
Abstract
The experimental autoimmune encephalitis (EAE) model is used for preclinical research into the pathogenesis of multiple sclerosis (MS), mostly in inbred, specific pathogen free (SPF)-raised laboratory mice. However, the naive state of the laboratory mouse immune system is considered a major hurdle in the translation of principles from the EAE model to the MS patient. Non-human primates (NHP) have an immune system harboring T- and B-cell memory against environmental antigens, similar as in humans. We sought to further refine existing NHP EAE models, which may help to bridge the gab between mouse EAE models and MS. We report here on new EAE models in three NHP species: rhesus monkeys (Macaca mulatta), cynomolgus monkeys (Macaca fascicularis) and common marmosets (Callithrix jacchus). EAE was induced with recombinant human myelin oligodendrocyte glycoprotein extracellular domain (1–125) (rhMOG) formulated in incomplete Freund’s adjuvant (IFA). IFA lacks the bacterial antigens that are present in complete Freund’s adjuvant (CFA), which are notorious for the induction of discomforting side effects. Clinically evident EAE could be induced in two out of five rhesus monkeys, six out of six cynomolgus monkeys and six out of six common marmosets. In each of these species, the presence of an early, high anti-rhMOG IgM response is correlated with EAE with an earlier onset and more severe disease course. Animals without an early high IgM response either did not develop disease (rhesus monkeys) or developed only mild signs of neurological deficit (marmoset and cynomolgus monkeys).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Experimental autoimmune encephalomyelitis (EAE) is the primary animal model of the human autoimmune neuroinflammatory disease multiple sclerosis (MS) (Hohlfeld and Wekerle 2004; Gold et al. 2006). EAE is also widely used as a generic animal model for autoimmunity, tolerance, and T helper subset function, notably Th1 and Th17. The validity of the EAE model for translational research into immunopathogenic mechanisms of MS and for the development of new treatments is often criticized, as in many cases experimental therapies effective in EAE models fail in MS patients (Sriram and Steiner 2005; Ransohoff 2006).
One of the fundamental differences between humans and inbred, specific pathogen free (SPF)-raised laboratory mice is that the immune system of these mice is essentially naive, while the human immune system receives continuous education by the daily encounter with foreign antigens (Sriram and Steiner 2005). Non-human primates (NHP) also have an antigen-experienced immune system, more closely resembling the human immune system than rodents do (Pitcher et al. 2002; Magalhaes et al. 2010).
A frequently expressed concern on the EAE model is the highly artificial way the disease is induced, i.e. via intradermal or subcutenaous injection of myelin antigen formulated in strong bacterial adjuvants such as complete Freund’s adjuvant (CFA) (Sriram and Steiner 2005; Ransohoff 2006). Most mouse EAE models require additional injection of pertussis toxin. The underlying theory is that mycobacterial components in the adjuvant provide the requisite “danger” context for the autoantigen to break tolerance and to activate signaling pathways in innate and adaptive immune cells that lead to the induction of an autoimmune process (Mills 2011). However, the bacterial antigens in CFA skew the response of T cells towards Th1, whereas in the absence of bacterial components, a Th2 response is observed (Billiau and Matthys 2001). CFA is also notorious for causing severe granulomatous skin reactions at the injection sites causing serious animal discomfort. These ethical concerns as well as conceptual arguments have prompted us to examine whether CFA can be replaced by the much milder incomplete Freund’s adjuvant (IFA). IFA provides an oil-in-water depot for the (auto) antigen, but lacks bacterial antigens. We tested this in three different NHP species from captive colonies: two Old World species, the rhesus monkey (Macaca mulatta) and the cynomolgus monkey (Macaca fascicularis), and one New World monkey species, the common marmoset (Callithrix jacchus).
Despite several attempts, an EAE model displaying satisfactory similarity with MS could not be established in rhesus macaques, as all immunization protocols tested thus far induced acute disease with seriously destructive CNS pathology, which more closely resembled acute disseminated encephalomyelitis (ADEM) than MS (reviewed in ('t Hart et al. 2005)). Although we have no prior experience with EAE in cynomolgus macaques, literature data suggest that they develop a similar type of acute inflammatory disease as rhesus macaques (Nam 2000), possibly slightly less acute (Ma et al. 2009).
In the past 15 years we have tested a variety of EAE induction protocols in the marmoset monkey (reviewed in ('t Hart and Massacesi 2009)). The clinical and pathological characteristics of these models have been unraveled in substantial detail (reviewed in ('t Hart and Massacesi 2009)) and were found to share relevant characteristics with MS. Recent studies in marmosets have all been performed with rhMOG or the immunodominant peptide MOG34−56. The aminoacid sequence of MOG is highly conserved between human, mouse, macaques and marmoset monkeys (Delarasse et al. 2006). Identical epitopes are found to be immunodominant in each of the species (von Budingen et al. 2004; Brok et al. 2007). rhMOG and MOG34−56 were used in combination with one of two adjuvants: CFA and IFA. The investigated combinations (rhMOG/CFA, MOG34−56/CFA, MOG34−56/IFA) all gave a consistent disease induction, with slight differences regarding disease onset and duration (Brok et al. 2000; Kap et al. 2008; Jagessar et al. 2010).
The finding that clinically evident EAE could be induced with MOG34−56/IFA (Jagessar et al. 2010) shows that the classical dogma for SPF mice that the induction of autoimmunity in animals depends on danger signals, may not apply to NHP. The primary aim of the current study was therefore to test whether EAE could be induced with the more complex rhMOG protein. We hypothesized that the rhMOG/IFA induction protocol would lead to anti-rhMOG T- and B-cells responses, whereas anti-rhMOG B-cell responses were absent in the MOG34−56/IFA model (Jagessar et al. 2010).
