
Abstract
Autism Spectrum Disorder (ASD) is associated with both (i) post-mortem and neuroimaging evidence of abnormal cortical development, and (ii) altered signalling in Brain Derived Neurotrophic Factor (BDNF) pathways - which regulate neuroproliferative and neuroplastic processes. In healthy controls genotype at a single nucleotide polymorphism that alters BDNF signalling (Val66met) has been related to regional cortical volume. It is not known however if this influence on brain development is intact in ASD. Therefore we compared the relationship between genotype and cortical anatomy (as measured using in vivo Magnetic Resonance Imaging) in 41 people with ASD and 30 healthy controls. We measured cortical volume, and its two sole determinants - cortical thickness and surface area - which reflect differing neurodevelopmental processes. We found “Group-by-Genotype” interactions for cortical volume in medial (caudal anterior cingulate, posterior cingulate) and lateral (rostral middle, lateral orbitofrontal, pars orbitalis and pars triangularis) frontal cortices. Furthermore, within (only) these regions “Group-by-Genotype” interactions were also found for surface area. No effects were found for cortical thickness in any region. Our preliminary findings suggest that people with ASD have differences from controls in the relationship between BDNF val66met genotype and regional (especially frontal) cortical volume and surface area, but not cortical thickness. Therefore alterations in the relationship between BDNF val66met genotype and surface area in ASD may drive the findings for volume. If correct, this suggests ASD is associated with a distorted relationship between BDNF val66met genotype and the determinants of regional cortical surface area – gyrification and/or sulcal positioning.
Similar content being viewed by others


Introduction
ASD is a group of increasingly recognised [1] lifespan persistent [2] neurodevelopmental disorders of early onset [3, 4] characterised by abnormalities in language, social interaction and a range of stereotyped and repetitive behaviours [5]. The biological basis of ASD remains poorly understood. However, autism (the paradigmatic ASD) is both highly heritable [6], and associated with alterations in both pre-natal brain development [7] and post-natal brain structure [8, 9]. Despite this, little is known regarding the influence of specific genes on brain anatomy in ASD with the exception of the serotonin transporter [10] and monoamine oxidase [11] genes which contain functional genetic variants that have been associated with cortical volume in children with ASD.
A strong and as yet unexamined candidate gene for modulating brain anatomy in ASD is Brain Derived Neurotrophic Factor (BDNF) which plays a key role in neurogenesis, cortical lamination, synaptic plasticity, and neuron survival [12]. Intracellular packaging and secretion of BDNF is altered by a common functional [13, 14] non-synonymous single nucleotide polymorphism (SNP) within the 5′ region of the gene (BDNF val66met) which codes for a valine to methionine substitution. In the healthy population, genotype at this polymorphism has been associated with differences in cortical plasticity [15] and regional cortical volume (CV) [16–21] (see Table 1). Many of the cortical areas where BDNF val66met genotype influences CV in typically developing individuals overlap with those where structural differences have been reported between people with ASD and typically developing controls.
For example three independent studies [16, 17, 19] have reported regional cortical CV reductions in BDNF met carriers compared to val homozygotes in regions such as the dorso-lateral and inferior frontal, superior parietal and precuneus cortices. These are cortical regions where our group [22], and others [23] that have shown structural differences between people with ASD and healthy controls.
Furthermore, all [24–28] but one [29] of the studies examining BDNF measures in ASD report abnormalities. Autism Spectrum Disorder has been associated with (i) elevated serum BDNF in infancy [24] childhood and adulthood [25, 26], (ii) elevated blood lymphocyte BDNF mRNA in young adults [30], and (iii) increased basal forebrain BDNF levels in post-mortem samples of cerebral cortex [28].
Therefore, it is possible that altered BDNF signalling in ASD contributes to the anatomical differences of people with ASD by disturbing the pathways through which the BDNF val66met polymorphism influences regional CV. It is not known however if the CV correlates of BDNF val66met genotype differ between people with ASD and typically developing controls. Therefore, we carried out a preliminary exploratory investigation in which we related BDNF val66met genotype to 33 regional measures of CV in 71 individuals – 41 of whom had an ASD diagnosis (35 with Asperger syndrome, four with high-functioning autism and two with other pervasive developmental disorders), and 30 typically developing controls. Furthermore, in those regions where there were significant differences between people with ASD and controls in the relationship between BDNF val66met genotype and CV – we also examined if these were accompanied by alterations in cortical thickness (CT) or surface area (SA). The volume of a cortical region is approximated by the product of the total surface area and the mean cortical thickness of that region.
