
Abstract
Regorafenib (BAY 73-4506, Stivarga® Bayer HealthCare Pharmaceutical Inc) is an oral multikinase inhibitor with a distinct and wide-ranging profile of tyrosine kinase inhibition, resulting in antiangiogenic and antiproliferative properties in tumors. Single-agent regorafenib administered as a 160-mg daily dose for the first 21 days of a 28-day cycle is approved for use in patients with pretreated metastatic colorectal cancer (mCRC) and gastrointestinal stromal tumor (GIST) progressing on imatinib and sunitinib, following publication of data from the phase III CORRECT and GRID studies respectively. Regorafenib is currently under phase III investigation in patients with hepatocellular carcinoma and is in several phase II studies in patients with gastrointestinal (GI) tumors. This review describes the clinical development of regorafenib in patients with GI cancers, and highlights the key issues important for the modern day clinical pharmacist who forms part of the multidisciplinary team ensuring safe and effective delivery of the drug to the patient. This information is considered of particular importance to the clinical pharmacist for the future development of regorafenib in this treatment setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal cancers comprise tumors of the gastrointestinal tract including the esophagus, bowel, and rectum, and the accompanying organs such as the stomach, liver, and pancreas. In 2008, there were approximately 3.9 million cases of gastrointestinal cancer worldwide and approximately 2.8 million deaths worldwide from the disease. Global incidence and mortality rates of 54.9 and 39.4 cases per 100,000 respectively, were reported [1].
The introduction of targeted agents either as monotherapy or in combination with standard chemotherapy regimens has improved clinical outcome in patients with gastrointestinal cancer where they have become standard of care. VEGF-targeting agents (monoclonal antibody [mAb] bevacizumab, and a VEGFR decoy receptor, aflibercept) and EGFR mAbs are approved for use in patients with metastatic colorectal cancer (mCRC) [2–4]. The tyrosine kinase inhibitor (TKI) imatinib targeted to activated KIT and PDGFRα receptors [5] and the multikinase inhibitor (MKI) sunitinib [6] are approved for use in patients with gastrointestinal stromal tumors (GIST), and the MKI sorafenib [7] is considered standard of care in previously untreated patients with hepatocellular carcinoma (HCC). However, there remains an unmet need in those patients whose tumors are intrinsically resistant, or that become resistant to the currently available treatments [2, 8].
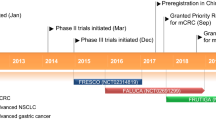
Regorafenib (BAY 73–4506, Stivarga®, Bayer HealthCare Pharmaceuticals Inc.) is a novel MKI with a distinct and wide-ranging profile of tyrosine kinase inhibition [9, 10]. It is the first MKI to be approved for use in mCRC patients and has also been approved in patients with advanced GIST progressing on imatinib and sunitinib. Approval was granted by the US Food and Drug Administration (FDA) for both these indications [11]. An application for marketing authorization has been submitted for the use of regorafenib in patients with advanced GIST, to the European Medicines Agency (EMA), following recent EU approval of regorafenib in the treatment of adult patients with mCRC who have been previously treated with, or are not considered candidates for, available therapies [12].
The modern day oncology clinical pharmacist is part of a multidisciplinary team (MDT) providing a safe continuum of care to the cancer patient in a new era of chemotherapy and biological agents. In addition to formulation, dispensing and disposing of cancer drugs, and the management of their cost and reimbursement, the clinical pharmacist plays an increasing role in the direct care of the patient. This includes the provision of counseling for treatment compliance, for possible drug-drug interactions, and for the potential side-effects and management of occurring side-effects, in addition to the provision of medicines during patient follow-up, and providing an education service for patients and health care providers [13–15]. With the inclusion of regorafenib to the available formulary for patients with gastrointestinal cancers this review describes the clinical development of regorafenib in this treatment setting, and highlights the key issues important for the clinical pharmacist to ensure safe and effective delivery of the drug to the patient.
Preclinical studies
Regorafenib is a fluoro-substituted omega-carboxylaryl diphenyl urea [9]. Its biochemical profile is similar to but distinct from sorafenib and it appears to be more pharmacologically potent than sorafenib [9, 16] (Table 1). The major metabolites, M-2 (N-oxide) and M-5 (N-oxide and N-desmethyl) reportedly exhibit similar efficacy compared with regorafenib in both in vitro and in vivo models [10, 17]. Analysis of the biochemical activity of regorafenib using in vitro kinase assays demonstrated the inhibition of a large set of kinases including angiogenic and stromal receptor tyrosine kinases (RTKs); VEGFR-1, -2, and -3, TIE2, FGFR-1, and PDGFR-β at IC50 concentrations of 4–311 nM; oncogenic RTKs, KIT, and RET, and the intracellular signaling kinases cRAF/RAF-1, and both the wild-type and mutant forms of BRAF (V600E) at IC50 concentrations of 1.5–28 nM [9]. Inhibition of additional kinases including DDR2, EphA2, PTK5, p38α, and β occurred at <100 nM concentrations. While inhibition was not demonstrated for other kinases (EGFR, PKC, MET, ERK1/2, and AKT) using these assays [9]. Cellular phosphorylation assays support the spectrum of tyrosine kinase inhibition by regorafenib including inhibition of MAPK, a component of an intracellular pathway important in epithelial cell signaling and tumorigenesis [9].
The antiangiogenic properties of regorafenib were demonstrated through the inhibition of cellular proliferation in stimulated vascular cell lines (human umbilical vascular endothelial cells), and through the recording of changes in tumor vasculature (using dynamic contrast enhanced magnetic resonance imaging, DCE-MRI) in rat glioblastoma xenografts [9]. Further inhibitory effects on tumor cell proliferation were shown in human hepatoma cell lines and in human cancer cell lines harboring mutations in RTK genes including, GIST (KIT K642E) and thyroid cancer (RET C634WT) cell lines [9], and in drug-resistant GIST cell lines harboring mutations in exon 11 of the KIT gene [18]. Inhibition of tumor growth and angiogenesis was further demonstrated in tumor xenograft models of a range of human tumors, including CRC, breast, and renal cell carcinoma (RCC) [9]. In these studies, no animal lethality was reported. Regorafenib was also shown to inhibit angiogenesis and metastasis in a highly aggressive orthotopic colon cancer model [19].
Clinical development
Based on the encouraging preclinical data, phase I and II studies were performed to establish a safe and potentially effective dose for single-agent regorafenib in patients with advanced solid tumors (summarized in Table 2).
The first-in-human phase I clinical trial in patients with advanced solid tumors (mainly CRC), performed by Mross et al., used a dose-escalation design to establish single-agent oral regorafenib at 160 mg as the recommended dose for future study in a schedule of daily administration for 21 days (3 weeks), with 7 days off (1 week), in repeated 28-day (4 week) cycles. This was based on dose-limiting toxicities and the pharmacokinetic and safety profile [20]. This regimen was confirmed as suitable in additional phase I and phase II studies which concluded that single-agent regorafenib was tolerable with promising activity in patients with refractory mCRC [21], advanced GIST progressing on imatinib and sunitinib [22, 23], and intermediate or advanced HCC progressing after first-line sorafenib [24] (Table 2).