We describe here the induction of EAE in the three NHP species, using rhMOG formulated in IFA. The studies were performed at two locations, namely the Biomedical Primate Research Centre (BPRC, Rijswijk, The Netherlands) and MIRCen (Fontenay-aux-Roses, France). The results show that clinically evident EAE could be induced in all three species presenting clinical and pathological features reminiscent of MS, although EAE developed at a different incidence and severity. Moreover, it is now possible to interrogate the non-human primate system for the presence and profile of encephalitogenic T cells without the bias of bacterial antigens skewing the response of activated T cells towards Th1 (Billiau and Matthys 2001).
Materials and methods
Innate immune stimulatory activity of rhMOG
rhMOG was produced in E. coli as an unglycosylated protein encompassing the extracellular domain (amino acids 1–125) of MOG and was purified as described previously (Kerlero de Rosbo et al. 1997). rhMOG was screened for contamination with innate immune antigen receptor ligands. Human endothelial kidney (HEK293) cell lines were stably transfected with the Toll-like receptors (TLR) and the nucleotide oligomerization domain receptor 2 (NOD2), as previously described (Jagessar et al. 2010). As a positive control for NF-κB-mediated activation 25 ng/ml TNF-α (Peprotech, London, UK) was used. Positive controls for TLR2 were 0.5 μg/ml synthetic diacylated lipoprotein (FSL-1) and 500 ng/ml LPS (L8274, Sigma-Aldrich, Zwijndrecht, The Netherlands), for TLR3: 0.25 μg/ml polyriboinosinic polyribocytidylic acid (Poly (I:C)), for TLR4: 500 ng/ml LPS, for TLR5: 0.5 μg/ml flagellin, for TLR7: 10 μg/ml of an adenine analogue (CL-264), for TLR8: 1 μg/ml of a thiozoloquinolone derivate (CL-075), for TLR9: 5 μM of synthetic oligonucleotides that contain unmethylated CpG (ODN2006), for NOD2: 1 μg/ml Muramyldipeptide (MDP). No ligands are known for TLR10, but TLR10 expression was confirmed by Western blot (data not shown). All ligands were obtained from InvivoGen (San Diego, CA), except LPS, which was obtained from Sigma-Aldrich.
Ethics statement and animals
The EAE experiments in rhesus macaques and marmosets were performed at the BPRC, (Rijswijk, The Netherlands). The EAE study in cynomolgus macaques was performed at the MIRCen NHP research facility (Fontenay-aux-Roses, France) with support from BPRC staff. Individual data of the monkeys used are listed in Table 1.
BPRC
Five adult male rhesus monkeys of Chinese origin were imported from a licensed breeder (R.C. Hartelust BV, Tilburg, The Netherlands), import permits were obtained and animals were housed at the BPRC. Six adult female marmosets were randomly selected from the purpose-bred colony housed at the BPRC. All procedures were performed in compliance with guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Biomedical Primate Research Centre (BPRC) in accordance with Dutch law. The rhesus monkey study was approved by the IACUC under permit number DEC#648. The marmoset monkey study was approved by the IACUC under permit number DEC#674. BPRC’s policy on the welfare of experimental animals can be found at http://www.bprc.nl/en/animal-welfare/. Before inclusion in the study, the monkeys underwent a complete physical, hematologic, and biochemical examination. They remained under veterinary care during the study. Rhesus monkeys were housed single, but all in one room, and were provided with cage enrichment, consisting of tennis balls, mirrors and toys. Marmoset monkeys were pair-housed in spacious cages enriched with branches and toys and with padded shelter provided on the floor. The daily diet consisted of commercial food pellets for, respectively, Old World monkeys (Sniff, Soest, Germany) or for New World monkeys (Special Diet Services, Witham, Essex, UK), each supplemented with rice, raisins, peanuts, and fresh fruit, divided over several times a day. Food treats were often provided in food puzzles. Marmosets additionally received biscuits, marshmallows, grasshoppers, and maggots. Drinking water was provided ad libitum. Animals were observed daily for signs of discomfort and suffering. When EAE symptoms were noted, the frequency of observation was increased to minimally three times per day. Rhesus monkey handling was performed under ketamine sedation. Most marmoset handlings were, after training of the animals, performed without sedation. When handling required sedation, alfaxalone was used. Animals were sacrificed by an overdose of barbiturate pentobarbital.
MIRCen
Six adult cynomolgus monkeys were selected from the MIRCen colony. MIRCen animals were purpose-bred animals from Chinese or Philippine origins raised in at MIRCen or imported F1 from a primate center on Mauritius (Noveprim, Le Tamarinier LTD, Mauritius). Following EC 2010/63 and French regulations the project was performed in an agreed user establishment (agreement nb 92 032 01). Institutional permission (personal authorization nb 75–739 was obtained for this project from the National Animal Welfare Office of the French Ministry of Agriculture after evaluation by the competent evaluation body of the ministry). All procedures were performed in compliance with animal welfare body of MIRCEN's user establishment. The monkeys underwent a complete physical, hematologic, and biochemical examination before inclusion in the study. They remained under animal welfare officer (veterinary) of MIRCen center survey and care during the study. They were housed single, but cages allowed visual, vocal and direct tactile contact favoring the social interactions. Cage enrichment was compatible with the conditions of the experiments and included the presence of different materials (woods and plastic toys) and structures (perching barrels). Feeding enrichment included the type of food supplied, and its spatial and temporal distribution. The daily diet consisted of commercial food pellets for Old World monkeys (SAFE, France), each supplemented with rice, peanuts, and fresh fruit, divided over several times a day in different parts of the cages. Drinking water was provided ad libitum. Animals were double blind observed daily for signs of discomfort and suffering. When EAE symptoms were noted, the frequency of observation was increased to minimally three times per day. Most handling was performed, after training without sedation. When handling required sedation, ketamin was used. Animals were sacrificed by an overdose of barbiturate pentobarbital.
