
Abstract
Wave intensity (WI) is a hemodynamic index, which can evaluate the working condition of the heart interacting with the arterial system. It can be defined at any site in the circulatory system and provides a great deal of information. However, we need simultaneous measurements of blood pressure and velocity to obtain wave intensity, which has limited the clinical application of wave intensity, in spite of its potential. To expand the application of wave intensity in the clinical setting, we developed a real-time non-invasive measurement system for wave intensity based on a combined color Doppler and echo-tracking system. We measured carotid arterial WI in normal subjects and patients with various cardiovascular diseases. In the coronary artery disease group, the magnitude of the first peak of carotid arterial WI (W 1) increased with LV max. dP/dt (r = 0.74, P < 0.001), and the amplitude of the second peak (W 2) decreased with an increase in the time constant of LV pressure decay (r = −0.77, P < 0.001). In the dilated cardiomyopathy group, the values of W 1 were much lower than those in the normal group (P < 0.0001). In the hypertrophic cardiomyopathy group, the values of W 2 were much smaller than those in the normal group (P < 0.0001). In mitral regurgitation before surgery, W 2 decreased or disappeared, but after surgery W 2 appeared clearly. In the hypertension group, the magnitude of reflection from the head was considerably greater than that in the normal group (P < 0.0001). We also evaluated hemodynamic effects of sublingual nitroglycerin in normal subjects. Nitroglycerin increased W 1 significantly (P < 0.001). WI can be obtained non-invasively using an echo-Doppler system in the clinical setting. This method will increase the clinical usefulness of wave intensity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Many theoretical and experimental studies have been made on the propagation of pulse waves in arteries during the past 200 years after Young [41]. Relatively recently, Parker et al. [28, 29] proposed a novel index related to wave transmission and named it wave intensity. Wave intensity can be defined at any site in the circulatory system. A positive value of wave intensity indicates that forward-traveling waves predominate while a negative value indicates that backward-traveling waves predominate.
Wave intensity was originally defined as the product of ΔP and ΔU, where ΔP and ΔU are the changes in blood pressure P and velocity U, respectively, during constant short time intervals [28]. It has the dimension of rate of energy flux per unit area (W/m2), which corresponds to acoustic intensity; hence it was named “wave intensity.” In the case of sound, the energy flux is given by pu, where p is the perturbation pressure and u the perturbation velocity in the sound wave. In the case of a pulse wave in an artery, a direct analogy with the acoustic energy flux may be the rate of flow work per unit cross-sectional area of the artery, PU. However, the wave intensity defined above (ΔPΔU) is a small quantity of the second order (ΔP and ΔU being of the first order). Therefore, it does not represent the ejecting power of the heart or the effects of afterload on the heart like PU. However, we should not underestimate the usefulness of wave intensity. The information about the heart which is provided by the wave intensity may be considered analogous to the information about an engine provided by the tone of the engine noise, although this represents only a small proportion of the total power generated.
The changes, ΔP and ΔU are easy to obtain from digitized signals by simple subtraction. However, the wave intensity thus obtained depends on the sampling interval of the signals. Although qualitative changes in wave intensity are unaffected by the sampling interval, comparisons between different investigators using different instruments operating at different sampling intervals are not possible. Nevertheless, the simple feature of this wave intensity, i.e. whether its value is positive or negative, revealed unanticipated behavior of the left ventricle (LV) near end-ejection. It was generally believed that reflected waves dominated aortic deceleration, in which case the wave intensity would have been negative. However, aortic wave intensity forms a sharp positive peak in late ejection, which means forward waves traveling from the heart towards the periphery are generated during this period. Both aortic pressure and velocity decrease during this period, hence these forward waves are expansion (suction) waves (‘expansion’ is a fluid dynamic terminology, which means ‘decreasing pressure.’ This could be confused with the medical terminology ‘dilatation.’ An expansion wave decreases arterial diameter. However, dilatation of an artery means an increase in diameter. Parker, the proposer of wave intensity, has recently suggested a change of nomenclature such that expansion waves might now be called decompression waves). This fact justifies the unexpected conclusion that the LV actively stops aortic blood flow near end-ejection by generating forward traveling suction waves [29].
Soon after Parker et al. [28, 29] proposed the concept of wave intensity, Jones et al. [12] and Sugawara et al. [14] suggested that the value of wave intensity has physiological meaning quantitatively, not only qualitatively, whether it was positive or negative. They performed animal experiments and found that the height of the first positive peak of aortic wave intensity, which appeared early in systole and coincided with rising pressure and acceleration, increased with an increase in cardiac contractility. This relation was also derived theoretically [21, 32].
In order for the values of wave intensity to be compared, they should not depend on sampling interval Δt. Thus we introduced “time-normalized” wave intensity [12, 32]. The normalized wave intensity (WI) was given by
For small values of Δt, WI can be written as
where dP/dt and dU/dt are the derivatives of P and U with respect to time.