An alternative dosing regimen of daily 100 mg single-agent regorafenib, administered continuously in 3-week cycles was also shown to be safe in patients with refractory solid tumors [25].
Pharmacokinetic analysis from phase I and phase II studies revealed a similar exposure at steady state for regorafenib (detailed below) and its pharmacologically active metabolites M-2 and M-5 [20, 21, 24].
The safety profile reported in many of these studies was typical for that reported with MKI. Adverse events were common but generally manageable with standard-of-care practices, with few permanent discontinuations from treatment, or deaths due to an adverse event reported. The most commonly occurring grade 3/4 treatment-related toxicities across studies included hand-foot skin reactions (HFSR), hypertension, rash, and diarrhea (Table 2).
Activity from phase I and phase II studies reported mainly prolonged stable disease with the disease control rates ranging from 66 % to 74 % (Table 2). Data from these studies formed the rationale for the pivotal phase III studies in mCRC and GIST and an ongoing phase III study in HCC.
mCRC
A phase 1b study by Strumberg et al. investigated oral regorafenib in 38 patients with chemorefractory mCRC, 15 received a dose escalation (60–220 mg) of regorafenib and 23 (expansion cohort) received a dose of 160 mg. Regorafenib was administered once daily for the first 3 weeks of each 4-week cycle (Table 2). The authors reported a tolerable toxicity profile and preliminary evidence of antitumor activity [21]. The disease control rate was 74 % (20 of 27 assessable patients). On the basis of these results, and the high unmet need in this population of patients, the decision was made to precede to a randomized phase III trial.
The phase III CORRECT study (patients with metastatic COloRectal cancer treated with REgorafenib or plaCebo after failure of standard Therapy, BAY 73-4506/14387, NCT01103323) investigated regorafenib in mCRC patients previously treated with 5-fluorouracil (5-FU), irinotecan, oxaliplatin, bevacizumab, and in patients with KRAS wild-type tumors an EGFR mAb [26]. This was a placebo-controlled double-blind trial at 114 centers in 16 countries worldwide. Patients were ≥18 years of age, had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1, and a life expectancy of >3 months with documented mCRC and progression during or within 3 months after the last standard therapy. Patients were randomly assigned (2:1 and stratified by previous treatment with VEGF-targeting drugs, time from diagnosis of metastatic disease, and geographical region) to receive best supportive care plus oral regorafenib 160 mg or placebo once daily, for the first 3 weeks of each 4-week cycle. No crossover between patient groups was allowed.
Between April 30, 2010 and March 22, 2011, a total of 760 patients were randomly assigned to receive regorafenib (n = 505) or placebo (n = 255), and 753 patients initiated treatment (500 with regorafenib and 253 with placebo; population for safety analyses). Baseline and disease characteristics were similar in the treatment arms except more patients in the placebo group had KRAS mutations (62 % vs 54 %). All patients had received prior anti-VEGF treatment.
The primary endpoint of overall survival was met at a preplanned interim analysis after 432 deaths at a data cutoff of July 21, 2011. Regorafenib demonstrated a significant 23 % reduction in the risk of death (Hazard ratio [HR] 0.77, 95 % CI 0.64–0.94; one-sided p = 0.0052). Median overall survival was 6.4 months in the regorafenib arm versus 5.0 months in the placebo arm. In the regorafenib compared with placebo arms the HR for progression-free survival (PFS) was 0.49 (95 % CI 0.42–0.58, p < 0.0001). The objective response rates (all partial responses) were comparable between the treatment arms (1.0 % vs 0.4 %, p = 0.19), however there was a significantly higher disease control (partial response plus stable disease after 6 weeks) in the regorafenib compared with placebo arm (41 % vs 15 %, p < 0.0001) [26].
Treatment-related adverse events of any grade occurred in 93 % of patients assigned to regorafenib and 61 % assigned to placebo. The most common grade ≥3 regorafenib-related reactions were HFSR (17 %), fatigue (10 %), diarrhea (7 %), hypertension (7 %), and rash or desquamation (6 %). Increased liver transaminases and bilirubin was higher in the regorafenib group than the placebo group. This was mainly due to grade 1 and 2 events. Eleven deaths were reported due to adverse events not associated with disease progression, eight (2 %) in patients treated with regorafenib (two for pneumonia and two for gastrointestinal bleeding, and one each for intestinal obstruction, pulmonary hemorrhage, seizure, and sudden death) and three (1 %) in those receiving placebo (two for pneumonia and one for sudden death). One fatal case associated with regorafenib-related liver injury was first reported 43 days after administration in a patient with liver metastases who died 6 weeks later. Dose reductions (38 % vs 3 %) and dose interruptions (61 % vs 22 %) were more common in patients receiving regorafenib compared with the placebo, mainly due to dermatological, gastrointestinal, constitutional, and metabolic or laboratory events.
Changes in patient quality of life (QoL), as assessed using the European Organization for the Research and Treatment of Cancer QoL-C30 questionnaire and the EuroQoL-five dimension health utility index, were reportedly not different between the regorafenib and placebo treatment arms over the treatment period [27].
Based on the CORRECT study data, the EMA approved the use of regorafenib in the treatment of adult patients with mCRC who have been previously treated with, or are not considered candidates for, available therapies (these include fluoropyrimidine-based chemotherapy, an anti-VEGF therapy and an anti-EGFR therapy) [10] with similar approval from the FDA [11].
GIST
A phase II study by George et al. investigated single-agent regorafenib in 33 patients with TKI-resistant GIST who received a median of 8 treatment cycles (range 2–17), with a median follow-up of 10.9 months [22] (Table 2). The clinical benefit rate (CBR, defined as the proportion of patients with a composite of complete response, partial response, and stable disease lasting ≥16 weeks) with regorafenib was 79 % (95 % CI 61–91), including four patients with partial response and 22 with stable disease lasting ≥16 weeks. Median PFS was 10.0 months (95 % CI 8.3–14.9). After a longer follow-up (median duration of 20 months), a CBR of 81 %, median PFS of 13 months (95 % CI 9–18), and a median overall survival of 27 months (95 % CI 16 to not reached) was reported [23].
On the basis of the preliminary data, and the preclinical rationale of targeting the pathogenic mutant RTKs with a structurally distinct small-molecule inhibitor, the phase III GRID study (GIST—Regorafenib In progressive Disease, NCT01271712) was performed to assess the efficacy and safety of regorafenib in patients with metastatic or unresectable GIST, progressing after failure of at least previous imatinib and sunitinib [28]. The double-blind placebo-controlled GRID study was conducted at 57 hospitals in 17 countries worldwide [28]. Patients with histologically confirmed metastatic or unresectable GIST with at least one measurable lesion, adequate organ function and ECOG PS ≤1, were randomly assigned to receive best supportive care and either oral regorafenib 160 mg daily or placebo for the first 3 weeks of a 4-week cycle.
