutamate receptors, mGlu1a and mGlu5a, couple to extracellular signal-regulated kinase (ERK) activation via distinct, but overlapping, signalling pathways. J Neurochem. 2002;83(5):1139–53.
Mao L, et al. Role of protein phosphatase 2A in mGluR5-regulated MEK/ERK phosphorylation in neurons. J Biol Chem. 2005;280(13):12602–10.
Weiler IJ, et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94(10):5395–400.
Job C, Eberwine J. Identification of sites for exponential translation in living dendrites. Proc Natl Acad Sci U S A. 2001;98(23):13037–42.
Gallagher SM, et al. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24(20):4859–64.
Banko JL, et al. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26(8):2167–73.
Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95(5):3291–5.
Abitbol M, et al. Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet. 1993;4(2):147–53.
Agulhon C, et al. Expression of FMR1, FXR1, and FXR2 genes in human prenatal tissues. J Neuropathol Exp Neurol. 1999;58(8):867–80.
Devys D, et al. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4(4):335–40.
Verheij C, et al. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature. 1993;363(6431):722–4.
Feng Y, et al. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17(5):1539–47.
Bakker CE, et al. Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse. Exp Cell Res. 2000;258(1):162–70.
Antar LN, et al. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24(11):2648–55.
Aschrafi A, et al. The fragile X mental retardation protein and group I metabotropic glutamate receptors regulate levels of mRNA granules in brain. Proc Natl Acad Sci U S A. 2005;102(6):2180–5.
Brown V, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107(4):477–87.
Zalfa F, Achsel T, Bagni C. mRNPs, polysomes or granules: FMRP in neuronal protein synthesis. Curr Opin Neurobiol. 2006;16(3):265–9.
Siomi MC, et al. Casein kinase II phosphorylates the fragile X mental retardation protein and modulates its biological properties. Mol Cell Biol. 2002;22(24):8438–47.
Schenck A, et al. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci U S A. 2001;98(15):8844–9.
Darnell JC, et al. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell. 2001;107(4):489–99.
Stefani G, et al. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci. 2004;24(33):7272–6.
Feng Y, et al. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1(1):109–18.
Khandjian EW, et al. A heterogeneous set of FMR1 proteins is widely distributed in mouse tissues and is modulated in cell culture. Hum Mol Genet. 1995;4(5):783–9.
Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2001;24:299–325.
Laggerbauer B, et al. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10(4):329–38.
Li Z, et al. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29(11):2276–83.
Qin M, et al. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005;25(20):5087–95.
Dolen G, et al. Correction of Fragile X Syndrome in Mice. Neuron. 2007;56(6):955–962.
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–7.
Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399(6731):66–70.
Marin-Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome. A Golgi study. J Comp Neurol. 1976;167(1):63–81.
Hayashi ML, et al. Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron. 2004;42(5):773–87.
Marin-Padilla M. Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res. 1972;44(2):625–9.
Belichenko PV, et al. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5(12):1509–13.
Irwin SA, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98(2):161–7.
Hinton VJ, et al. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41(3):289–94.
Vanderklish PW, Edelman GM. Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2002;99(3):1639–44.
Snyder EM, et al. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4(11):1079–85.
Nakamoto M, et al. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007;104(39):15537–42.
Comery TA, et al. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94(10):5401–4.
Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10(10):1038–44.
Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21(14):5139–46.
Irwin SA, et al. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet. 2002;111(2):140–6.
McKinney BC, et al. Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am J Med Genet B Neuropsychiatr Genet. 2005;136(1):98–102.
Galvez R, Greenough WT. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A. 2005;135(2):155–60.
Grossman AW, et al. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084(1):158–64.
Hubel DH, Wiesel TN. Effects of Monocular Deprivation in Kittens. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1964;248:492–7.
Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol. 1970;206(2):419–36.