At this stage, the analogy between the wave intensity and acoustic intensity was lost in that wave intensity, WI, no longer has the same dimensions as acoustic intensity. It seems that there are no physical quantities which have the same dimension as the newly defined wave intensity. This is regrettable from the viewpoint of dimensional analysis. The acoustic intensity, pu, is defined at each instant and does not depend on the sampling interval of the signals. The rate of flow work, PU, is also defined at each instant and does not depend on the sampling interval. However, to obtain ΔP and ΔU, segmentation of the large-amplitude waveforms P and U is required, which makes ΔPΔU necessarily depend on the sampling interval. Nevertheless, the time-normalized wave intensity (WI) has the same property as the original wave intensity: if WI > 0, the changes in pressure and velocity caused by the forward wave are greater than those caused by the backward wave, and vice versa [21]. In addition, defining wave intensity as (dP/dt)(dU/dt) is perhaps more intuitive to cardiovascular physiologists because it represents the product of the familiar parameters of rate of change in pressure and flow acceleration. Thus, we developed a non-invasive method of measuring carotid arterial wave intensity and applied it in the clinical setting.
2 Non-invasive implementation of clinical measurements of wave intensity
2.1 Methods
2.1.1 Measurements of pressure waveforms
To calculate wave intensity, we need a blood pressure waveform. Many studies using dogs have demonstrated that arterial pressure waveforms and diameter-change waveforms are similar [1, 30]. We reported the similarity between carotid arterial pressure waveforms measured with a catheter-tipped micromanometer and carotid arterial diameter-change waveforms measured by echo tracking in humans [34]. Thus, using systolic and diastolic pressure measured with a cuff-type manometer applied to the upper arm, we calibrated the maximum and minimum values of a diameter-change waveform and used it as a surrogate for a blood pressure waveform.
2.1.2 Ultrasonic measurements of wave intensity
We developed a real-time measurement system for WI, which simultaneously measured carotid arterial blood flow velocity and diameter, and calculated wave intensity. This system was incorporated in ultrasonic diagnostic equipment (SSD-6500, Aloka, Tokyo, Japan), which had a color-Doppler system for blood flow velocity measurements and an echo-tracking subsystem for diameter-change measurements with a linear array probe (5 MHz for color Doppler, 10 or 13 MHz for echo-tracking). We reported the details and reproducibility of this system elsewhere [22]. Briefly, by setting the tracking positions (displayed as small bars on the ultrasound beam line for echo tracking (line A in Fig. 1), to the near and far arterial walls), the echo-tracking subsystem automatically starts measurements of arterial diameter change with a precision of one-sixteenth of the ultrasound wavelength (i.e., 7.4 μm); the data are updated at a rate of 1 kHz. The blood flow velocity averaged along the Doppler beam (line B in Fig. 1) crossing the artery is measured by using range-gated color Doppler signals. This system uses different ultrasound beams for diameter change and for blood flow velocity measurements that can be manipulated independently. The two beams intersect so that the center of the two tracking lines on the diameter-measuring beam and the center of the range gate of the velocity-measuring beam are superimposed. The diameter-change waveform, which is calculated by subtracting the distance to the near wall from that to the far wall, and the blood flow velocity waveform are displayed on the M-mode view. The data can be saved for 1–20 s. The sphygmomanometer-measured blood pressure data are entered for calibration. Five consecutive beats are ensemble-averaged to obtain a representative waveform. The maximum and minimum values of the diameter-change waveform are calibrated by systolic and diastolic blood pressure and WI indices are calculated automatically. Figure 2 shows representative recordings of WI. We derived two indices of intensity, W 1 and W 2, and two indices of time, the interval between the R wave of the ECG and the peak of W 1 (R−W 1) and the interval between the peaks of W 1 and W 2 (W 1−W 2). The interpretation of the indices of time is straightforward; R−W 1 corresponds to pre-ejection period, and W 1−W 2 corresponds to ejection time. NA, which is the negative area between W 1 and W 2, indicates the effects of reflected waves. We also measured the height of the mid-systolic positive wave (X) when it appeared.

Simultaneous measurements of diameter change wave form and blood flow velocity. View on the monitor during the measurements Left color Doppler/B-mode long-axis view of the common carotid artery. Lines A and B indicate the ultrasonic beam for echo-tracking and for blood flow velocity measurement, respectively. By setting the tracking positions displayed as small orange bars on the echo tracking beam (line A) to arterial walls, echo-tracking automatically starts. The blood flow velocity averaged along the Doppler beam (line B) crossing the artery was measured using range-gated color Doppler signals (color of the blood flow velocity is removed to indicate the tracking bars clearly). Right the diameter change waveform, which is calculated by subtracting the distance to the near wall from that to the far wall, and the velocity waveform are displayed on the M-mode view. (From [23] with permission)

Representative recordings of pressure (diameter) and blood flow velocity, and calculated wave intensity obtained from the common carotid artery of a normal human. Intensive indices (W 1, W 2, NA, X) and temporal indices (R−W 1, W 1−W 2) are derived from wave intensity. (From [23] with permission)
2.1.3 Properties of carotid arterial wave intensity derived from clinical measurements
Ohte et al. used carotid arterial WI to study 64 consecutive patients (55 men and 9 women) with suspected coronary artery disease who underwent diagnostic cardiac catheterization with a catheter-tipped micro-manometer [26]. Carotid arterial WI was measured with the above-mentioned combined color Doppler and echo-tracking system. Diagnostic cardiac catheterization was performed within 2 h after the cardiac and carotid echo studies. LV pressure was obtained with a catheter-tipped micro-manometer and recorded on a digital data recorder. From the recorded pressure waves, the maximum rate of LV pressure rise (max. dP/dt) and that of LV pressure decay (min. dP/dt) were computed in each patient. The time constant of LV pressure decay during isovolumic relaxation, τ, was also calculated from the LV pressure—dP/dt relationship (phase loop) according to the method proposed by Sugawara et al. [33]. From the phase loop, an index grading inertial force of late systolic aortic flow was also calculated [33]. Then the patients were divided into two groups: patients having the inertial force (n = 36) and those not having the inertial force (calculated inertial force = 0) (n = 28).