From January 4 to August 18, 2011, 199 patients were randomly assigned to best supportive care plus regorafenib (n = 133) or placebo (n = 66), one patient in the regorafenib arm did not receive treatment. The primary endpoint was PFS and secondary endpoints included overall survival, time to progression, objective response rate and disease control rate. At the data cutoff (January 26, 2012), PFS was significantly longer in the regorafenib compared with the placebo arm (median 4.8 vs 0.9 months, HR 0.27, 95 % CI 0.19–0.39, p < 0.0001). Following progression, 85 % of patients assigned to placebo crossed over to regorafenib and this may have accounted for the lack of a significant difference in overall survival between the regorafenib and placebo arms (29 [22 %] vs 17 [17 %] events, HR 0.77, 95 % CI 0.42–1.41, p = 0.199). Overall response rates were 4.5 % and 1.5 % (all partial responses), and stable disease at any time and of any duration occurred in 71.4 % and 33.3 % of patients in the regorafenib and placebo arms, with reported disease control rates of 52.6 and 9.1 % respectively (p < 0.0001).
The most common drug-related adverse events of any grade in patients treated with regorafenib (56 %) and placebo (14 %) were HFSR. Any grade ≥3 adverse events occurred in 61 % and 14 % of patients respectively, with the most common related to regorafenib being hypertension (23 %) and diarrhea (5 %). There were ten deaths reported as related to adverse events, three were deemed drug-related, two receiving regorafenib (cardiac arrest and hepatic failures), and one treated with placebo (fatigue). Adverse events leading to dose modifications were more frequent in patients treated with regorafenib (72 %) than with placebo (26 %), although the occurrence of adverse events leading to permanent discontinuation was similar (6 % vs 8 %) between the groups.
Data from the GRID study, in particular the fivefold increase in PFS and the excellent disease control rate in patients treated with regorafenib, led to the approval by the FDA of regorafenib in treating patients with locally advanced, unresectable, or metastatic GIST who have previously been treated with imatinib mesylate and sunitinib malate [11].
HCC
A phase II study by Bruix et al. assessed safety (primary endpoint) and efficacy (secondary endpoints) of single-agent regorafenib in 36 patients with HCC and preserved to mildly impaired liver function (Child-Pugh A), progressing on previous treatment with sorafenib (Table 2) [24]. The median treatment duration was 19.5 weeks (range 2–103). At the data cutoff, three patients remained on treatment. Reasons for discontinuation were adverse events (56 %), disease progression (28 %), consent withdrawal (6 %) and death (3 %). Among the 18 patients who stopped treatment due to adverse events, 7 (19 %) were reported as drug-related. Seventeen patients (47 %) required dose reductions (mostly for adverse events [n = 15]), 35 (97 %) patients had treatment interruption (mostly for adverse events [n = 32] or patient error [n = 11]). The most frequent (>5 %) treatment-related grade ≥3 adverse events were fatigue (17 %), HFSR (14 %), diarrhea (6 %), hyperbilirubinemia (6 %) hypophosphatemia (6 %). Disease control was achieved in 26 patients (partial response n = 1; stable disease n = 25). Median time to progression was 4.3 months (95 % CI 2.9–13.1) and median overall survival was 13.8 months (95 % CI 9.3–18.3). Accounting for the fact that these patients had tolerated prior treatment with sorafenib, the authors found regorafenib to have acceptable and manageable toxicity profile, comparable with those reported for sorafenib in this setting [29]. No dose adjustment was recommended in patients with mild or moderate hepatic impairment [11, 24].
This phase II study provided the rationale for the phase III RESORCE study (REgorafenib after SORafenib in patients with hepatoCEllular carcinoma, NCT01774344) of regorafenib after sorafenib in patients with hepatocellular carcinoma [30]. The RESORCE study is a randomized, double-blind, placebo-controlled multicenter phase III study of regorafenib in patients with HCC after sorafenib. The study is currently recruiting patients, of whom approximately 530 who meet the entry criteria [30] will be randomly assigned in a 2:1 ratio to regorafenib or placebo. The primary endpoint of the study is overall survival. Secondary endpoints include objective tumor response, PFS, time to tumor progression and disease control. Study treatment arms are regorafenib 160 mg orally every day for 3 weeks of every 4-week cycle plus best supportive care and placebo for 3 weeks of every 4-week cycle (i.e. 3 weeks on, 1 week off) plus best supportive care. The estimated primary completion date is October 2015 (final completion October 2016).
Clinical pharmacology of regorafenib
Mode of action and delivery
Regorafenib inhibits a wider spectrum of tyrosine kinase targets involved in regulating angiogenesis and oncogenic signaling compared with other inhibitors of angiogenesis such as bevacizumab and aflibercept or other MKI such as sorafenib.
Bevacizumab is a humanized monoclonal antibody to VEGF-A and is indicated for use in a number of solid tumors including mCRC, locally advanced or metastatic non-small-cell lung cancer (NSCLC), metastatic breast cancer, metastatic RCC and glioblastoma [31, 32]. Aflibercept (ziv-aflibercept in the US) is a recombinant fusion protein consisting of the second immunoglobulin (Ig) domain of VEGFR-1 and the third Ig domain of VEGFR-2 fused to human Ig and exhibits affinity for VEGF-A, VEGF-B and placental growth factor [33]. Aflibercept is indicated for use in combination with FOLFIRI in patients with mCRC progressing on oxaliplatin-based chemotherapy [34, 35]. In comparison with these agents, regorafenib inhibits a number of angiogenic specific targets including VEGFR-1 -2, and -3, and TIE2 and stromal RTKs PDGFRβ and FGFR-1. Furthermore, regorafenib appears to be a more potent inhibitor of oncogenic kinases (BRAF V600E, RET, RAF-1 and KIT) than sorafenib [16, 21].
Regorafenib film-coated tablets should be swallowed whole with water after a low-fat meal at the same time every day (breakfast, <30 % fat) [10]. An example of a low-fat meal would include one portion of cereal (about 30 g), one glass of skimmed milk, one slice of toast with jam, one glass of apple juice, and one cup of coffee or tea (520 cal, 2 g of fat). In a food-effect study, 24 healthy men received a single 160 mg dose of regorafenib on three separate occasions: under a fasted state, with a high-fat meal and with a low-fat meal. A high-fat meal (945 cal and 54.6 g fat) increased the mean area under the plasma concentration vs time curve (AUC) of regorafenib by 48 % and decreased the mean AUC of the M-2 and M-5 metabolites by 20 and 51 %, respectively, compared with the fasted state. A low-fat meal (319 cal and 8.2 g fat) increased the mean AUC of regorafenib, M-2, and M-5 by 36, 40, and 23 %, respectively compared with fasted conditions [11].
As described previously the recommended dose and regimen is 160 mg (4 tablets of 40 mg) once daily for 21 days out of 28 cycles. Treatment should be administered until disease progression or the occurrence of unacceptable toxicity [10, 11].
Pharmacokinetic properties of regorafenib
Regorafenib is metabolized primarily in the liver by CYP3A4 (oxidative metabolism—special attention must be paid when administering to patients receiving CYP3A4 inhibitors/inducers as described below) and UGT1A9 (glucuronidation). The main circulating metabolites of regorafenib in human plasma, M-2 and M-5, are both pharmacologically active and have similar concentrations to regorafenib at steady state. The metabolites may be reduced or hydrolyzed in the gastrointestinal tract by microbial flora, allowing reabsorption of the unconjugated active substances (enterohepatic circulation). The pharmacokinetic data for regorafenib are based primarily on preclinical data and data from phase 1 studies [10, 11, 20, 21].