EAE induction and disease monitoring
IFA was purchased from Difco Laboratories (Detroit MI) for immunization of rhesus monkeys and marmosets and from Sigma-Aldrich (St Quentin Fallavier, France) for immunization of cynomolgus monkeys. The immunogen preparations for immunization of a rhesus or cynomolgus macaque were prepared by dissolving 300 μg rhMOG in 300 μl PBS, emulsified with an equal volume of IFA. The emulsion was injected into the dorsal skin (intradermal route) distributed over 6 spots of 100 μl each. The immunogen preparation for marmosets was prepared by dissolving 100 μg rhMOG in 200 μl PBS and emulsified with an equal volume of IFA. The 400 μl emulsion was injected into the dorsal skin (intradermal route) distributed over 4 spots of 100 μl each. All rhMOG-adjuvant emulsions were prepared by gently stirring for at least 1 h. Immunizations were repeated approximately every 28 days until clinical signs were diagnosed (Table 2). Effectively, rhesus monkey R1 was immunized on day 0, R2 and R3 on days 0 and 28, and monkeys R4 and R5 were additionally immunized on post immunization day (pid) 56 and 84. Cynomolgus monkeys C1 and C2 were immunized on days 0, 28, 56 and 188; C2 was additionally immunized on pid 273. C3 and C4 were immunized only once (day 0) and C5 and C6 were immunized on days 0 and 26. Marmosets M1, M2, M3 and M4 were immunized on days 0 and 28 and M5 and M6 were additionally immunized on pid 84.
Monkeys were observed at least once daily in their home cage, starting before immunization until the end of the experiment. Clinical signs were scored using a semi-quantitative scale (Kerlero de Rosbo et al. 2000): 0 = no clinical signs; 0.5 = loss of appetite, vomiting; 1 = substantial reduction of general condition; 2 = ataxia, sensory loss and/or visual problems; 2.5 = incomplete paralysis of one (hemiparesis) or two sides (paraparesis); 3 = complete paralysis of one (hemiplegia) of two sides (paraplegia); 4 = complete paralysis (quadriplegia); 5 = moribund. Maximum cumulative discomfort scores have been defined as the end of the experimentation (euthanasia). Termination of an animal was decided when it showed signs of clinical grade 2.5 or 3 during more than 3 days, signs of clinical grade 4 during more than 18 h and clinical signs of grade 5 for more than 1 h.
Histology
Rhesus and marmoset monkey brains and spinal cords were immersion-fixed in 4 % buffered formalin and then transferred to PBS. Marmoset brains were sectioned in 6–7 slices while from rhesus brains three samples were excised from standardized regions of each brain. Next, brain slices were embedded in paraffin. Cynomolgus monkey brains were sampled after intracardiac perfusion with PBS and 4 % PFA and were post-fixed by overnight immersion in 4 % PFA and then transferred to PBS and embedded in paraffin. The brain of the animal found dead (C5) was immersed in 4 % buffered formalin during 3 weeks and then transferred to PBS until processing into paraffin.
For histochemical analysis 3–5 μm-thick paraffin sections were cut, deparaffinized and stained with the following histochemical stains: Haematoxylin/Eosin (HE; inflammation), Luxol Fast Blue/Periodic Acid Schiff (LFB/PAS; demyelination).
Immunohistology
Immunohistochemical staining on paraffin sections of regions of interest were then performed to characterize lymphocyte subtypes (CD3, CD20) and macrophages (MRP8 or MRP14). Tissues from all three species were stained with anti-CD3 (A0452, Dako; dilution 1/100) and anti-CD20 (M0755, Dako; dilution 1/200). Rhesus and marmoset tissues were additionally stained with MRP-14 (BMA Biomedicals, Switzerland) and anti-PLP (Serotec, MCA839G, UK). Cynomolgus tissues were additionally stained with anti-MRP8 (AB92331, Abcam; dilution 1/1000), and anti-PLP (Sc-58571, Santa-Cruz, dilution 1/100).
Anti-rhMOG antibodies
Serum or plasma samples were collected prior to EAE induction, at several time points thereafter, and at necropsy. Samples were tested by ELISA in 96-well microtiter plates for the presence of antibodies against rhMOG. Plates were filled with 100 μl per well of rhMOG (5 μg/ml) and incubated overnight at 4 °C. After washing and blocking with PBS/1 % BSA the wells were incubated in duplicate with 1:100 or 1:1000 diluted sample. Bound rhesus or cynomolgus monkey antibodies were detected with alkaline phosphate-labeled goat-anti-human IgG (1:2000, AHI1305, Invitrogen/Life Technologies, Bleiswijk, The Netherlands) or alkaline phosphate-labeled goat-anti-human IgM (1:10000, A9794, Sigma, Zwijndrecht, The Netherlands). Marmoset antibodies were detected with alkaline phosphatase-labeled rabbit-anti-human IgG (1:2000, Abcam, Cambridge, UK) or alkaline phosphatase-labeled goat-anti-monkey IgM (1:2000, Rockland Immunochemicals, Gelbertsville, PA). Conjugate binding was quantified with p-nitrophenyl phosphate (Sigma). OD values of rhesus and cynomolgus data were converted to arbitrary units (AU) using the same positive control on all plates as reference. Marmoset OD values are expressed as fold increase relative to pre-immune plasma of the same monkey.