The magnitude of the first peak of carotid arterial WI (W 1) correlated significantly with LV max. dP/dt (r = 0.74, P < 0.001) (Fig. 3). The amplitude of the second peak of carotid arterial WI (W 2) correlated significantly with LV min. dP/dt (r = − 0.61, P < 0.001) and with τ (r = − 0.77, P < 0.001) (Fig. 4). W 2 was significantly higher in patients with the inertial force than in those without the inertial force (3,080 ± 1,740 vs. 1,890 ± 1,290 mmHg ms−3, P < 0.01). (If not mentioned in particular, values are expressed as mean ± SD hereafter.) A close relation between the first peak of aortic WI and LV max. dP/dt was found in dogs [14], and the same relation was predicted from theoretical considerations [21, 32]. Here, clinical measurements have confirmed a similar relation between carotid arterial WI and LV max. dP/dt [26].

Relationship between the magnitude of the first peak of carotid arterial wave intensity (W 1) and the maximum rate of LV pressure rise (max. dP/dt). A significant positive correlation is observed. (From [26] with permission)

Relationship between the amplitude of the second peak of carotid arterial wave intensity (W 2) and the time constant (τ) of LV pressure decay. A significant negative correlation is found. (From [26] with permission)
2.1.4 Separation of wave intensity into the forward and backward components
On condition that the regional pulse wave speed in the artery, c, is available and is constant during each cardiac cycle (linear approximation), the forward and backward components of pressure waveform, P f and P b, and those of velocity waveform, U f and U b, are given as follows [38, 39]:
where P and U are measured instantaneous pressure and velocity, P 0 is end-diastolic pressure in the artery, and ρ is the density of blood. The forward and backward components of wave intensity, WIf and WIb, are given by
and
Several different methods of obtaining regional pulse wave velocity, c, in an artery have been reported by Khir et al. [16, 17], Davies et al. [6], and ourselves [10]. Our wave intensity measurement system uses the two methods proposed by ourselves. The principle of one method is to measure blood pressure and velocity at a point, and use the water-hammer equation for forward traveling waves. The principle of the other method is to derive pulse wave velocity from the stiffness parameter of the artery. Using these two pulse wave velocities, our wave intensity measurement system can automatically calculate and display the forward and the backward components of wave intensity. However, in the clinical application of wave intensity up to now, we have not used this facility for the following reason: we have been mainly interested in the first and the second peaks of wave intensity (W 1 and W 2). During the periods of the rising limbs of W 1 and W 2, the backward component of wave intensity, WIb, is practically zero and WI is mainly composed of WIf in the carotid artery. The magnitude of W 1 and W 2, and the timing of the appearance of their peaks are not affected by backward components. Therefore, we did not separate wave intensity into the forward and backward components. We considered that at the first stage of development of clinical applications of wave intensity, simpler indices would be more widely accepted.
There are phenomena in the circulation which can be explained only by the separation of wave intensity into forward and backward components. The most important one may be coronary blood flow. Sun et al. [35, 36] reported the results of wave intensity separation analysis in dogs, and Davies et al. [7] reported those in humans.
2.1.5 Other methods of clinical measurement of wave intensity
Using a multi-sensor catheter which contained a pressure sensor and a velocity probe energized by an electromagnetic flowmeter, Koh et al. [18] measured wave intensity in the ascending aorta in patients undergoing coronary artery surgery. Using applanation tonometry and pulsed Doppler, Zambanini et al. [42] measured wave intensity in the carotid, brachial, and radial arteries in healthy nonsmokers. Using the same method, Curtis et al. [4] measured wave intensity in the carotid artery in normal subjects and patients with heart failure. Using intracoronary wires, Davies et al. [7] measured pressure and Doppler velocity simultaneously in the coronary artery in patients after a normal coronary arteriogram, and performed wave intensity analysis.
3 Results of ultrasonic measurements
3.1 Dilated cardiomyopathy
In dilated cardiomyopathy (DCM) systolic cardiac performance is impaired. We measured carotid arterial WI in 36 patients with DCM and 170 normal volunteers, and compared the data (unpublished). In the normal group, W 1 did not significantly depend on age. In the DCM group, W 1 decreased with age (deviation of the slope from zero: P = 0.0033), which suggests deterioration of cardiac contractility with age. The values of W 1 in the DCM group were much lower than those in the normal group (mean ± SEM: 4,320 ± 420 vs. 9,060 ± 330 mmHg ms−3, P < 0.0001). The values of W 2 in both groups did not differ significantly.
3.2 Hypertrophic cardiomyopathy
In hypertrophic cardiomyopathy (HCM) diastolic function is impaired. We measured carotid arterial WI in 27 patients with HCM and 170 normal volunteers, and compared the data (unpublished). There was no significant difference in values of W 1 between the two groups. However, W 1 in the HCM group decreased significantly with age, which suggests deterioration with age. W 2 did not depend on age in both groups. The values of W 2 in the HCM group were much smaller than those in the normal group (mean ± SEM: 875 ± 98 vs. 1,763 ± 57 mmHg ms−3, P < 0.0001).