Absorption
Following a single oral dose of 160 mg given to patients with solid tumors, regorafenib reaches a geometric mean peak plasma level (C max) of 2.5 μg/mL at a median time of 4 h, and a mean AUC of 70.4 μg h/mL.
Distribution
Plasma concentration–time profiles for regorafenib and M-2 and M-5 showed multiple peaks across the 24-h dosing interval (attributable to enterohepatic circulation). In vitro protein binding of regorafenib, M-2, and M-5 to human plasma proteins is high at 99.5, 99.8, and 99.95 %, respectively.
Elimination
Following oral administration (160 mg), the mean elimination half-lives for regorafenib and M-2 in plasma are similar at 28 h (range 14–58) and 25 h (range 14–32), respectively. The mean elimination half-life for M-5 is longer at 51 h (range 32–70). Studies using a radiolabeled oral solution of regorafenib (120 mg) showed that approximately 90 % of the radioactive dose was recovered within 12 days of administration, with about 71 % of the dose excreted in feces (47 % as parent compound, 24 % as metabolites), and about 19 % of the dose excreted in urine as glucuronides. Urinary excretion of glucuronides decreased below 10 % under steady-state conditions.
Linearity/non-linearity
Systemic exposure of regorafenib at steady-state increases in a dose proportional manner up to 60 mg doses, but at doses >60 mg there is a lack of dose proportional increases. Accumulation of regorafenib at steady state results in an approximately two-fold increase in plasma concentrations, which is consistent with the elimination half-life and dosing frequency. After oral administration of 160 mg regorafenib, at steady state, regorafenib reaches mean peak plasma levels of approximately 3.9 μg/mL (8.1 μM) and a geometric mean AUC of 58.3 μg h/mL. The coefficient of variation for AUC and C max is between 35 and 44 %. The peak-to-trough ratio of mean plasma concentrations is <2. Both M-2 and M-5 metabolites exhibit non-linear accumulation, which might be caused by enterohepatic recycling, or saturation of the UGT1A9 pathway. Whereas plasma concentrations of M-2 and M-5 after a single dose of regorafenib are much lower than those of the parent compound, the steady-state plasma concentrations of M-2 (3.3 μg/mL) and M-5 (2.9 μg/mL) are comparable with those of regorafenib.
The Strumberg phase 1 study in mCRC reported a similar pharmacokinetic profile for regorafenib and its metabolites as described. In the expansion cohort of patients treated with 160 mg of regorafenib, at steady state, systemic exposure of M-2 (48 μg h/mL) and M-5 (65–79 μg h/mL) were similar to that recorded for parent regorafenib (45–50 μg h/mL) [21].
A population pharmacokinetic model was used to investigate the effects of patient demographics (sex, weight, body mass index, height, and ethnic group) and baseline parameters (kidney and liver function test, protein levels and hematocrit/hemoglobin levels) on drug exposure in the CORRECT study [36]. The model for regorafenib, M-2, and M-5 was developed with NONMEM, based on pharmacokinetic data from a densely sampled, open-label, phase I dose-escalation study of regorafenib in 67 patients with advanced solid tumors. This model was subsequently applied to the sparsely sampled CORRECT study. The pharmacokinetics of regorafenib, M-2, and M-5 were described by a two-compartmental model that provided an adequate fit of the plasma concentration–time profiles from both studies. Derived pharmacokinetic parameter estimates were used to calculate the individual exposure (Cavmd) of regorafenib, M-2, and M-5 in the CORRECT study. Overall, a moderate to high variability in the regorafenib pharmacokinetics was observed in the CORRECT study, with a coefficient of variance of clearance of 44 % and even higher variability in exposure to M-2 and M-5. Analysis of the CORRECT study showed that higher bilirubin levels at baseline were associated with higher individual exposure of regorafenib and M-2 (Cavmd −14 to +18 %) when comparing the 5th and 95th percentiles of the observed bilirubin distribution to the median baseline bilirubin levels, the overall variability was −48.7 to +79.9 %. Higher body weight was associated with lower exposure of M-2 and M-5, and exposure of M-5 was higher in women (Cavmd 77 %) than in men (the overall variability of M-2 and M-5 were −69.8 to +149 % and −89.2 to +348 %, respectively). The magnitude of observed covariate effects however was small compared with the overall high variability in exposure. The other tested covariates did not have an impact. The authors concluded that the effects of the covariates tested in population pharmacokinetic analysis of the CORRECT data were not considered clinically relevant in light of the overall high variability in exposure [36].
Drug-drug interactions of regorafenib
There is potential for drug-drug interactions when administering regorafenib [10, 11]. Co-administration with strong inducers of cytochrome P-450 (CYP) 3A4 such as rifampicin, phenytoin, carbamazepine, phenobarbital, and St John’s Wort should be avoided. Co-administration with rifampicin (600 mg for 9 days in 22 healthy male volunteers) with a single dose of regorafenib (160 mg on day 7) was associated with a reduction in the AUC of regorafenib of approximately 50 % and a three- to four-fold increase in exposure to the M-5 metabolite, but with no change in exposure to the M-2 metabolite.
Administration of ketoconazole (400 mg for 18 days), a strong CYP3A4 inhibitor, with a single dose of regorafenib (160 mg on day 5) in 18 healthy male volunteers resulted in an increase in the mean exposure (AUC) of regorafenib of approximately 33 %, and a decrease in mean exposure to the M-2 and M-5 metabolites of approximately 90 %. It is recommended that the concomitant use of strong inhibitors of CYP3A4 activity (e.g., clarithromycin, grapefruit juice, itraconazole, ketoconazole, posaconazole, telithromycin, and voriconazole) is avoided as their influence on the steady-state exposure of regorafenib and its metabolites has not been studied.
In vitro study data also suggest an interaction between regorafenib and warfarin. In patients with advanced solid tumors administered a single oral dose of 10 mg of warfarin (substrate of CYP2C9) 1 week before and 2 weeks after regorafenib (160 mg once daily) the mean AUC of warfarin increased by 25 %.
Data from in vitro studies indicate that regorafenib and M-2 inhibit glucuronidation mediated by UGT1A1 and UGT1A9 whereas M-5 only inhibits UGT1A1 at concentrations which are achieved in vivo at steady state [10]. In a phase Ib study of regorafenib in combination with FOLFIRI or FOLFOX in patients with mCRC, administration of regorafenib with a 4-day break prior to administration of irinotecan resulted in a significant increase in irinotecan and its metabolite SN-38 (a substrate of UGT1A1). For irinotecan and SN-38, the AUC following exposure to regorafenib in cycle-2 was significantly higher than prior to regorafenib dosing in cycle-1, the ratios of AUC values (cycle-2:cycle-1) were 1.28 (95 % CI 1.06–1.54) and 1.44 (95 % CI 1.12–1.85), respectively [37]. This suggests that co-administration of regorafenib may increase systemic exposure to UGT1A1 and UGT1A9 substrates.