Results
Innate immune stimulatory activity of rhMOG
Innate immune stimulatory activity in the rhMOG preparation was assayed by testing its ability to stimulate HEK293 cells expressing human TLRs (TLR2, 3, 4, 5, 7, 8, 9, and 10) or the NOD2 receptor (Fig. 1). The activation of these receptors was determined by increased expression of the NF-κB-driven luciferase reporter gene in HEK293 transfected cells. The rhMOG preparation showed dose-dependent stimulation via TLR2, and a slight stimulation (fold increase of ±2.4) via TLR4 by the highest rhMOG concentration was observed. The remaining TLRs, TLR3, 5, 7, 8, 9, and 10, and the NOD2 receptor did not show a response upon exposure to rhMOG. IFA did not provide any ligands for TLR2, 3, 4, 5, 7, 8, 9, and 10 (Jagessar et al. 2010). These data suggest that the rhMOG/IFA immunization protocol provides TLR2-restricted innate immune stimulation. However, the serious granuloma formation normally seen with CFA was not observed with the current protocol.

Stimulation of innate immune activity by rhMOG. TLR and NOD2 stimulatory activity of rhMOG was tested in HEK293 cell lines stably transfected with human TLR2, 3, 4, 5, 7, 8, 9, and 10 or NOD2. Cell lines were incubated with a dose titration of rhMOG. Positive controls comprised stimulation with the specific TLR and NOD2 ligands. Data is expressed as fold increase of bioluminescence (mean ± SEM) relative to unstimulated cells. Fold increase above 2 was considered positive
Clinical symptoms in actively induced EAE
Rhesus monkeys
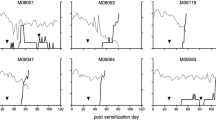
Five unrelated rhesus monkeys (Table 1) were immunized with rhMOG/IFA (Table 2). Overt clinical EAE was observed only in two animals (R2 and R3). EAE symptoms (visual problems and lethargy, EAE score 2, Table 2) were first observed in animal R2 on pid 40, which was 12 days after the second immunization. The EAE symptoms worsened at the same fast rate as in the rhMOG/CFA model. Therefore we chose to euthanize this monkey within 24 h (Fig. 2a). On the day of euthanasia (pid 41) R2 showed symptoms of complete paralysis of all 4 limbs (score 4). Animal R3 showed the first clinical signs of EAE (left sided incomplete paralysis, score 2.5) on pid 46. Its condition remained stable during the next day, but worsened on pid 48, reaching complete paralysis from below the waist (score 3) and euthanasia was indicated. One animal without EAE symptoms (R1) did not recover from sedation on day 28. The remaining two monkeys (R4 and R5) never displayed clinical symptoms of EAE even after 4 immunizations, and were euthanized around pid 110.

Clinical expression of EAE induced by rhMOG/IFA in three NHP species. EAE was induced in (a) rhesus monkeys (n = 5), (b) cynomolgus monkeys (n = 6) and (c) common marmosets (n = 6) by immunization with rhMOG emulsified in IFA. EAE symptoms were observed in only two rhesus monkeys. One animal died at anesthesia on pid 28. All six cynomolgus monkeys and all six marmoset monkeys developed EAE (score ≥2)
Cynomolgus monkeys
Six unrelated cynomolgus monkeys (Table 1) were immunized with rhMOG/IFA (Table 2) in two separate experiments, 8 months apart and all developed EAE. In the first experiment, monkeys C1 and C2 developed a protracted EAE course (Fig. 2b). Animal C1 showed the first clinical signs of EAE (paresis) on pid 211, 23 days after the fourth immunization. The severity of the clinical signs remained mild during 5 days (from grade 2 to grade 2.5). Although the clinical state did not worsen, we decided to euthanize this animal for histological examination. Animal C2 developed a first EAE episode on pid 67 (11 days after the third immunization) with ataxia and tremors (grade 2). The clinical signs remained stable for 16 days but then disappeared. Seventeen days after onset this animal had recovered completely. A second bout of clinical signs was observed on pid 286 (13 days after the fifth immunization) with paresis (score 2) progressing within 2 days to a moribund state.
In the second experiment, EAE was induced in four additional monkeys, which all developed a similar acute EAE beginning either after the first immunization for C3 (pid 11) and C4 (pid 25) or after the second immunization for C5 (pid 30, 4 days after the second immunization) and C6 (pid 41, 15 days after the second immunization). The clinical signs in these animals started with ataxia (C4) or paresis (C3, C5) or both (C6) progressing in 2 or 3 days to quadriplegia or paralysis leading to euthanasia (C3, C4, C6) or death (C5) when the process evolved too fast.
Common marmosets
Six unrelated common marmosets were immunized at 28-day intervals with rhMOG/IFA. Overt clinical EAE was observed in all six animals (Fig. 2c). Four monkeys received a total of two immunizations, after which all developed EAE (M1, M2, M3, M4). The disease durations from onset to the ethical end-point clinical score 3.0 (paralysis) in these animals were respectively: M1 pid 50–57, M2 pid 54–57, M3 pid, 56–65 and M4 pid 72–81. Monkey M5 developed neurological signs after three immunizations (pid 107), reaching a maximum EAE score of 2.5 (paresis of two hind limbs). Noteworthy is that the disease presentation in M5 differed from the other 5 monkeys. The hind limbs displayed signs of spastic paresis, and the fore limbs were weak. M5 was euthanised with score 2.5 (hemiparesis) at pid 110. Monkey M6 displayed only a short period of clinically evident EAE (ataxia, score 2) after the second immunization (pid 52) and went into remission. A third immunization with the same antigen preparation did not induce a relapse; this monkey was sacrificed without clinical symptoms at pid 114.