3.3 Mitral regurgitation
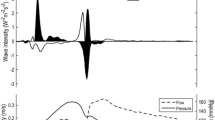
From the viewpoint of wave intensity analysis, mitral regurgitation (MR) is a particularly interesting case. The details will be described in Sect. 4. Figure 5 shows wave intensity in the right common carotid artery recorded before and after mitral valvuloplasty. Before surgery, no second peak was seen. After surgery, the second peak appeared clearly. We studied 11 patients with MR, and compared wave intensity indices before and after surgery [21]. W 1, R−W 1, and W 1−W 2 did not change significantly. However, W 2 changed dramatically from 140 ± 130 to 1,090 ± 580 mmHg ms− 3.

Recordings of wave intensity in the right common carotid artery representative of before and after valvuloplasty on the mitral valve. Before surgery, no second peak was seen. After surgery, the second peak appeared clearly. (Modified from [21] with permission)
3.4 Hypertension
It is generally accepted that nearly all tissues have an intrinsic ability to regulate the local blood flow, which is termed autoregulation [9]. Autoregulation in a region may be manifested as characteristics of pulse wave reflection from that region. There may be differences in autoregulation, hence in pulse wave reflection, between hypertensive and normotensive subjects. The increase in strength of pulse wave reflections from the lower body in patients with hypertension is well documented. However, wave reflections from the cerebral circulation in hypertension remains poorly reported because of difficulty in assessment. We measured common carotid arterial WI in 74 hypertensive patients (HT) (systolic pressure >135 or diastolic pressure >85 mmHg) and in 59 normal subjects (Norm) (ages matched with HT) (unpublished). The area, NA (see Fig. 2), of the negative part of carotid arterial WI is attributable to integrated reflections from downstream of the common carotid artery in the head and neck. There were no significant differences in W 1 and W 2 between the two groups. Both in Norm and HT, magnitude of reflection (NA) did not depend on age. Although reflections from the head do not greatly augment aortic pressure, NA was considerably greater in HT than in Norm (P < 0.0001). This suggests enhanced cerebral vasomotor tone in hypertensive subjects. Figure 6 shows the relation between NA and the maximum blood velocity (max U) in the common carotid artery in both groups. NA increased with max U in HT, but was independent of it in Norm. The association of NA with max U indicates more sensitive autoregulation of blood flow in hypertensive subjects.

Relation between the strength of wave reflection (NA) and the maximum blood velocity (max U) in the common carotid artery in the normal (Norm) and hypertensive (HT) groups. NA increased with max U in HT, but was independent of it in Norm
3.5 Effects of sublingual nitroglycerin
The effects of nitroglycerin (NTG) on the vascular system are well-known. However, the effects of NTG on the heart are still obscure, because the effects are modified by those on the vascular system, and vice versa. Therefore, to evaluate hemodynamic effects of NTG, it is important to understand the interaction between the heart and the vascular system. We studied effects of NTG on the cardiovascular system using WI [23]. We measured carotid arterial WI in 13 normal subjects (9 men, 4 women; age, 47 ± 10 years) in the supine position before and after sublingual NTG. W1 and the mid-systolic positive wave, X (see Fig. 2), increased (W 1: from 9.1 ± 4.3 to 12.3 ± 5.5 × 103 mmHg ms−3, P < 0.001, X: from 105 ± 185 to 345 ± 370 mmHg ms−3, P < 0.05). Although W 1 is mainly and markedly affected by changes in the contractile state, it is also slightly increased by an increase in preload as is max. dP/dt. However, NTG decreases preload. Therefore, the increase in W 1 after NTG was not caused by the change in preload. During the period of W 1, there are no effects of reflected waves. Therefore, the change in W 1 was not caused by reduction of reflected waves. We consider that the increase in W 1 suggests enhanced cardiac contractility. NTG made the X wave prominent. The X-wave does not necessarily appear or is not prominent in most of the subjects before NTG administration. However, in 63% of the cases without the X-wave before NTG, the X-wave appeared after NTG. During the period of this wave, pressure and blood flow velocity decrease. Therefore, this is a suction wave. The mechanism of the appearance of the X-wave will be discussed in Sect. 4.