Because these two sole determinants of CV reflect distinct evolutionary and developmental processes [31, 32] – it is important to disambiguate their relative contribution to any differences in the CV correlates of BDNF val66met genotype in people with ASD and controls. For example, SA is influenced by division of progenitor cells in the embryological periventricular area and varies as a function of brain volume and cortical folding (gyrification). In contrast, CT is likely to reflect dendritic arborization/pruning within the grey matter [33] or myelination at the grey/white matter interface [34].
Materials and methods
Subjects
We included 41 subjects with ASD and 30 healthy controls. All people with ASD were recruited through a hospital-based national clinic and a university department specialising in the assessment of ASD with infrastructure support from the Medical Research Council United Kingdom Autism Imaging Multicentre Study (MRC UK A.I.M.S) program. All cases were diagnosed clinically with Asperger syndrome (n = 35), autism (n = 4), or Pervasive-Developmental Disorder-not otherwise specified (n = 2) according to DSM-IV criteria. Diagnoses were made by a team of senior clinicians trained in the Autism Diagnostic Interview-Revised (ADI-R) (Le Couteur et al. 1989) and the Autism Diagnostic Observation Schedule (ADOS-G) (Lord et al. 2000). Twenty-four of the participants with ASD also agreed to confirmation of clinical diagnosis using the ADI-R or ADOS. Typically developing healthy controls were recruited through advertisement in the community.
Participants in the study underwent structured physical and psychiatric examination (for the presence of a co-morbid DSM-IV Axis I or II disorder). Participants did not have a history of physical or psychiatric disorder affecting brain function (e.g., epilepsy or psychosis), or a genetic disorder putatively associated with autistic spectrum disorders (e.g., Fragile X syndrome). All participants had full-scale intelligence quotients above 70. All subjects (or guardians where applicable) gave informed and written consent.
There were no statistically significant differences in mean age between cases and controls or between BDNF val homozygotes and BDNF met carriers. Demographic and cognitive details for all participants are given in Table 2.
Genotyping
The BDNF val66met genotype was determined using a TaqMan allele specific assay method (Applied Biosystems) according to the manufacturer’s protocols. Polymerase chain reaction (PCR) amplifications were performed on an ABI PRISM® 7000 Sequence Detection System (Applied Biosystems) with the reaction mixture in a total volume of 2 μl, consisting of 5 ng of genomic DNA, 2 × Universal PCR Master Mix (ABGENE), 40× TaqMan SNP Genotyping Assay Mix (Applied Biosystems), and deionized H2O. After denaturing at 95°C for 15 min, 50 cycles of PCR were performed under the following conditions: 92°C for 15 s and 60°C for 90 s. All genotypes were reported with the allelic discrimination program using the Applied Biosystems Sequence Detection System (SDS).
Magnetic resonance imaging
Brain MRIs were acquired using a GE Signa 1.5 T neuro-optimised MR system (General Electric, Milwaukee WI, USA). “Freesurfer” freeware (http://surfer.nmr.mgh.harvard.edu/fswiki) was used to derive models of the cortical sheet in each T1-weighted image and parcellate out the cortex into 33 regions. These well-validated [35, 36] and fully automated procedures have been extensively described elsewhere [37, 38], and we will only provide a brief description here. Firstly, a surface reconstruction was made for each scan [39]. This involved intensity normalisation, the removal of extra-cerebral tissues using a “skull stripping” algorithm, and image segmentation using a connected components algorithm. The output of this stage is a single filled white-matter volume for each cerebral hemisphere. A surface tessellation was then generated for each white- matter volume by fitting a deformable template. The grey matter/CSF surface was also modelled using a similar process. Given explicit models for the white/grey and grey/CSF surfaces, the measure of absolute cortical thickness at any given point on the white/grey matter surface is then taken to be the shortest distance between that point and the grey/CSF surface. This measurement is made at approximately 150,000 points across each hemisphere for each scan.
In order to achieve automated parcellation of each individual cortical hemispheric sheet into 33 regions [38], a probabilistic atlas was placed within a surface-based co-ordinate system to which each scan was aligned. Parcellation generated a measure of total CV, total SA and average CT for each sub-region within each hemisphere. For each sub-region values for left and right hemispheres were combined, resulting in a total of three measures (SA, CT and GV) for each of 33 sub-regions.