In conclusion, in all three species a variable clinical presentation of EAE induced with rhMOG/IFA was observed. Clinical non-responders were only observed in rhesus monkeys.
Histology of the brain and spinal cord
Rhesus monkeys (Fig. 3)
Similar to the EAE models induced with myelin, rhMBP or rhMOG in CFA, pathological changes observed after rhMOG/IFA immunizations were characterized by edema, severe inflammation and demyelination, mainly localized in the brain (Fig. 3a–c, g–j). In the brains of the two monkeys that did present clinically evident EAE (R2 and R3) large-sized demyelinated areas were observed. Lesion infiltrates contained T and B lymphocytes (Fig. 3d, e) mainly organized in perivascular cuffs as well as MRP14+ monocytes and granulocytes (Fig. 3f) infiltrating the CNS parenchyma. Some of these lesions were hemorrhagic and contained mainly neutrophilic granulocytes (PMNs, Fig. 3g–j). The spinal cord of these monkeys contained some smaller-sized infiltrates (not shown). In monkey R1, which did not recover from anesthesia at pid 28, small perivascular infiltrates were found in the white matter parenchyma of brain and spinal cord (not shown). In the two rhesus monkeys that did not develop overt clinical signs (R4 and R5), no histological signs of inflammation or demyelination could be found (not shown).

Histology of the rhMOGF/IFA model in rhesus monkeys. a–f Early demyelinating lesion in the brain of rhesus monkey R3. a Staining for LFB/PAS (bar: 5 mm). Border of the lesion is indicated by red dots. The rectangle frames part of the lesion enlarged in panel B. b The border of the lesion is indicated by red dots. A large blood vessel is seen in the center (bar: 500 μm). Left from the vessel is a grey matter area (GM) while the area on the right displays extensive edema. The rectangle shows the area enlarged in panel c. c This panel shows the center of the demyelinating lesion (bar: 100 μm). Arrowheads point at macrophages with LFB + myelin degradation products. The insert shows an HE stain of cells in the perivascular cuff. Arrowheads point at eosinophilic granulocytes. d–f The same blood vessel as seen in panel c is shown. d Staining for CD3 shows lymphocytes in the perivascular space and in the surrounding parenchyma (arrowheads) (bar: 50 μm). e Staining for CD20 shows B cells in the perivascular space (bar: 50 μm) but not in the surrounding parenchyma. f Staining for MRP14 shows macrophages and activated microglia in the perivascular space and deeply infiltrated in the surrounding parenchyma (bar: 50 μm). g–j Large hemorrhagic lesions are visible in the brain of R2. g LFB/PAS stains (bar: 5 mm). Lesions are indicated by the rectangles. h Lesions can be seen more clearly in the HE staining (bar: 5 mm). The area in the rectangle is enlarged in panel i. i The lesion consists of large numbers of PMN (blue) mixed with erythrocytes (red, bar 500 μm). In the insert, individual PMN and erythrocytes can be seen. j MRP14 staining demonstrating both MRP14+ PMN and macrophages (bar 50 μm)
Cynomolgus monkeys (Fig. 4)
Different from rhesus monkeys, EAE-affected cynomolgus monkeys displayed various types of lesions in the brain and the spinal cord. Monkey C1 which displayed late onset EAE (pid 211), presented a unilateral subacute demyelinated lesion in the subcortical white matter (frontal lobe) (Fig. 4a–f), characterized by the presence of perivascular macrophages and a low number of CD3+ and CD20+ lymphocytes. At the periphery of the lesion, MRP8+ macrophages containing intracytoplasmic LFB+/PLP + myelin degradation products were also found. Macrophages in the lesion center contained either PAS+, and/or LFB + myelin degradation products associated with loss of MRP8 reactivity, indicating early active to late active demyelination. Monkey C2 presented two types of lesions (Fig. 4g–k). First, a large subacute lesion was observed in the left frontal and parietal lobe associated with edema and swelling of the left hemisphere. This lesion was composed of a large demyelinated center containing LFB+, PAS + and MRP8- macrophages indicating early active to late active demyelination. Near the fornix, large coalescing perivascular cuffs of neutrophils and macrophages associated with few CD3 and CD20 lymphocytes were observed. In the cerebellar white matter a large demyelinated lesion was observed, surrounded by a few sparse pale and thin myelin sheaths, suggestive of remyelination. The presence of PAS, PLP and LFB negative macrophages indicated that this was an inactive demyelinated lesion. At the periphery of the lesion, two encapsulated granulomas containing PAS + macrophages were noted.