4 Discussion
4.1 The meaning of the second peak
The physiological and pathophysiological meanings of the second positive peak, W 2, are rather complicated. During the period of W 2, as mentioned above, the LV actively stops aortic blood flow by generating forward-traveling suction waves. The generation of suction waves is caused by rapid fall in LV pressure. The decline in tension bearing ability of cardiac muscle might account for the rapid fall of LV pressure. However, the mechanism is not so simple. There are two puzzling facts that are known to physiologists and cardiologists with expertise in the simultaneous measurement of LV and aortic pressures. It used to be believed that LV pressure and aortic pressure coincide at the time of aortic valve closure. However, LV pressure is always lower than aortic pressure at this instant, frequently ~30 mmHg lower [24]. Furthermore, the time course of the decay in LV pressure before aortic valve closure is different from that during the period of isovolumic relaxation after aortic valve closure. Before aortic valve closure, the rate of fall of LV pressure (–dP/dt) increases and reaches its maximum immediately before aortic valve closure. On the other hand, –dP/dt decreases exponentially with time during isovolumic relaxation, a decline often characterized by a time constant, τ. High-fidelity LV pressure recordings therefore show a point of inflection at the time of aortic valve closure when –dP/dt changes discontinuously [13]. If the decline in LV pressure before aortic valve closure is caused purely by the decline in tension bearing ability of the LV, as must be the case during isovolumic relaxation, the maximum (–dP/dt) which occurs immediately before aortic valve closure should vary with the time constant τ of isovolumic relaxation. However, the correlation between maximum (–dP/dt) and τ is rather poor and that between maximum (–dP/dt) and the deceleration rate of aortic flow near end-ejection is much higher. Based on these data, Sugawara et al. [33] proposed a concept that the momentum (inertial force) of the blood flowing out of the LV toward the aorta, which must be decelerated to a standstill, causes a rapid decrease in pressure and generates suction waves in the LV near end-ejection. They also devised a method of grading the effect of inertial force, and showed that the strength of inertial force had a significant correlation with LV end-diastolic pressure, cardiac index, stroke volume index, ejection fraction, and maximum (−dP/dt). An increase or decrease in the inertial force corresponds to improvement or deterioration of the heart [33]. In conclusion, suction wave generation by the LV can be a consequence of both the decline in tension bearing ability of cardiac muscle and the inertial force of aortic blood flow, but a better heart utilizes the inertial force more effectively.
How the LV can have the ability to stop the blood flowing out of it is a remaining issue. As shown in Fig. 2B of the paper by Brutsaert and Sys [2], the shortening velocity of an isolated cardiac muscle in the physiological relaxation sequence decreases to zero before the force starts to decay. Concerning this time difference, European physiologists of an earlier era [11] subdivided LV systole into three phases: Anspannungszeit (isometric contraction), Austreibungszeit (ejection) and Zeit der rückständigen Contraction (remaining contraction). Patterson et al. [31] and Wiggers [40] referred to this idea and mentioned that the latter portion of the systole of the ventricles represents a “Verharrungszeit” or “rückständige Contraction”, i.e. a condition in which the ventricles remain contracted without forcing out any blood. During this rückständige contraction, blood still has a tendency to flow out of the ventricle into the aorta under its own momentum. This inertial force is considered to cause a rapid fall in LV pressure, hence cause a forward-traveling suction wave.
By assessing intraventricular flow dynamics, Ohte et al. demonstrated a mechanism whereby the LV actively stops blood flow ejection [25]. During isovolumic relaxation apically directed intraventricular flow was observed by using high-frame-rate, two-dimensional, color-Doppler imaging. The apically directed intraventricular flow appeared first near the LV apex before aortic valve closure, when ejection flow still continued in the outflow tract. A moment later, apically directed intraventricular flow was observed in almost the whole LV, except below the mitral valve. These findings suggest that before the end of ejection the apically directed intraventricular flow plays an important role to suck the blood from the aorta to the LV cavity, closing the aortic valve. The inertia of the blood flowing out of the LV causes rapid end-systolic unloading of the LV, tending to reduce LV end-systolic volume and increase the force of elastic recoil. Thus, the inertial force of aortic flow produces a greater W 2. They also found that most patients with apically directed intraventricular flow during isovolumic relaxation had an inertial force and a relatively small time constant of LV relaxation. W 2 was significantly higher in the patients with inertial force than in those without inertial force. Thus, the amplitude of expansion wave (W 2) should have a tight relation to LV function during late systolic ejection, and isovolumic relaxation (through τ).
4.2 Disappearance of the second peak in mitral regurgitation
In MR, the LV continues ejection towards the left atrium even after aortic valve closure, i.e. the cardiac muscle does not stop shortening near end-ejection. In other words, MR has not a clear “Verharrungszeit”. Therefore, the LV does not show the ability to stop aortic blood flow actively. Thus, it is expected that the second peak of wave intensity, W 2, will not appear or will be decreased in patients with MR (see Fig. 5).
The recovery of W 2 after surgery suggests that the LV had recovered the ability to generate suction waves. The generation of suction waves is associated with the rapid decrease in LV pressure near end-ejection, which is considered to be caused by the momentum of flowing blood and which results in rapid late-systolic unloading of the heart. Gillebert et al. [8] showed that late-systolic unloading makes isometric relaxation of the cardiac muscle faster and makes the subsequent lengthening of the cardiac muscle more rapid, all of which enhance diastolic function.
4.3 Appearance of the X-wave after sublingual nitroglycerin
After NTG administration, the momentum of blood generated during initial ejection may often become too large (max. U increased after NTG) for ventricular wall motion caused by myocardial shortening to keep matching the movement of blood. The mismatch of the motion of the wall and the movement of blood generates an expansion wave in the same way as the generation of W 2. The mismatch for the generation of W 2 is caused when the myocardium actively stops shortening just before isometric relaxation starts (slowing and cessation of myocardial shortening). On the other hand, the mismatch for generation of the X-wave is caused when the myocardium continues to shorten, but the momentum of blood has been increased too much by the initial ejection. This results in systolic unloading for the ventricle. Therefore, the increase in the X-wave is one of the beneficial effects of NTG. WI analysis elucidated that NTG increased W 1 and the mid-systolic suction wave X, which suggests enhanced cardiac power in the initial ejection and mid-systolic unloading. These results are new findings [23] related to the effects of NTG that should be taken into consideration in addition to the widely known late systolic unloading and preload reduction when using NTG in a clinical setting.