Statistical analyses
Analyses of covariance (ANCOVAs) were used to model the effects of Group (ASD vs controls), BDNF genotype (Val vs Met carriers) and the interaction of these two terms on regional measures of CV. In order to limit the number of statistical tests, we combined measures of CV for each of 33 cortical regions per hemisphere into total bilateral CV for that region. Given the exploratory nature of this analysis we adopted an uncorrected p value of 0.05. For those regions showing significant “Group by Genotype” interaction effects, we carried out further ANCOVA’s for CT and SA in order to establish which of these parameters may be contributing to “Group-by-Genotype” effects for CV (again, applying an uncorrected p value of 0.05). Cortical volume, SA and CT were not transformed before being used as dependent variables in these analyses.
Within this cross-sectional dataset, we were not able to identify the non-linear age effects reported in much larger, longitudinal studies[40]. Linear age effect were however seen and given the wide age-range of our sample, age was included as a co-variate to remove variance not related to our independent variables of interest. As there were no significant differences between groups in gender, or intracranial volume (ICV) – we did not co-vary for these in analyses. Furthermore, the relationships between ICV and CV, SA/gyrification and CT are complex, non-linear [41] and may differ with age and gender [42].
Results
Neither group, genotype, not the interaction of these two terms was associated with total intracranial volume, total cortical volume, total cortical surface area or mean cortical thickness.
There was a significant “Group by Genotype” interaction for CV in six cortical areas - posterior cingulate, caudal anterior cingulate, rostral middle frontal, lateral orbitofrontal, pars orbitalis and pars triangularis. Further, within all of these regions, significant “Group by Genotype” interactions were also found for SA, but not CT (Table 3). In order to determine how specific “Group-by-Genotype” interactions for SA were to those regions also showing interactions for CV, we next ran ANCOVAs for SA in all cortical regions. No other areas showed a “Group-by-Genotype” interaction for SA (results not shown but available).
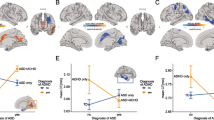
In all six regions “Group by Genotype” interactions for both CV and SA reflected the same phenomenon – lower CV/SA in control BDNF met allele carriers compared to control BDNF val homozygotes, but higher CV/SA in ASD BDNF met allele carriers as compared to ASD BDNF val homozygotes. The same pattern was seen in all six regions and is illustrated in Fig. 1 using the caudal anterior cingulated as an example.

Caudal anterior and posterior cingulate CV and SA by Group and BDNF genotype. Bars represent 95% confidence intervals for mean CV/SA
Discussion
The results from our exploratory study suggest that the relationship between cortical anatomy and BDNF genotype may differ between people with ASD and typically developing controls. Given the preliminary nature of our investigation we did not correct for multiple comparisons, and so replication is required. Nevertheless, our findings do lead to testable hypotheses.
Firstly, rather than being generalised, “Group-by-Genotype” interactions may be most marked in particular cortical regions, i.e the caudal anterior cingulate, posterior cingulate, rostral middle frontal, lateral orbitofrontal, pars orbitalis and pars triangularis. These regions are not scattered across the cortex, but describe two larger contiguous frontal areas – one medial and one lateral.
Secondly, many of the cortical regions where we identified “Group-by-Genotype” interactions for CV/SA overlap with those where BDNF has been shown to modulate CV in typically developing by structural MRI [eg in the medial, middle and inferior frontal [16, 17, 19]], and N-acetylaspartate levels (a marker of neuronal integrity) in healthy controls by Magnetic Resonance Spectroscopy [anterior cingulate [43]]. In our sample the relationship between CV and BDNF val66met genotype in controls was in the same direction and similar frontal distribution as that reported in other samples of typically developing individuals [16, 17]. That is in controls CV was reduced in BDNF met allele carriers compared to BDNF val allele homozygotes. This relationship was reversed in people with ASD. Furthermore, cortical regions where we identified “Group-by-Genotype” interactions for CV/SA also overlap with those where compared to controls, people with ASD have been reported as showing alterations in CV [23], cortical folding [44] and magnetic resonance spectroscopy measures of neuronal integrity [anterior cingulate [45]]. This suggests that differences in cortical structure between people with ASD and typically developing controls may be due to differences in BDNF val66met genotype frequency and/or function.
Thirdly, in those (and only those) regions showing significant “Group x Genotype” interactions for CV, we also found significant “Group x Genotype” interactions for SA, but not CT. This suggests that the CV findings are driven by “Group x Genotype” interactions for SA. The SA within a given cortical region may be altered in three ways; (i) alterations in radial brain size, (ii) displacement of the landmarks (eg sulcal landmarks) used to define regional boundaries, or (ii) increased gyrification of the cortical sheet within unaltered regional boundaries.