Histology of the rhMOGF/IFA model in cynomolgus monkeys. a–f Lesions in the brain of cynomolgus monkey C1. a Overview with HE stain (magnification x0.8). The rectangle indicates features of pallor and inflammation. b Overview with LFB/PAS stain (x0.8). The rectangle indicates a demyelinated area. c Macrophagic perivascular inflammation (HE stain, x100). Insert: HE stain, x400. d Macrophages containing intracytoplasmic LFB + myelin degradation products (x400). Insert: PLP stain, macrophages. e, f A perivascular cuff is shown (both x400). e CD3 stain. f CD20 stain. g–k Lesions in the brain of cynomolgus monkey C2. g Overview with HE stain (x0.8). The rectangle indicates features of pallor and inflammation. h Overview with LFB/PAS stain (x0.8). The rectangle indicates a demyelinated area. i Macrophagic infiltration (HE stain x400). Note the vacuolated cytoplasm of the macrophages. Insert: Macrophages containing PAS + and LFB + myelin degradation products. j Overview of cerebellar white matter (LFB/PAS stain, x10). Demyelinated area is outlined in black. Insert: Pale luxol staining associated with remyelination. k Vacuolated macrophages (HE stain, x100). Insert: PLP immunostaining. Absence of myelin. l Brain of cynomolgus monkey C4. Macrophagic and neutrophilic dense tissue infiltrate (HE stain, x100). Insert: MRP8 immunostaining
Monkeys immunized in the second experiment all presented large areas of neutrophil infiltration, necrosis, hemorrhages and demyelination (Fig. 3i). These lesions contained perivascular and MRP8+ macrophages, which contained PLP + and Luxol + myelin degradation products (active demyelination) associated with a lower number of CD3+ T lymphocytes and very few CD20+ B lymphocytes. In these animals a limited number of large lesions with the same features as in the brain were also observed in the spinal cord.
Common marmosets (Fig. 5)
In all five monkeys that presented clinically evident EAE at the time of necropsy, we observed presence of inflammation and demyelination in the brain and spinal cord, although the severity varied between individual cases. In the CNS, demyelinating lesions were present in large white matter tracts such as corpus callosum and optic tract (Fig. 5a–d). In addition demyelination was seen in cortical regions. All three lesion types defined in MS (subpial, intracortical and leukocortical) were found. Subpial lesions in these animals were inactive and showed minor PLP + degradation products in microglial cells at the border of the lesions. Similar to MS subpial demyelinating lesions (Lucchinetti et al. 2011), the corresponding meninges contained dense infiltrates of lymphocytes and macrophages (Fig. 5e). Intracortical lesions with an inactive core and late active rim with PLP + macrophages were regularly found (Fig. 5f). In the one monkey that displayed only a short period of ataxic problems but was healthy at the time of necropsy, no histological abnormalities were found (not shown). Thus, a close correlation exists between the presence of histological and neurological abnormalities.

Histology of the rhMOG/IFA model in marmoset monkeys. a–f Lesions in the brain of marmoset monkey M4. a Right hemisphere overview (bar: 2.5 mm). Staining for LFB/PAS. Rectangle 1 shows a large demyelinating lesion in the corpus callosum enlarged in panel B. Rectangle 2 shows a cortical lesion enlarged in panel E. b Large late active/inactive demyelination lesion in the corpus callosum (bar: 250 μm). c HE staining shows the same lesion (bar: 250 μm). Insert shows CD3+ T cells present at the border of the lesion indicated by the rectangle. d Staining for PLP demonstrates that the center of the lesion is devoid of myelin and myelin degradation products (bar: 250 μm). The rectangle at the border of the lesion shows an area with macrophages with PLP + degradation products (enlarged in the insert). e PLP staining shows inactive subpial demyelination (bar: 200 μm). The rectangle at the border of the lesion shows the area enlarged in the insert: a microglial cell with weak PLP + degradation products. M: arrows point at meningeal infiltrates. CC: corpus callosum. f PLP staining shows an intracortical demyelinating lesion (bar: 250 μm). Rectangle indicates the area with multiple PLP + macrophages which are enlarged in the insert
Anti-rhMOG antibodies
We determined anti-rhMOG antibody levels in serum or plasma. To this end, samples collected at regular intervals and at necropsy were tested for the presence of IgM and IgG binding to rhMOG (Fig. 6), being the most relevant specificity for mediating demyelination (Menge et al. 2007).

Early and/or high level anti-MOG IgM antibodies correspond with EAE onset and severity. Anti-rhMOG IgM (a, c, e) and IgG (b, d, f) antibodies. Anti-rhMOG antibodies in (a, b) rhesus monkeys, (c, d) cynomolgus monkeys, and (e, f) marmoset monkeys. Antibodies in rhesus and cynomolgus monkeys are expressed as arbitrary units (AU), which are identical for both species. Antibodies in marmosets are expressed as fold increase relative to pre-immune plasma. All monkeys have IgG antibodies against rhMOG. In rhesus and cynomolgus monkeys, early, high titers of anti-rhMOG IgM antibodies are limited to animals with acute onset and/or disease progression (R2, R3, C3, C4, C5, C6). Monkeys with early, high anti-rhMOG IgG antibodies also have early, high IgM antibodies. In marmoset monkeys this correlation is less clear, but here, the longest surviving animals (M5 and M6) also have no IgM antibodies
Rhesus monkeys
A clear dichotomy between the animals that developed EAE and those that did not was observed for anti-rhMOG IgM and IgG levels in the serum. High levels of IgM antibodies were detected already on pid 14 in monkeys that displayed clinical signs of EAE (R2 and R3), which exceeded the titers measured in the other animals several fold (Fig. 6a). The IgG levels were also already higher in these two monkeys on pid 14 as compared to the other monkeys, peaking around disease onset and exceeding the levels in the disease free monkeys between 10 and 500 fold (Fig. 6b). Monkey R1, which died at anesthesia on pid 28 had similar IgM and IgG levels as the other two monkeys without EAE.
Cynomolgus monkeys
Similar to what was observed in the rhesus monkeys, anti-rhMOG IgM rose sharply in the case of acute outcome in the cynomolgus model as well, but remained low or absent in cases with protracted EAE. Early high plasma levels of anti-rhMOG IgM antibodies were present only in the four monkeys from experiment 2 (C3, C4 and C5, C6), which developed early EAE, i.e. within 50 days (Fig. 6c). The monkeys from experiment 1, that developed remitting (C2) or late (C1) EAE after several immunizations, displayed low-level IgM antibodies, without an early peak. Unlike in rhesus monkey, in cynomolgus macaques, variations in the levels of anti-rhMOG IgG did not seem to correlate with the course of EAE as elevated anti-rhMOG IgG levels were measured in C1, C2 and C5 at euthanasia.