4.4 Calibration of the blood pressure waveform
The use of upper arm pressure for calibrating carotid arterial pressure (diameter) might be criticized. However, there is a method of obtaining carotid arterial pressure from upper arm pressure [15, 37]. The arterial pressure is amplified in transit from the ascending aorta to peripheral sites particularly in young subjects. Although the peak pressure is altered as the pressure wave travels, it is observed that mean blood pressure is constant throughout the extensive arterial tree and that diastolic pressure (the minimum pressure) does not change substantially. Van Bortel et al. [37] defined a K factor as
where mean pressure is the pressure averaged over a cardiac cycle. In the ascending aorta, K usually shows a value of about 1/2. K decreases as the pressure is amplified. Since the diameter change waveform is considered to be similar to the pressure waveform, K can be measured using echo-tracking.
We measured carotid arterial K (K c) and brachial K (K b), and calculated K b/K c in a study group (n = 15, age = 64.3 ± 11.8 years). K b/K c is equal to the ratio of carotid arterial pulse pressure to brachial pulse pressure. The coefficient of variation of K b/K c for the study group was 13.4%. This value gives the amount of error in the use of brachial pulse pressure as a surrogate for carotid pulse pressure in the study group. Calculation of wave intensity also contains the same error as pulse pressure. This method is feasible, but time consuming.
Recently, quite a few investigators have measured radial blood pressure using applanation tonometry and, via a general transfer function, calculated central aortic pressure. However, direct comparisons between central aortic pressure calculated by radial applanation tonometry and pressure measured with a catheter-tipped micro-manometer are lacking. Several investigators reported direct comparisons of the two aortic pressures [3, 5], but they are critical of the accuracy of aortic pressure obtained by using radial arterial applanation tonometry with the transfer function.
The process of formation of the peak of pressure wave in the upper arm is different from that in the aorta. The peak in the upper arm is formed due to summation of reflected waves returning from the upper limb (mainly from the hand). Since reflection sites in the upper arm are anatomically closer, the timing of the formation of pressure peak is considerably earlier than that in the aorta. In the upper arm, the peak appears in very early systole. In contrast to pulse wave transmission along the aorta to and from the lower body, aging has little effect on pulse wave velocity in the upper limb [19]. Therefore, aging has little effect on the timing of the formation of pressure peak in the upper arm. In young adults, this peak in the upper arm is considerably higher than aortic pressure peak. Therefore, amplification of pressure in the upper arm in young adults is considerable. However, in the elderly, augmentation of aortic pressure caused by early return of reflected waves from the lower body becomes prominent, which increases aortic peak pressure (late systolic peaking), and eventually makes the difference between aortic peak pressure and upper arm peak pressure less significant.
Ohte et al. studied the relationship between blood pressure obtained from the upper arm and central blood pressure measured with a catheter-tipped micro-manometer in 82 consecutive patients (65 men and 17 women; age 64.3 ± 9.4 years) with suspected coronary artery disease (CAD) who underwent cardiac catheterization [27]. Aortic pressure waveforms were obtained using a catheter-tipped micro-manometer placed approximately 10 cm above the aortic valve in the ascending aorta and, at the same time, systolic and diastolic pressures were measured one time (single measurement) from the right upper arm using a cuff-type sphygmomanometer and the oscillometric technique. No significant systematic difference (bias) was found between the peak pressure in the ascending aorta and the systolic pressure from the right upper arm (134 ± 25 vs. 132 ± 22 mmHg, NS). Bland–Altman analysis showed only a small bias of +1.8 mmHg and the limits of agreement were 25.4 and −21.8 mmHg. In contrast, the diastolic pressure in the ascending aorta was significantly lower compared with the diastolic pressure from the upper arm (69 ± 11 vs. 73 ± 12 mmHg, P < 0.0001). Bland–Altman analysis showed a small but significant bias of −4.5 mmHg and the limits of agreement were 14.1 and −23.1 mmHg. The observed biases seemed to remain within practical range. However, random variation in the two measurements was rather large. This is considered to be caused by the random error in the single measurement with the cuff-type sphygmomanometer. Therefore, measurement by sphygmomanometry should be repeated as many times as practically possible (e.g. three times) to obtain proper values of pressure. The difference between aortic and carotid arterial pressure waveforms is little. Therefore, peak systolic and bottom diastolic pressures in the carotid artery can be directly estimated from systolic and diastolic pressures measured from the upper arm using an automated cuff-type sphygmomanometer in an elderly study group.
4.5 Accuracy in sphygmomanometry
In the above-mentioned Ohte et al.’s measurements, nearly half of the points show a positive value of the difference between aortic and brachial systolic pressure. This does not indicate that in these subjects central systolic pressure is actually higher than brachial systolic pressure. Positive values of difference between aortic and brachial systolic pressure are attributed to random variation in values obtained by sphygmomanometry. It should be noted that errors in sphygmomanometry often reaches over 20 mmHg [20].
Theoretically, aortic and upper arm diastolic pressures should be equal. However, the auscultatory method using Korotkoff sounds tends to give diastolic pressures that are higher than direct intra-arterial measurements. Most of oscillometric devices are designed to give the same systolic and diastolic pressures as the auscultatory technique. Because of this, aortic diastolic pressure was lower than upper arm diastolic pressure, and aortic pulse pressure was greater than upper arm pulse pressure in Ohte et al.’s data, although the difference was small (4.5 mmHg).