Hubel DH, Wiesel TN, LeVay S. Plasticity of ocular dominance columns in monkey striate cortex. Philos Trans R Soc Lond B Biol Sci. 1977;278(961):377–409.
Normann C, et al. Long-term plasticity of visually evoked potentials in humans is altered in major depression. Biol Psychiatry. 2007;62(5):373–80.
Antal A, Nitsche MA, Paulus W. Transcranial direct current stimulation and the visual cortex. Brain Res Bull. 2006;68(6):459–63.
Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336(6197):348–52.
Lu YM, et al. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17(13):5196–205.
Drager UC. Observations on monocular deprivation in mice. J Neurophysiol. 1978;41(1):28–42.
Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16(10):3274–86.
Porciatti V, Pizzorusso T, Maffei L. The visual physiology of the wild type mouse determined with pattern VEPs. Vision Res. 1999;39(18):3071–81.
Sawtell NB, et al. NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron. 2003;38(6):977–85.
Prusky GT, Douglas RM. Developmental plasticity of mouse visual acuity. Eur J Neurosci. 2003;17(1):167–73.
Frenkel MY, Bear MF. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron. 2004;44(6):917–23.
Taha S, Stryker MP. Rapid ocular dominance plasticity requires cortical but not geniculate protein synthesis. Neuron. 2002;34(3):425–36.
Di Cristo G, et al. Requirement of ERK activation for visual cortical plasticity. Science. 2001;292(5525):2337–40.
Tuchman R, Rapin I. Epilepsy in autism. Lancet Neurol. 2002;1(6):352–8.
Fombonne E. The epidemiology of autism: a review. Psychol Med. 1999;29(4):769–86.
Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord. 2003;33(4):365–82.
In addition to autism, the PDD category (also called the autistic spectrum or ASD), includes less severe forms of autism without cognitive impairment, like Asperger’s disease; therefore estimates of relative rates of cognitive impairment are necessarily lower in ASD cohorts [98].
Kawamura Y, Takahashi O, Ishii T. Reevaluating the incidence of pervasive developmental disorders: impact of elevated rates of detection through implementation of an integrated system of screening in Toyota. Japan. Psychiatry Clin Neurosci. 2008;62(2):152–9.
Berry-Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44(11):724–8.
Musumeci SA, et al. Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia. 1999;40(8):1092–9.
Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103(4):1043–50.
Musumeci SA, et al. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia. 2000;41(1):19–23.
Qin M, Kang J, Smith CB. A null, mutation for Fmr1 in female mice: effects on regional cerebral metabolic rate for glucose and relationship to behavior. Neuroscience. 2005;135(3):999–1009.
Yan QJ, et al. A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav. 2004;3(6):337–59.
Yan QJ, et al. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49(7):1053–66.
Godfraind JM, et al. Long-term potentiation in the hippocampus of fragile X knockout mice. Am J Med Genet. 1996;64(2):246–51.
Paradee W, et al. Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience. 1999;94(1):185–92.
Tizzano JP, Griffey KI, Schoepp DD. Induction or protection of limbic seizures in mice by mGluR subtype selective agonists. Neuropharmacology. 1995;34(8):1063–7.
Thomsen C, et al. (S)-4-carboxy-3-hydroxyphenylglycine, an antagonist of metabotropic glutamate receptor (mGluR) 1a and an agonist of mGluR2, protects against audiogenic seizures in DBA/2 mice. J Neurochem. 1994;62(6):2492–5.
Chapman AG, et al. Anticonvulsant activity of two metabotropic glutamate group I antagonists selective for the mGlu5 receptor: 2-methyl-6-(phenylethynyl)-pyridine (MPEP), and (E)-6-methyl-2-styryl-pyridine (SIB 1893). Neuropharmacology. 2000;39(9):1567–74.
Merlin LR, Bergold PJ, Wong RK. Requirement of protein synthesis for group I mGluR-mediated induction of epileptiform discharges. J Neurophysiol. 1998;80(2):989–93.