In vitro data suggest that regorafenib is an inhibitor of breast cancer resistance protein (BCRP) (ABCG2, IC50 values 40–70 nM) and P-glycoprotein (ABCB1, IC50 value approximately 2 μM). Co-administration of regorafenib may increase the plasma concentrations of concomitant BCRP substrates, such as methotrexate, or P-glycoprotein substrates, such as digoxin.
Tolerability and monitoring
The overall safety profile of regorafenib is based on data from more than 1,200 cancer patients (all types of cancer) treated in clinical trials [10, 11]. The most frequently observed drug-related adverse events (≥20 %) are asthenia/fatigue, decreased appetite and food intake, HFSR, diarrhea, weight loss, infection, hypertension and dysphonia. The most serious adverse events are severe liver injury, hemorrhage, and gastrointestinal perforation. Adverse events that carry a specific warning and/or specify precautions in the regorafenib prescribing information, which may require monitoring leading to dose modifications or treatment discontinuation, are summarized in Table 3.
Data from the CORRECT study report that the most common adverse events including diarrhea, fatigue, HFSR, hypertension, and rash occurred early during treatment and that close early monitoring of adverse events and management by dose modification is recommended [38].
The incidence of HFSR is commonly increased in cancer patients treated with regorafenib. A meta-analysis [39] of 1,078 patients treated with regorafenib from four studies [24, 26, 28, 40] reported that the relative risk (RR) of all grade (RR = 5.4, 95 % CI 3.76–7.76, p < 0.001) and high grade (RR = 41.99, 95 % CI 5.88–299.93, p < 0.001) HFSR was significantly increased in patients treated with regorafenib compared with controls. In this analysis, the incidence of HFSR varied with tumor type; 71.4 % for RCC, 60.2 % for GIST, 50 % for HCC and 46.6 % mCRC. The mechanism for HFSR in patients treated with regorafenib is not known but the occurrence of HFSR appears to be a class effect for MKI [39]. Clinically, HFSR patients initially present with erythema and dysesthesia on the palms and soles (areas of mechanical stress) followed by painful sharply demarcated erythema and callus-like thickening. Large, painful blisters may also form in more severe cases. HFSR can be managed by the prophylactic and therapeutic use of cotton socks and gloves, gel inserts, soft footwear and pedicures for calluses. The treatment of symptoms often includes the use of topical applications with moisturizing creams and soaps, keratolytics (e.g., urea, salicylic acid), and the use of cold immersions and pain relief medication to ease the pain. Severe reactions may require dose modifications, interruptions, or even discontinuation (Table 3). HFSR appears to be one of the most common reasons for dose reductions of regorafenib [22, 40, 41] however; dose re-escalations appear to be tolerated [22].
Biochemical (hypophosphatemia, hypocalcemia, hyponatremia, and hypokalemia) and metabolic laboratory abnormalities (increases in thyroid stimulating hormone, lipase and amylase) have been reported in patients treated with regorafenib [22, 26, 40]. It is recommended to monitor biochemical and metabolic parameters during regorafenib treatment and to apply appropriate replacement therapy if required. Dose reduction or modification, or in severe or persistent cases discontinuation, should be considered.
Instances where regorafenib doses should be interrupted, reduced, or discontinued are summarized in Table 4.
For populations that might be less tolerant to exposure to regorafenib, animal studies have reported regorafenib to be embryolethal, and teratogenic, with increased incidences of cardiovascular, genitourinary and skeletal abnormalities found in the fetuses from animals exposed to the drug. Advice of these risks should be provided to patients of reproductive age, and special appraisal of the harm to the fetus should be undertaken if the patients become pregnant during treatment.
No clinically important differences in the mean exposure of regorafenib or the active metabolites M-2 and M-5 were observed in patients with HCC and mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment compared with patients with normal hepatic function. No dose adjustment is recommended in patients with mild or moderate hepatic impairment [11, 24]. However, it is recommended that patients with hepatic impairment are closely monitored for adverse reactions. Regorafenib is not recommended for use in patients with severe hepatic impairment (Child-Pugh C), as it has not been studied in this population.
No clinically relevant differences in the mean exposure of regorafenib and the active metabolites M-2 and M-5 were observed in patients with mild renal impairment (CLcr 60–89 mL/min) compared to patients with normal renal function following regorafenib 160 mg daily for 21 days. No dose adjustment is recommended for patients with mild renal impairment. Limited pharmacokinetic data are available from patients with moderate renal impairment (CLcr 30–59 mL/min). Regorafenib has not been studied in patients with severe renal impairment or end-stage renal disease.
In the CORRECT and GRID studies in subgroup analyses, no differences in safety and efficacy of regorafenib were observed in patients <65 years compared with patients ≥65 years of age, or in those of different ethnic origins [28, 38, 42].
Perspectives
In the current treatment landscape in which regorafenib is approved for use, there are specific considerations for the clinical pharmacist to ensure its safe and effective use.
Pharmacists should provide advice on drug administration, including patient counseling regarding strict adherence to the regorafenib oral regimen, where tablets are to be taken every morning with a low-fat meal. Regorafenib tablets should be swallowed whole, not crushed or chewed, and patients should avoid overdosing if a previous dose is missed. Advice should also be provided by the pharmacist about potential drug-drug interactions with regorafenib, and a thorough medication profile review of the patient should be undertaken. Regorafenib may increase exposure of drugs metabolized by specific CYP450 enzymes (2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) and UGT1A1 and 1A9 enzymes. Regorafenib concentrations may be increased or decreased by CYP3A4 inhibitors or inducers. Patients should be advised to alert their health care provider if they take any new medications, vitamins or herbal products, and food (especially grapefruit juice).
Patients should also be counseled on the risk of serious adverse events associated with regorafenib and advice should be give to health care providers on patient monitoring. There are specific warnings and precautions for the use of regorafenib (detailed in Table 3). In patients with HCC, liver failure is a major concern and severe drug-related toxicities have been reported in trials of other MKI in HCC [43]. However, in the phase II study by Bruix et al. regorafenib was tolerated in patients with mild- or moderate hepatic impairment [11, 24]. Recommendations are that hepatic toxicity should be monitored at baseline and throughout treatment.
Regorafenib-related HFSR is manageable by prophylactic measures, the treatment of symptoms, and the use of treatment interruptions and dose reductions. Patients appear to tolerate resumption of treatment and escalation of dose. Patients should be advised for symptoms of hypertension that include severe headache, light-headedness, and neurological symptoms, and their blood pressure should be monitored throughout treatment. Patients should be counseled on the increased risk of bleeding/hemorrhage and should be advised to inform their healthcare provider to any surgical procedure while on regorafenib, or if they have recently undergone a surgical procedure while initiating regorafenib. The increased risk of cardiac toxicity, myocardial ischemia, and/or infarction, should be highlighted with dizziness, chest pain, or dyspnea indicated as symptoms.