We did not identify any “Group-by-Genotype” interactions for intracranial volume or total cerebral SA (data not shown but available), which argues against the first of these explanations. The remaining two explanations are compatible with existing reports of altered sulcal positioning [46] and gyrification [47] in people with ASD compared to controls. As both of these cortical characteristics can be quantified from MRI scans – their relative contributions to putative differences in the SA correlates of BDNF val66met genotype between people with ASD and controls can be directly examined in future work.
The mechanisms that could underlie differences between people with ASD and controls in the CV/SA correlates of BDNF val66met genotype are unclear. Such differences could be due to abnormal BDNF signalling in ASD causing disruption of the pathways through which the BDNF val66met polymorphism might influence cortical structure during typical development such as neuronal migration [48] and/or experience dependent plasticity [49]. Alternatively, our findings may not be due to abnormal BDNF signalling in ASD per se, but reflect the fact that brain structure is so atypical in ASD that the subtle differences accounted for by any given single genetic polymorphisms are “drowned out” (Raznahan et al, in press). A further possibility is that due to dysfunction within molecular pathways that interact with BDNF to influence brain development – the typical downstream effects of differences in BDNF signalling due to val66met genotype are not seen in ASD. Autism Spectrum Disorder has been associated with reports of abnormalities within several discrete molecular systems that all share the feature of directly impacting on or interacting with BDNF transcription and function. These include serotonin (5HT) [50, 51], Calcium-dependent activator protein for secretion 2 (CAPS2) [52, 53], methyl-CpG binding protein 2 (MeCP2) [14], and Jumonji AT-rich interactive domain 1C (JARID1C) [54].
Our study has several limitations. Firstly, our sample size is relatively small. Our study does however represent the first examination of the relationship between BDNF and brain anatomy in ASD, and is equivalent in sample size to some of the smaller studies that have been published to date on BDNF vall66met and brain structure in typically developing controls [55, 56]. Secondly we were not able to obtain ADI-R/ADOS confirmation of clinical ASD diagnosis in all volunteers in our ASD group. However, this confirmation was available for the majority of people with ASD, and all ASD diagnoses were made by a multidisciplinary team trained in the ADI-R and ADOS. Thirdly, almost all participants with ASD in our study had a diagnosis of Asperger syndrome, and all had a FSIQ greater than 70. Further studies in diagnostically homogeneous samples of people with other ASD diagnoses and cognitive profiles will be required to establish the generalisability of our findings across the autism spectrum. Fourthly, people with ASD and controls differed significantly in general cognitive ability. Although this means that our findings may be driven my differences in FSIQ rather than ASD diagnosis per se, the strong association between ASD and learning disability complicates the issue of matching for IQ in study design [57]. Furthermore, in our sample, FSIQ did not correlate with any of the regional cortical measures showing group-by-genotype interactions. Fifthly, because our sample covered a wide age-range, we co-varied for age in all analyses and were not therefore able to directly model possible “Group-by-Genotype-by-Age” effects on cortical anatomy. The age range of our sample and the predominance of individuals with Asperger syndrome diagnoses may also account for the fact that we did not observe the increased cortical volumes commonly reported in autism [58, 59]. Finally, as our sample included both males and females we were not able to determine the influence of gender on our findings.
In summary, our results provide preliminary evidence that in medial and lateral frontal cortices the relationship between BDNF val66met genotype and cortical volume is not the same in people with ASD as it is in typically developing populations. This may be due to differences between people with ASD and typically developing populations in either (i) BDNF val66met genotype frequencies, or (ii) the mechanisms linking this genetic polymorphism to regional variations in cortical surface area.
References
Baird G, Simonoff E, Pickles A, et al. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the Special Needs and Autism Project (SNAP). Lancet. 2006:368(9531):210–5.
Howlin P, Goode S, Hutton J, Rutter M. Adult outcome for children with autism. Journal of Child Psychology & Psychiatry & Allied Disciplines. 2004;45(2):212–29.
World Health O. Mental disorders; a glossary and guide to their classification in accordance with the 10th revision of the international classification of
Zwaigenbaum L, Bryson S, Rogers T, Roberts W, Brian J, Szatmari P. Behavioral manifestations of autism in the first year of life. Int J Dev Neurosci. 2005;23:143–52.
Volkmar FR, Lord C, Bailey A, Schultz RT, Klin A. Autism and pervasive developmental disorders. J Child Psychol Psychiatry & Allied Disciplines. 2004;45:135–70.