Common marmosets
IgM antibodies peaked between pid 14 and 28. A correlation between the IgM titer and survival in marmosets was also observed, similar to both macaque models. Animals M5 and M6 with the longest survival times (111 and 114 days), showed no IgM peak between pid 14 and 28. High IgM plasma levels measured in M1, M2 and M4 were instead associated with a faster disease progression. There was one exception (M3), which did not display an IgM peak, but was already sacrificed with neurological symptoms at day 65. Anti-rhMOG IgG levels increased in all animals from pid 28 onwards, but no correlation between IgG plasma levels and the EAE course was found. Remarkably, the highest IgG autoantibody reactivity was measured in the monkey with the atypical EAE presentation (M6).
In conclusion, the acute form of EAE in macaques and marmosets seems to be characterized by high anti-rhMOG IgM levels preceding the onset of EAE, whereas this is absent in animals with the more chronic form of EAE (C1, C2, M5 and M6), or without EAE (R4, R5, and possibly R1).
Discussion
NHP models are valid for translational research into the immunological mechanisms that drive MS pathogenesis ('t Hart and Amor 2003). They are particularly useful for the evaluation of new therapies, which, due to species specificity cannot be tested in the more commonly available rodent models ('t Hart et al. 2004). To fulfill the need for a preclinical model in which new therapies can be evaluated, EAE models equivalent to those developed in rodents have been previously implemented in three captive-bred NHP species, i.e. rhesus and cynomolgus macaques and common marmosets (for review: ('t Hart et al. 2005)).
Traditionally, EAE has been induced using a myelin antigen formulated in CFA, which in mice is often supplemented with injection of pertussis toxin. The increasing criticism against the usage of CFA for the establishment of disease models in NHP, with regard to the acute fatal outcome and the serious ulcerative skin lesions formed at the inoculation sites, motivated us to examine whether EAE could also be induced with the less immunogenic and less discomforting IFA.
TLR and NOD-like receptors are two major forms of the innate immune mechanism that supply immediate responses against pathogenic disruption e.g. of tissue injury. The activation of innate immune mechanisms by microbial danger signals from the adjuvant is commonly regarded as indispensable for EAE induction in rodent models (Darabi et al. 2004; Visser et al. 2005). We are aware of only one exception to this dogma, i.e. the EAE model in DA rats (Lorentzen et al. 1995; Lenz et al. 1999).
Our previous work showed that IFA does not contain ligands for TLRs (Jagessar et al. 2010). In the current study we tested whether rhMOG contains ligands for TLR2, 3, 4, 5, 7, 8, 9, and 10, and NOD2. We found that rhMOG stimulates HEK293 cells expressing TLR2 and 4 transgenes in a dose dependent manner. This TLR2 and 4 activation may be explained by the fact that rhMOG was produced in E.coli. Although rhMOG was purified by a TALON® metal affinity resin (Clontech Laboratories, Inc. CA) and exhaustively dialyzed, it could contain traces of LPS or peptidoglycan. Responses of the remaining tested TLRs (TLR 3, 5, 7, 8, 9, and 10) and NOD2 transfected cells to stimulation with rhMOG were absent.
Previous studies in the marmoset showed that replacement of CFA by IFA did not affect the possibility to elicit EAE with MOG34−56 (Jagessar et al. 2010). In these studies minor differences in clinical presentation of the diseases induced with CFA or IFA were observed, although analysis of the involved T-cell subsets indicated that the MOG34−56/IFA protocol induced a more Th17 driven disease (Jagessar et al. 2010), while the MOG34−56/CFA is a more Th1 driven disease (Kap et al. 2008).
The results reported in this study show that the replacement of CFA by IFA substantially reduced either the severity of EAE in at least part of the cynomolgus monkeys and the EAE incidence in rhesus monkeys. In common marmosets, the disease is very similar to the MOG34−56/IFA model. However, skin reactions at IFA injection sites were much milder than observed with CFA. Histological examination of the injection sites in cynomolgus monkey showed only low grade eosinophilic granuloma-formation containing rhMOG without skin ulceration (data not shown).
The incidence of EAE induced with rhMOG/IFA was reduced in rhesus monkeys as compared to the rhMOG/CFA model (Kerlero de Rosbo et al. 2000; Haanstra et al. 2013), but the clinical and pathological features were very similar with both protocols. In contrast to rhesus monkeys but similar to marmosets, all immunized cynomolgus monkeys developed EAE although clinical presentations were variable, with fulminant and milder forms. Acute fulminant forms were observed with the same clinical presentation as in rhesus monkeys while milder presentations of either a late onset disease or a relapsing-remitting form were observed in C1 and C2, respectively. Relapsing-remitting forms of EAE in macaques were previously described with immunomodulatory treatment (Rose et al. 1987; Shaw et al. 1988; Rose et al. 1991) but never as a spontaneous course of the disease. Thus, the substitution of CFA by IFA seems to permit the appearance of milder chronic forms of EAE. The reasons of variability of EAE outcome among the cynomolgus monkeys remain unknown. It could be due to several factors as genotype, gender, previous infections, antigen repertoire, or individual microbiome that alone or in combination can determine onset and modulate EAE or MS development (Ehlers et al. 2010; Berer et al. 2011). In cynomolgus macaques gender does not explain variability, as acute and mild forms of EAE were observed in both males and females. Difference of incidence between rhesus and cynomolgus can be due to genetic factors favoring resistance to rhMOG/IFA immunization. It could also be explained by shorter duration of the rhesus monkey experiment, although the number of immunizations is similar. One possible explanation for the difference between marmosets and rhesus monkeys could be the different expression of complement regulatory proteins within lesions (van Beek et al. 2005). The expression of decay-accelerating factor (DAF, CD55) was not studied in cynomolgus monkeys.