In specific patient populations, advice should be given to patients of reproductive age to avoid pregnancy. Regorafenib appears to be safe and effective in patients <65 and ≥65 years of age, [26, 28, 42], but there are little data in patients ≥75 years old. Regorafenib is effective in Asian patients in subgroup analyses of randomized clinical trials [26, 28]. The CONCUR clinical trial is currently investigating the efficacy and safety of regorafenib in an Asian population [44].
Future development of regorafenib in gastrointestinal cancers
A number of studies are investigating the positioning of single-agent regorafenib in patients with gastrointestinal cancers. In patients with mCRC in the CORRECT study, subgroup analysis demonstrated regorafenib to be active irrespective of the KRAS or BRAF tumor mutation status [26]. An ongoing trial is investigating regorafenib in combination with FOLFIRI in mCRC patients whose tumors harbor mutations at codons 12 and 13 (exon 2) of the KRAS gene or codon 600 (exon 15) of the BRAF gene, previously treated with FOLFOX [45]. If this is proven to be effective, this would have important implications for the use of regorafenib in patients with KRAS exon 2 mutated tumors where EGFR mAbs have previously been proven to be ineffective [2–4]. Recent data also indicate that patients with tumors harboring any RAS mutations (any mutations at specific codons within exons 2–4 of the KRAS or the NRAS gene) do not benefit from treatment with EGFR mAbs [46].
Previous reports from the ML18147 (with bevacizumab) and VELOUR (with aflibercept) studies [34, 47] suggest that post progression use of anti-angiogenesis therapy appears to be a successful strategy for treating chemorefractory mCRC. Analysis from the CORRECT and GRID studies appear to support the use of regorafenib in patients progressing on previous anti-angiogenesis therapy in mCRC (progressing on bevacizumab) and GIST (progressing on sunitinib), respectively.
In the adjuvant treatment setting, the COAST study is a randomized double-blind placebo-controlled phase III study (currently registered, not yet open for recruitment) investigating regorafenib as adjuvant therapy for CRC with resected liver metastases. This trial aims to evaluate the efficacy and safety of regorafenib (160 mg for 3 weeks of a 4-week cycle) compared with placebo in CRC patients after curative resection of their liver metastases and completion of all planned chemotherapy. The primary endpoint is disease-free survival. The enrollment of 750 patients is estimated with an expected completion date of September 2021 [48].
There are currently no predictive biomarkers to identify those patients who will benefit from regorafenib in gastrointestinal cancers. Prespecified biomarker analysis is planned for the CONCUR study of best supportive care plus regorafenib or placebo in Asian mCRC patients who have progressed on standard therapy [44]. A phase II study is underway to determine the efficacy of single-agent regorafenib, and to identify biomarkers in mCRC patients who have failed one prior treatment for mCRC [49]. The estimated enrollment is for 52 patients with a final completion date December 2017 (primary date March 2017).
The REFRAME study is currently recruiting to investigate the efficacy and safety of single-agent regorafenib in previously untreated patients with mCRC who are frail or unsuitable for polychemotherapy regimens. The primary endpoint is PFS at 6 months [50] and the estimated primary completion date is June 2015.
Regorafenib dose and combination with standard chemotherapy regimens is also under investigation. Promising efficacy and safety has been reported with 100 mg daily regorafenib in a continuous schedule [25, 41]. The flexibility of regorafenib dosing is apparent when used in combination with irinotecan- or oxaliplatin-based chemotherapy [37, 45, 51].
FOLFIRI and FOLFOX are standard chemotherapy regimens in CRC [3]. A phase 1b study by Schultheis et al. investigated regorafenib in combination with FOLFOX or FOLFIRI in patients with mCRC [37]. Safety and pharmacokinetics were primary objectives and tumor response was a secondary objective. Forty-five patients were treated with FOLFOX or FOLFIRI every 2 weeks; on days 4–10, patients received regorafenib 160 mg orally once daily. The median duration of treatment was 108 days (range 2–345 days). Treatment was stopped for adverse events or death (17 patients), disease progression (11 patients), and consent withdrawal or investigator decision (11 patients). Six patients remained on regorafenib at data cutoff (two without chemotherapy). Drug-related adverse events occurred in 44 patients (grade ≥3 in 32 patients: mostly neutropenia [17 patients] and leukopenia, HFSR, and hypophosphatemia [four patients each]). Thirty-three patients (87 %) achieved disease control (partial response or stable disease) for a median of 126 (range 42–281) days. Regorafenib had acceptable tolerability in combination with chemotherapy, with increased exposure of irinotecan and SN-38 but no significant effect on 5-fluorouracil or oxaliplatin pharmacokinetics. This study provided the rational for a randomized placebo-controlled phase II trial of regorafenib plus FOLFIRI compared with placebo plus FOLFIRI in patients with mCRC progressing on oxaliplatin-based chemotherapy. The primary endpoint is PFS, with expected enrolment of 240 patients and completion date of February 2021 [45].
The CORDIAL study was a single-arm phase II study which investigated regorafenib in combination with mFOLFOX6 in patients with previously untreated mCRC (primary completion date March 2012, final completion date April 2014) [51, 52]. The treatment combination had an acceptable toxicity profile with some efficacy. Fifty three patients received mFOLFOX6 on days 1 and 15, and regorafenib (160 mg) on days 4–10 and 18–24 of each 4-week cycle. The best overall response rate (in 41 evaluable) patients was 44 % (95 % CI 28–60), all were partial responses and 17 patients had stable disease with a median duration of 7.6 months (95 % CI 5.5–8.5). The disease control rate was 85 % (95 % CI 71–94). Median PFS in the ITT population (n = 54) was 8.5 months (95 % CI 7.4–11.3). At the time of the data cutoff (November 15, 2012) median overall survival had not been reached. The most common reported grade ≥3 adverse events were decreased neutrophil count (40 %), hypertension (28 %), diarrhea (23 %), increased serum lipase (19 %), and hypophosphatemia and peripheral neuropathy (13 %). Regorafenib was permanently discontinued in four patients with dose reductions and dose interruptions recorded in 24 (45 %) and in 47 (89 %) patients respectively.
Regorafenib is in clinical development in other gastrointestinal-related cancers. As described previously, following encouraging phase II study data [24], the phase III RESORCE study is investigating single-agent regorafenib in patients with advanced HCC [30]. Single-agent regorafenib is also under investigation in the randomized phase II INTEGRATE study of regorafenib versus placebo in patients with advanced esophagogastric cancer performed by the Australian Gastrointestinal Trials Group [53]. Finally, a randomized phase II study is investigating regorafenib in combination with FOLFOX in patients with unresectable or metastatic esophagogastric cancer (estimated primary completion date July 2015) [54].
Summary
Single-agent regorafenib is an oral multikinase inhibitor approved for use in the continuum of care in patients with mCRC and GIST. It is administered at 160 mg daily for the first 21 days of a 28-day cycle. It is currently being investigated in phase III study of patients with HCC, and in phase II studies in patients with advanced unresectable or metastatic esophagogastric cancer. Understanding the pharmacokinetics, efficacy, and safety profile of single-agent regorafenib will ensure that the oncology clinical pharmacist is able to contribute in the MDT to the safe and efficient delivery of regorafenib in gastrointestinal cancer patients. This information is important to the clinical pharmacist for the future development of regorafenib in this treatment setting.