4.6 Limitations of the study
We estimated peak systolic and bottom diastolic pressures in the carotid artery directly from systolic and diastolic upper-arm pressures measured with a sphygmomanometer. This method is only applicable to elderly study groups. In young adults, the difference between aortic peak pressure and upper arm peak pressure is considerably large. Therefore, in young study groups, calibration using K value (described above) is required even if it is time consuming. When using sphygmomanometry, particular attention should be paid to random variation of measured systolic and diastolic blood pressures. It often exceeds 20 mmHg. Regrettably, this is the present accuracy of shygmomanometry [20].
5 Conclusions
Cardiac function should be evaluated together with the condition of the arterial system, which consistently affects the working condition of the heart. WI, which is derived from pressure and blood flow velocity, provides quantitative and detailed information about the heart interacting with the arterial system. WI can be obtained non-invasively using an echo-Doppler system in the clinical setting. This method will increase the clinical usefulness of wave intensity.
References
Barnett GO, Mallos AJ, Shapiro A (1961) Relationship of aortic pressure and diameter in the dog. J Appl Physiol 16:545–548
Brutsaert DL, Sys SU (1989) Relaxation and diastole of the heart. Physiol Rev 69(4):1228–1351
Cloud GC, Rajkumar C, Kooner J, Cooke J, Bulpitt CJ (2003) Estimation of central aortic pressure by SphygmoCor® requires intra-arterial peripheral pressures. Clin Sci 105:219–225. doi:10.1042/CS20030012
Curtis SL, Zambanini A, Mayet J, McG Thom SA, Foale R, Parker KH et al (2007) Reduced systolic wave generation and increased peripheral wave reflection in chronic heart failure. Am J Physiol Heart Circ Physiol 293:H557–H562. doi:10.1152/ajpheart.01095.2006
Davies JI, Band MM, Pringle S, Ogston S, Struthers A (2003) Peripheral blood pressure measurement is as good as applanation tonometry at predicting ascending aortic blood pressure. J Hypertens 21:571–576. doi:10.1097/00004872-200303000-00023
Davies JE, Whinnett ZI, Francis DP, Willson K, Foale RA, Malik IS et al (2006) Use of simultaneous pressure and velocity measurements to estimate arterial wave speed at a single site in humans. Am J Physiol Heart Circ Physiol 290:H878–H885. doi:10.1152/ajpheart.00751.2005
Davies JE, Whinnett ZI, Francis DP, Manisty CH, Aguado-Sierra J, Willson K et al (2006) Evidence of a dominant backward-propagating “suction” wave responsible for diastolic coronary filling in humans, attenuated in left ventricular hypertrophy. Circulation 113:1768–1778. doi:10.1161/CIRCULATIONAHA.105.603050
Gillebert TC, Sys SU, Brutsaert DL (1989) Influences of loading patterns on peak length–tension relation and on relaxation in cardiac muscle. J Am Coll Cardiol 13(2):483–490
Guyton AC (1963) Circulatory physiology: cardiac output and its regulation. W·B. Saunders, Philadelphia
Harada A, Okada T, Niki K, Chang D, Sugawara M (2002) On-line noninvasive one-point measurements of pulse wave velocity. Heart Vessels 17:61–68. doi:10.1007/s003800200045
Huerthle K (1891) Beitraege zur Haemodynamik. Pflugers Arch 49:29–104. doi:10.1007/BF01661834
Jones CJH, Sugawara M, Davies RH, Kondoh Y, Uchida K, Parker KH (1994) Arterial wave intensity: physical meaning and physiological significance. In: Hosoda H, Yaginuma T, Sugawara M, Taylor MG, Caro CG (eds) Recent progress in cardiovascular mechanics. Harwood Academic Publishers, Chur, pp 129–148
Jones CJH, Sugawara M (1993) “Wavefronts” in the aorta—implications for mechanisms of left ventricular ejection and aortic valve closure. Cardiovasc Res 27:1902–1905. doi:10.1093/cvr/27.11.1902
Jones CJH, Sugawara M, Kondoh Y, Uchida K, Parker KH (2002) Compression and expansion wavefront travel in canine ascending aortic flow: wave intensity analysis. Heart Vessels 16:91–98. doi:10.1007/s003800200002
Kelly R, Fitchett D (1992) Noninvasive determination of aortic input impedance and external left ventricular power output: a validation and repeatability study of a new technique. J Am Coll Cardiol 20:952–963
Khir AW, Zambanini A, Parker KH (2004) Local and regional wave speed in the aorta: effects of arterial occlusion. Med Eng Phys 26:23–29. doi:10.1016/j.medengphy.2003.08.004
Khir AW, Swalen MJ, Feng J, Parker KH (2007) Simultaneous determination of wave speed and arrival time of reflected waves using the pressure–velocity loop. Med Biol Eng Comput 45:1201–1210. doi:10.1007/s11517-007-0241-7
Koh TW, Pepper JR, DeSouza AC, Parker KH (1998) Analysis of wave reflections in the arterial system using wave intensity: a novel method for predicting the timing and amplitude of reflected waves. Heart Vessels 13:103–113. doi:10.1007/BF01747827