Bailey A, Le Couteur A, Gottesman I, Bolton P. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol Med. 1995;25:63–77.
Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: A review and future directions. Int J Dev Neurosci. 2005;23:183–87.
Redcay E, Courchesne E. When is the brain enlarged in autism? A meta-analysis of all brain size reports. Biol Psychiatry. 2005;58:1–9.
Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–45.
Wassink TH, Hazlett HC, Epping EA, et al. Cerebral cortical gray matter overgrowth and functional variation of the serotonin transporter gene in autism. Arch Gen Psychiatry. 2007;64:709–17.
Davis LK, Hazlett HC, Librant AL, et al. Cortical enlargement in autism is associated with a functional VNTR in the monoamine oxidase A gene. Am.J.Med.Genet.B Neuropsychiatr.Genet. 2008.
Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22:123–31.
Egan MF, Kojima M, Callicott JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function.[see comment]. Cell. 2003;112(2):257–69.
Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2.[see comment]. Science. 2003;302(5646):885–9.
Cheeran B, Talelli P, Mori F, et al. A common polymorphism in the brain-derived neurotrophic factor gene (BDNF) modulates human cortical plasticity and the response to rTMS. J Physiol. 2008;586:5717–25.
Pezawas L, Verchinski BA, Mattay VS, et al. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci. 2004;24(45):10099–102.
Ho BC, Milev P, O'Leary DS, Librant A, Andreasen NC, Wassink TH. Cognitive and magnetic resonance imaging brain morphometric correlates of brain-derived neurotrophic factor Val66Met gene polymorphism in patients with schizophrenia and healthy volunteers. Arch Gen Psychiatry. 2006;63:731–40.
Agartz I, Sedvall GC, Terenius L, et al. BDNF gene variants and brain morphology in schizophrenia. Am J Med Genet.Part B, Neuropsychiatric Genetics: the Official Publication of the International Society of Psychiatric Genetics. 2006;141(5):513–23.
Nemoto K, Ohnishi T, Mori T, et al. The Val66Met polymorphism of the brain-derived neurotrophic factor gene affects age-related brain morphology. Neuroscience Letters.397(1–2):25–9. 2006:17.
Varnas K, Lawyer G, Jonsson EG, et al. Brain-derived neurotrophic factor polymorphisms and frontal cortex morphology in schizophrenia. Psychiatr Genet. 2008;18:177–83.
Schofield PR, Williams LM, Paul RH, et al. Disturbances in selective information processing associated with the BDNF Val66Met polymorphism: Evidence from cognition, the P300 and fronto-hippocampal systems. Biol Psychol. Sep 16 2008.
McAlonan GM, Cheung V, Cheung C, et al. Mapping the brain in autism. A voxel-based MRI study of volumetric differences and intercorrelations in autism. Brain. 2005;128(Pt 2):268–76.
Hadjikhani N, Joseph R, Snyder J, Tager-Flusberg H. Anatomical Differences in the Mirror Neuron System and Social Cognition Network in Autism. Cerebral Cortex. 2005:1–7.
Nelson KB, Grether JK, Croen LA, et al. Neuropeptides and neurotrophins in neonatal blood of children with autism or mental retardation. Ann Neurol. 2001;49(5):597–606.
Connolly AM, Chez M, Streif EM, et al. Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau-Kleffner syndrome, and epilepsy. Biol Psychiatry. 2006;59(4):354–63.
Miyazaki K, Narita N, Sakuta R, et al. Serum neurotrophin concentrations in autism and mental retardation: a pilot study. Brain Dev. 2004;26(5):292–5.
Nishimura K, Nakamura K, Anitha A, et al. Genetic analyses of the brain-derived neurotrophic factor (BDNF) gene in autism. Biochem Biophys Res Commun. 2007;356(1):200–6.
Perry EK, Lee ML, Martin-Ruiz CM, et al. Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am J Psychiatry. 2001;158(7):1058–66.
Croen LA, Goines P, Braunschweig D, et al. Brief Report: Brain-Derived Neurotrophic Factor and Autism: Maternal and Infant Peripheral Blood Levels in the Early Markers for Autism (EMA) Study. Autism Res. 2008;1:130–37.
Nishimura K, Nakamura K, Anitha A, et al. Genetic analyses of the brain-derived neurotrophic factor (BDNF) gene in autism. Biochem Biophys Res Commun. 2007;356:200–6.