References
Hu QD, Zhang Q, Chen W, Bai XL, Liang TB (2013) Human development index is associated with mortality-to-incidence ratios of gastrointestinal cancers. World J Gastroenterol 19:5261–5270
Prenen H, Vecchione L, Van Cutsem E (2013) Role of targeted agents in metastatic colorectal cancer. Target Oncol 8:83–96
Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, Nordlinger B, van de Velde CJ, Balmana J, Regula J, Nagtegaal ID, Beets-Tan RG, Arnold D, Ciardiello F, Hoff P, Kerr D, Kohne CH, Labianca R, Price T, Scheithauer W, Sobrero A, Tabernero J, Aderka D, Barroso S, Bodoky G, Douillard JY, El Ghazaly H, Gallardo J, Garin A, Glynne-Jones R, Jordan K, Meshcheryakov A, Papamichail D, Pfeiffer P, Souglakos I, Turhal S, Cervantes A (2012) ESMO Consensus Guidelines for management of patients with colon and rectal cancer. a personalized approach to clinical decision making. Ann Oncol 23:2479–2516
National Comprehensive Cancer Network Guidelines Colon Cancer Version 3.2013 http://www.nccn.org/professionals/physician_gls/f_guidelines.asp
Glivec® summary of product characteristics http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000406/WC500022207.pdf
SUTENT® summary of product characteristics http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000687/WC500057737.pdf
Nexavar® summary of product characterstics http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000690/WC500027704.pdf
Gossage L, Eisen T (2010) Targeting multiple kinase pathways: a change in paradigm. Clin Cancer Res 16:1973–1978
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, Thierauch KH, Zopf D (2011) Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 129:245–255
Stivarga® summary of product characteristics http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002573/WC500149164.pdf
Stivarga® (regorafenib) label- accessdata FDA http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204369lbl.pdf
EMA summary of opinion initial authorization—Stivarga® (regorafenib) http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/002573/WC500144844.pdf
Sessions JK, Valgus J, Barbour SY, Iacovelli L (2010) Role of oncology clinical pharmacists in light of the oncology workforce study. J Oncol Pract 6:270–272
Al-Quteimat OM, Al-Badaineh MA (2013) Role of oncology clinical pharmacist: a case of life-saving interventions. Int J Basic Clin Pharmacol 2:655–658
The NHS cancer plan and the pharmacy contribution to cancer care (January 2001) http://www.bopawebsite.org/contentimages/publications/OncologyPharmacy.pdf
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA (2004) BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64:7099–7109
Zopf D, Heinig R, Schutz G, Thierauch K-H, Hirth-Dietrich C, Hafner F-T, Christensen O, Lin T, Wilhelm S, Radtke M (2010) Regorafenib (BAY 73–4506): identification of clinically relevant metabolites and their preclinical pharmacology. 101st Annual Meeting of the American Association for Cancer Research, Washington, DC, April 17–21 Abstract 1666
Serrano-Garcia C, Heinrich MC, Zhu M, Raut CP, Eilers G, Ravegnini G, Demetri GD, Bauer S, Fletcher JA, George S (2013) In vitro and in vivo activity of regorafenib (REGO) in drug-resistant gastrointestinal stromal tumors (GIST). J Clin Oncol 31(Suppl):Abstract 10510
Abou-Elkacem L, Arns S, Brix G, Gremse F, Zopf D, Kiessling F, Lederle W (2013) Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol Cancer Ther 12:1322–1331
Mross K, Frost A, Steinbild S, Hedbom S, Buchert M, Fasol U, Unger C, Kratzschmar J, Heinig R, Boix O, Christensen O (2012) A phase I dose-escalation study of regorafenib (BAY 73–4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res 18:2658–2667
Strumberg D, Scheulen ME, Schultheis B, Richly H, Frost A, Buchert M, Christensen O, Jeffers M, Heinig R, Boix O, Mross K (2012) Regorafenib (BAY 73–4506) in advanced colorectal cancer: a phase I study. Br J Cancer 106:1722–1727
George S, Wang Q, Heinrich MC, Corless CL, Zhu M, Butrynski JE, Morgan JA, Wagner AJ, Choy E, Tap WD, Yap JT, Van den Abbeele AD, Manola JB, Solomon SM, Fletcher JA, von Mehren M, Demetri GD (2012) Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol 30(19):2401–2407
George S, Feng Y, von Mehren M, Choy E, Corless CL, Hornick JL, Butrynski JE, Wagner AJ, Solomon S, Morgan JA, Heinrich MC, Demetri GD (2013) Prolonged survival and disease control in the academic phase II trial of regorafenib in GIST: Response based on genotype. J Clin Oncol 31 (Suppl):Abstract 10511
Bruix J, Tak WY, Gasbarrini A, Santoro A, Colombo M, Lim HY, Mazzaferro V, Wiest R, Reig M, Wagner A, Bolondi L (2013) Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: Multicentre, open-label, phase II safety study. Eur J Cancer 49(16):3412–3419
Shimizu T, Tolcher AW, Patnaik A, Papadopoulos K, Christensen O, Lin T, R. BJG (2010) Phase I dose-escalation study of continuously administered regorafenib (BAY 73–4506), an inhibitor of oncogenic and angiogenic kinases, in patients with advanced solid tumors. J Clin Oncol 28 (15S):abstr 3035
Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz HJ, Goldberg RM, Sargent DJ, Cihon F, Cupit L, Wagner A, Laurent D, Group CS (2013) Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381:303–312
Siena S, Grothey A, Sobrero A, Falcone A, Ychou M, Lenz HJ, Yoshino T, Cihon F, Pawar V, Van Cutsem E (2013) Effects of regorafenib therapy on health-related quality of life in patients with metastatic colorectal cancer in the phase III CORRECT study. Eur J Cancer 49 (Suppl):Abstract 2156
Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H, Badalamenti G, Blackstein M, Le Cesne A, Schoffski P, Maki RG, Bauer S, Nguyen BB, Xu J, Nishida T, Chung J, Kappeler C, Kuss I, Laurent D, Casali PG, investigators Gs (2013) Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381:295–302
Abou-Alfa GK, Schwartz L, Ricci S, Amadori D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz B, Taylor I, Moscovici M, Saltz LB (2006) Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol 24:4293–4300
Study of regorafenib after sorafenib in patients with hepatocellular carcinoma (RESORCE). ClinicalTrials.gov Identifier: NCT01774344 http://clinicaltrials.gov/ct2/show/record/NCT01774344 Accessed November 2013
National Cancer Institute FDA approval bevacizumab http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab
Avastin® summary of product characteristics http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000582/WC500029271.pdf
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Radziejewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS (2002) VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A 99:11393–11398
Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, Ruff P, van Hazel GA, Moiseyenko V, Ferry D, McKendrick J, Polikoff J, Tellier A, Castan R, Allegra C (2012) Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 30:3499–3506
ZALTRAP® (ziv-aflibercept) label access data FDA http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125418s000lbl.pdf
Trnkova ZJ, Grothey A, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, Lenz HJ, Cihon F, Wagner A, Reif S, Smeets J, Diefenbach K, Laurent D, Van Cutsem E (2013) Population pharmacokinetics analysis of regorafenib and its active metabolites from the phase III CORRECT study of metastatic colorectal cancer. Ann Oncol 24(Suppl 4):iv37
Schultheis B, Folprecht G, Kuhlmann J, Ehrenberg R, Hacker UT, Kohne CH, Kornacker M, Boix O, Lettieri J, Krauss J, Fischer R, Hamann S, Strumberg D, Mross KB (2013) Regorafenib in combination with FOLFOX or FOLFIRI as first- or second-line treatment of colorectal cancer: results of a multicenter, phase Ib study. Ann Oncol 24:1560–1567
Grothey A, Sobrero AF, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Argiles G, Yoshino T, Lenz H-J, Goldberg RM, Sargent DJ, Cihon F, Wagner A, Cupit L, Laurent D, Van Cutsem E (2013) Time profile of adverse events (AEs) from regorafenib (REG) treatment for metastatic colorectal cancer (mCRC) in the phase III CORRECT study. J Clin Oncol 31 (Supp):Abstract 3637.