Nichols WW, O’Rourke MF (2005) McDonald’s blood flow in arteries, 5edn. Hodder Arnold, London, p 351
Nichols WW, O’Rourke MF (2005) McDonald’s blood flow in arteries, 5th edn. Hodder Arnold, London, p 390–393
Niki K, Sugawara M, Uchida K, Tanaka R, Tanimoto K, Imamura H et al (1999) A noninvasive method of measuring wave intensity, a new hemodynamic index: application to the carotid artery in patients with mitral regurgitation before and after surgery. Heart Vessels 14:263–271. doi:10.1007/BF02481746
Niki K, Sugawara M, Chang D, Harada A, Okada T, Sakai R et al (2002) A new noninvasive measurement system for wave intensity: evaluation of carotid arterial wave intensity and reproducibility. Heart Vessels 17:12–21. doi:10.1007/s003800200037
Niki K, Sugawara M, Chang D, Harada A, Okada T, Tanaka R (2005) Effects of sublingual nitroglycerin on working conditions of the heart and arterial system: analysis using wave intensity. J Med Ultrason 32:145–152. doi:10.1007/s10396-005-0057-8
Noble MIM (1968) The contribution of blood momentum to left ventriclar ejection in the dog. Circ Res 23(11):663–670
Ohte N, Narita H, Akita S, Kurokawa K, Hayano J, Sugawara M et al (2002) The mechanism of emergence and clinical significance of apically directed intraventricular flow during isovolumic relaxation. J Am Soc Echocardiogr 15:715–722. doi:10.1067/mje.2002.119113
Ohte N, Narita H, Sugawara M, Niki K, Okada T, Harada A et al (2003) Clinical usefulness of carotid arterial wave intensity in assessing left ventricular systolic and early diastolic performance. Heart Vessels 18:107–111. doi:10.1007/s00380-003-0700-5
Ohte N, Saeki T, Miyabe H, Sakata S, Mukai S, Hayano J et al (2007) Relationship between blood pressure obtained from the upper arm with a cuff-type sphygmomanometer and central blood pressure measured with a catheter-tipped micromanometer. Heart Vessels 22:410–415. doi:10.1007/s00380-007-0998-5
Parker KH, Jones CJH (1990) Forward and backward running waves in the arteries: analysis using the method of characteristics. ASME J Biomech Eng 112(August):322–326. doi:10.1115/1.2891191
Parker KH, Jones CJH, Dawson JR, Gibson DG (1988) What stops the flow of blood from the heart? Heart Vessels 4:241–245. doi:10.1007/BF02058593
Patel DJ, de Freitas FM, Greenfield JCJ, Fry DL (1963) Relationship of radius to pressure along the aorta in living dogs. J Appl Physiol 18:1111–1117
Patterson SW, Piper H, Starling EH (1914) The regulation of the heart beat. J Physiol 48:465–513
Ramsey MW, Sugawara M (1997) Arterial wave intensity and ventriculoarterial interaction. Heart Vessels (suppl) 12:128–134
Sugawara M, Uchida K, Kondoh Y, Magosaki N, Niki K, Jones C et al (1997) Aortic blood momentum—the more the better for the ejecting heart in vivo? Cardiovasc Res 33:433–446. doi:10.1016/S0008-6363(96)00241-6
Sugawara M, Niki K, Furuhata H, Ohnishi S, Suzuki S (2000) Relationship between the pressure and diameter of the carotid artery in humans. Heart Vessels 15:49–51. doi:10.1007/PL00007261
Sun YH, Anderson TJ, Parker KH, Tyberg JV (2000) Wave-intensity analysis: a new approach to coronary hemodynamics. J Appl Physiol 89:1636–1644
Sun YH, Anderson TJ, Parker KH, Tyberg JV (2004) Effects of left ventricular contractility and coronary vascular resistance on coronary dynamics. Am J Physiol Heart Circ Physiol 286:H1590–H1595. doi:10.1152/ajpheart.01100.2001
Van Bortel LM, Balkestein EJ, van der Heijden-Spek JJ et al (2001) Non-invasive assessment of local arterial pulse pressure: comparison of applanation tonometry and echo-tracking. J Hypertens 19:1037–1044. doi:10.1097/00004872-200106000-00007
Westerhof N, Sipkema P, van den Bos CG, Elzinga G (1972) Forward and backward waves in the arterial system. Cardiovasc Res 6:648–656. doi:10.1093/cvr/6.6.648
Westerhof N, van den Bos CG, Laxminarayan S (1978) Arterial reflection. In: Bauer RD, Busse R (eds) The arterial system. Springer, Berlin, pp 48–62
Wiggers CJ (1921) Studies on the consecutive phases of the cardiac cycle. Am J Physiol 56:415–438
Young T (1808) Hydraulic investigations, subservient to an intended Croonian lecture on the motion of blood. Philos Trans R Soc Lond 98:164–186. doi:10.1098/rstl.1808.0014
Zambanini A, Cunningham SL, Parker KH, Khir AW, McG Thom SA, Hughes AD (2005) Wave-energy patterns in carotid, brachial, and radial arteries: a noninvasive approach using wave-intensity analysis. Am J Physiol Heart Circ Physiol 289:H270–H276. doi:10.1152/ajpheart.00636.2003
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sugawara, M., Niki, K., Ohte, N. et al. Clinical usefulness of wave intensity analysis. Med Biol Eng Comput 47, 197–206 (2009). https://doi.org/10.1007/s11517-008-0388-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11517-008-0388-x