Rubenstein JL, Rakic P. Genetic control of cortical development. Cereb Cortex. 1999;9:521–3.
Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–9.
Huttenlocher PR. Morphometric study of human cerebral cortex development. Neuropsychologia. 1990;28(6):517–27.
Sowell ER, Thompson PM, Leonard CM, Welcome SE, Kan E, Toga AW. Longitudinal mapping of cortical thickness and brain growth in normal children. J Neurosci. 2004;24(38):8223–31.
Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968–80.
Han X, Jovicich J, Salat D, et al. Reliability of MRI-derived measurements of human cerebral cortical thickness: the effects of field strength, scanner upgrade and manufacturer. Neuroimage. 2006;32(1):180–94.
Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA. 2000;97:11050–5.
Fischl B, Van Der KA, Destrieux C, et al. Automatically Parcellating the Human Cerebral Cortex. Cereb Cortex. 2004;14(1):11–22.
Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9(2):179–94.
Shaw P, Kabani NJ, Lerch JP, et al. Neurodevelopmental trajectories of the human cerebral cortex. J Neurosci. 2008;28:3586–94.
Toro R, Perron M, Pike B, et al. Brain Size and Folding of the Human Cerebral Cortex. Cerebral Cortex. 2008:bhm261.
Luders E, Narr KL, Thompson PM, et al. Gender differences in cortical complexity. Nat Neurosci. 2004;7(8):799–800.
Lang UE, Hellweg R, Seifert F, Schubert F, Gallinat J. Correlation Between Serum Brain-Derived Neurotrophic Factor Level and An In Vivo Marker of Cortical Integrity. Biol Psychiatry. 2007;62:530–5.
Nordahl CW, Dierker D, Mostafavi I, et al. Cortical Folding Abnormalities in Autism Revealed by Surface-Based Morphometry. J Neurosci. 2007;27:11725–35.
Murphy DGM, Critchley HD, Schmitz N, et al. Asperger syndrome: A proton magnetic resonance spectroscopy study of brain. [References]. Arch Gen Psychiatry. 2002;59:885–92.
Levitt JG, Blanton RE, Smalley S, et al. Cortical Sulcal Maps in Autism. Cereb Cortex. 2003;13:728–35.
Hardan AY, Jou RJ, Keshavan MS, Varma R, Minshew NJ. Increased frontal cortical folding in autism: A preliminary MRI study. Neuroimaging. 2004;131:263–8.
Zhou P, Porcionatto M, Pilapil M, et al. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 2007;55:53–68. Jul 5.
Kuipers SD, Bramham CR. Brain-derived neurotrophic factor mechanisms and function in adult synaptic plasticity: new insights and implications for therapy. Curr Opin Drug Discov Devel. 2006;9:580–6.
Lam KSL, Aman MG, Arnold LE. Neurochemical correlates of autistic disorder: A review of the literature. Res Dev Disabil. 2006;27:254–89.
Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–94.
Sadakata T, Washida M, Iwayama Y, et al. Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. J.Clin Invest. 2007;117:931–43.
Sadakata T, Kakegawa W, Mizoguchi A, et al. Impaired cerebellar development and function in mice lacking CAPS2, a protein involved in neurotrophin release. J Neurosci. 2007;27:2472–82.
Adegbola A, Gao H, Sommer S, Browning M. A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD). Am J Med Genet. 2008;146A:505–11.
Frodl T, Schule C, Schmitt G, et al. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch Gen Psychiatry. 2007;64:410–6.
Sublette ME, Baca-Garcia E, Parsey RV, et al. Effect of BDNF val66met polymorphism on age-related amygdala volume changes in healthy subjects. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2008.
Szatmari P, Zwaigenbaum L, Bryson S. Conducting Genetic Epidemiology Studies of Autism Spectrum Disorders: Issues in Matching. J Autism Dev Disorders. 2004;34:49–57.
Hazlett HC, Poe MD, Gerig G, Smith RG, Piven J. Cortical gray and white brain tissue volume in adolescents and adults with autism. Biol Psychiatry. 2006;59(1):1–6.
Carper RA, Moses P, Tigue ZD, Courchesne E. Cerebral lobes in autism: early hyperplasia and abnormal age effects. Neuroimage. 2002;16:1038–51.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Raznahan, A., Toro, R., Proitsi, P. et al. A functional polymorphism of the brain derived neurotrophic factor gene and cortical anatomy in autism spectrum disorder. J Neurodevelop Disord 1, 215–223 (2009). https://doi.org/10.1007/s11689-009-9012-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11689-009-9012-0