Belum VR, Wu S, Lacouture ME (2013) Risk of hand-foot skin reaction with the novel multikinase inhibitor regorafenib: a meta-analysis. Invest New Drugs 31:1078–1086
Eisen T, Joensuu H, Nathan PD, Harper PG, Wojtukiewicz MZ, Nicholson S, Bahl A, Tomczak P, Pyrhonen S, Fife K, Bono P, Boxall J, Wagner A, Jeffers M, Lin T, Quinn DI (2012) Regorafenib for patients with previously untreated metastatic or unresectable renal-cell carcinoma: a single-group phase 2 trial. Lancet Oncol 13(10):1055–1062
Kies MS, Blumenschein Jr. GR, Christensen O, Lin T, Tolcher AW (2010) Phase I study of regorafenib (BAY 73–4506), an inhibitor of oncogenic and angiogenic kinases, administered continuously in patients (pts) with advanced refractory non-small cell lung cancer (NSCLC). J Clin Oncol 28 (15 Suppl):abstr 7585
Van Cutsem E, Alberto A, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Argilés G, Yoshino T, Lenz H-J, Goldberg RM, Sargent DJ, Cihon F, Wagner A, Laurent D, Cupit L, Grothey A (2013) Regorafenib (REG) in progressive metastatic colorectal cancer (mCRC): Analysis of age subgroups in the phase III CORRECT trial. J Clin Oncol 31 (Suppl):Abstract 3636
Faivre S, Raymond E, Boucher E, Douillard J, Lim HY, Kim JS, Zappa M, Lanzalone S, Lin X, Deprimo S, Harmon C, Ruiz-Garcia A, Lechuga MJ, Cheng AL (2009) Safety and efficacy of sunitinib in patients with advanced hepatocellular carcinoma: an open-label, multicentre, phase II study. Lancet Oncol 10:794–800
Asian subjects with metastatic colorectal cancer treated with regorafenib or placebo after failure of standard therapy (CONCUR) ClinicalTrials.gov Identifier: NCT01584830. http://clinicaltrials.gov/show/NCT01584830. Accessed November 2013
Regorafenib + FOLFIRI versus placebo + FOLFIRI as 2nd line Tx in metastatic colorectal cancer ClinicalTrials.gov Identifier: NCT01298570 http://clinicaltrials.gov/show/NCT01298570 Accessed November 2013
Douillard JY, Rong A, Sidhu R (2013) RAS mutations in colorectal cancer. N Engl J Med 369:2159–2160
Bennouna J, Sastre J, Arnold D, Osterlund P, Greil R, Van Cutsem E, von Moos R, Vieitez JM, Bouche O, Borg C, Steffens CC, Alonso-Orduna V, Schlichting C, Reyes-Rivera I, Bendahmane B, Andre T, Kubicka S, Investigators MLS (2013) Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol 14:29–37
Regorafenib as adjuvant therapy for colorectal cancer (CRC) with resected liver metastases (COAST). ClinicalTrials.gov Identifier: NCT01939223. http://clinicaltrials.gov/show/NCT01939223 Accessed November 2013
Study to determine the efficacy of regorafenib in metastatic colorectal cancer patients and to discover biomarkers. ClinicalTrials.gov Identifier: NCT01949194. http://clinicaltrials.gov/show/NCT01949194 Accessed November 2013.
Regorafenib in frail and/or unfit for chemotherapy patients with metastatic colorectal cancer (REFRAME). ClinicalTrials.gov Identifier: NCT01875380. http://clinicaltrials.gov/show/NCT01875380 Accessed November 2013.
Argiles Martinez G, Troiani T, Rivera F, Sobrero A, Benson A, Guillen C, Garosi VL, Wagner A, Saunders M (2013) First-line treatment with regorafenib (REG) in combination with mFOLFOX6 (folinic acid + 5-fluorouracil [5-FU] + oxaliplatin) for metastatic colorectal cancer (mCRC): A single-arm, open-label phase II clinical trial. Eur J Cancer 49 (Supplement 2):Abstract 2367
First line treatment of metastatic colorectal cancer with mFOLFOX6 in combination with regorafenib. ClinicalTrials.gov Identifier: NCT01289821. http://clinicaltrials.gov/show/NCT01289821 Accessed November 2013
A randomised phase II double-blind study of regorafenib or placebo in refractory advanced oesophago-gastric cancer (AOGC). ACTRN.org Identifier: ACTRN12612000239864. https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12612000239864 Accessed November 2013
FOLFOX plus regorafenib in patients with unresectable or metastatic esophagogastric cancer. ClinicalTrials.gov Identifier: NCT01913639. http://clinicaltrials.gov/show/NCT01913639 Accessed November 2013.
Acknowledgments
Dr Paul Hoban of Cancer Communications & Consultancy Ltd (Knutsford, UK) provided medical writing services on behalf of the authors, which were funded by Bayer Consumer Care AG. These included drafting and subsequent revision of the manuscript under the guidance of the authors, who had final responsibility for the manuscript content.
Conflicts of interest
Jean-Baptiste Rey reports personal fees from Amgen, Archimedes Pharma, Boehringer-Ingelheim, Eisai, IDIS, Leo Pharma, Merck Serono, MSD, Munidpharma Pfizer, Roche, Vifor Pharma, grants from Amgen, Boehringer-Ingelheim, Eisai, Janssen, LFB, Leo Pharma, Mundipharma, Roche, non-financial support from Institut National du Cancer, and a drugs ‘good use contract’ with ARS Champagne-Ardenne, all outside of the submitted work; Christophe Tournigand reports personal fees from Bayer, outside of the submitted work; Vincent Launay-Vacher reports no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Rey, JB., Launay-Vacher, V. & Tournigand, C. Regorafenib as a single-agent in the treatment of patients with gastrointestinal tumors: an overview for pharmacists. Targ Oncol 10, 199–213 (2015). https://doi.org/10.1007/s11523-014-0333-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-014-0333-x