Wong RK, et al. Role of metabotropic glutamate receptors in epilepsy. Adv Neurol. 1999;79:685–98.
Stoop R, et al. Activation of metabotropic glutamate 5 and NMDA receptors underlies the induction of persistent bursting and associated long-lasting changes in CA3 recurrent connections. J Neurosci. 2003;23(13):5634–44.
Freund LS, Reiss AL. Cognitive profiles associated with the fra(X) syndrome in males and females. Am J Med Genet. 1991;38(4):542–7.
Van Dam D, et al. Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav Brain Res. 2000;117(1–2):127–36.
Kooy RF, et al. Transgenic mouse model for the fragile X syndrome. Am J Med Genet. 1996;64(2):241–5.
D'Hooge R, et al. Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience. 1997;76(2):367–76.
Gold PE. The use of avoidance training in studies of modulation of memory storage. Behav Neural Biol. 1986;46(1):87–98.
Power AE, et al. Anisomycin infused into the hippocampus fails to block “reconsolidation” but impairs extinction: the role of re-exposure duration. Learn Mem. 2006;13(1):27–34.
Whitlock JR, et al. Learning induces long-term potentiation in the hippocampus. Science. 2006;313(5790):1093–7.
Szatmari P, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39(3):319–28.
Kim HG, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82(1):199–207.
Moessner R, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81(6):1289–97.
Durand CM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39(1):25–7.
Chubykin AA, et al. Dissection of synapse induction by neuroligins: effect of a neuroligin mutation associated with autism. J Biol Chem. 2005;280(23):22365–74.
Jamain S, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–9.
Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14(4):483–92.
Kwon CH, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50(3):377–88.
Peters SU, et al. Autism in Angelman syndrome: implications for autism research. Clin Genet. 2004;66(6):530–6.
Smalley SL. Autism and tuberous sclerosis. J Autism Dev Disord. 1998;28(5):407–14.
Tavazoie SF, et al. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8(12):1727–34.
van Woerden GM, et al. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10(3):280–2.
Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–55.
Guy J, et al. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315(5815):1143–7.
Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28(11):573–6.
Tu JC, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23(3):583–92.
Giuffrida R, et al. A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J Neurosci. 2005;25(39):8908–16.
Zalfa F, et al. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007;10(5):578–587.
Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3(3):175–90.
Muddashetty RS, et al. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27(20):5338–48.
Zhou Z, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52(2):255–69.
Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004;11(2):172–8.
Castren M, et al. BDNF regulates the expression of fragile X mental retardation protein mRNA in the hippocampus. Neurobiol Dis. 2002;11(1):221–9.
Bear MF, et al. Fragile X: translation in action. Neuropsychopharmacology. 2008;33(1):84–7.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Dölen, G., Bear, M.F. Fragile x syndrome and autism: from disease model to therapeutic targets. J Neurodevelop Disord 1, 133–140 (2009). https://doi.org/10.1007/s11689-009-9015-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11689-009-9015-x
Keywords
- Fragile X
- FXS
- Metabotropic
- Glutamate
- Receptor
- mglur
- mglur5
- FMRP
- Fragile x mental retardation protein
- Synaptic plasticity
- Long term depression
- LTD
- Protein synthesis
- Translation
- Ocular
- Dominance
- Plasticity
- Visual
- Cortex
- Hippocampus
- Inhibitory avoidance
- Passive avoidance
- Extinction
- Autism
- HOMER
- SHANK
- Neuroligin
- Neurexin
- Tuberous sclerosis
- TSC
- TSC1
- TSC2
- Rett
- MeCP
- BDNF
- PTEN
- Hamartoma
- Angelman
- UBE3
- Dendritic spine
- Synapse
- Development
- Synapsopathy
- Audiogenic seizure
- Seizure
- Mental retardation
- Cognitive
- Impairment