We observed a remarkable association between EAE susceptibility and high serum levels of anti-rhMOG IgM antibodies in all three species. Acute responders to the immunization, characterized by high anti-rhMOG IgM serum or plasma levels, developed clinical EAE earlier than monkeys with low or no detectable anti-rhMOG IgM in their serum. In the rhesus monkey model, early onset of EAE was also correlated with high IgG titers, which has been described for MS patients and marmoset monkeys immunized with myelin in CFA as well (Lalive et al. 2006). Furthermore, anti-MOG antibodies are more frequently found in patients with ADEM than with MS (Mader et al. 2011). Moreover, high titers of anti-MOG autoantibodies are frequently found in children with early onset inflammatory/demyelinating disease (McLaughlin et al. 2009).
Association of autoimmune disease susceptibility with high IgM autoantibody serum levels has also been found in a well-established autoimmune arthritis model in rhesus monkeys, namely bovine type II collagen (bCII)-induced arthritis (CIA) (Bakker et al. 1991). In this CIA model we were able to demonstrate a causal relation between IgM autoantibodies against collagen and disease, because inability to produce these autoantibodies made the monkeys tolerant to CIA induction with native bCII in CFA ('t Hart et al. 1993). The inability to produce anti-collagen IgM autoantibodies was due to one particular MHC class I serotype (Mamu-B26) in monkeys producing low level anti-bCII IgM autoantibodies (Bakker et al. 1992). Such an association between MHC alleles and forms of EAE has not been described.
In conclusion, we present new and refined EAE models in three different NHP species. The models are induced with IFA, which does not contain the mycobacterial components that are present in CFA. These models are therefore more animal friendly than the corresponding models induced with CFA. These CFA-free models are also useful for investigating subtle adaptive immune mechanisms of disease susceptibility or resistance.
Abbreviations
- CFA:
-
Complete Freund’s adjuvant
- CNS:
-
Central nervous system
- EAE:
-
Experimental autoimmune encephalomyelitis
- IFA:
-
Incomplete Freund’s adjuvant
- MNC:
-
Mononuclear cell(s)
- MOG:
-
Myelin oligodendrocyte glycoprotein
- MOG34−56 :
-
Amino acids 34–56 of MOG
- MS:
-
Multiple sclerosis
- NHP:
-
Non-human primate
- NOD2:
-
Nucleotide oligomerization domain receptor 2
- PBMC:
-
Peripheral blood mononuclear cells
- pid:
-
Post immunization day
- rhMOG:
-
Recombinant human myelin oligodendrocyte glycoprotein
- SPF:
-
Specific pathogen free
- TLR:
-
Toll-like receptor
References
Bakker NP, van Erck MG, Botman CA, Jonker M, 't Hart BA (1991) Collagen-induced arthritis in an outbred group of rhesus monkeys comprising responder and nonresponder animals. Relationship between the course of arthritis and collagen-specific immunity. Arthritis Rheum 34:616–624
Bakker NP, van Erck MG, Otting N, Lardy NM, Noort RC, 't Hart BA, Jonker M, Bontrop RE (1992) Resistance to collagen-induced arthritis in a nonhuman primate species maps to the major histocompatibility complex class I region. J Exp Med 175:933–937
Berer K, Wekerle H, Krishnamoorthy G (2011) B cells in spontaneous autoimmune diseases of the central nervous system. Mol Immunol 48:1332–1337
Billiau A, Matthys P (2001) Modes of action of Freund's adjuvants in experimental models of autoimmune diseases. J Leukoc Biol 70:849–860
Brok HP, Uccelli A, Kerlero De Rosbo N, Bontrop RE, Roccatagliata L, de Groot NG, Capello E, Laman JD, Nicolay K, Mancardi GL, Ben-Nun A, Hart BA (2000) Myelin/oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis in common marmosets: the encephalitogenic T cell epitope pMOG24-36 is presented by a monomorphic MHC class II molecule. J Immunol 165:1093–1101
Brok HP, Boven L, van Meurs M, Kerlero de Rosbo N, Celebi-Paul L, Kap YS, Jagessar A, Hintzen RQ, Keir G, Bajramovic J, Ben-Nun A, Bauer J, Laman JD, Amor S, 't Hart BA (2007) The human CMV-UL86 peptide 981–1003 shares a crossreactive T-cell epitope with the encephalitogenic MOG peptide 34–56, but lacks the capacity to induce EAE in rhesus monkeys. J Neuroimmunol 182:135–152
Darabi K, Karulin AY, Boehm BO, Hofstetter HH, Fabry Z, LaManna JC, Chavez JC, Tary-Lehmann M, Lehmann PV (2004) The third signal in T cell-mediated autoimmune disease? J Immunol 173:92–99
Delarasse C, Della Gaspera B, Lu CW, Lachapelle F, Gelot A, Rodriguez D, Dautigny A, Genain C, Pham-Dinh D (2006) Complex alternative splicing of the myelin oligodendrocyte glycoprotein gene is unique to human and non-human primates. J Neurochem 98:1707–1717
Ehlers S, Kaufmann SH, Participants of the 99 Dahlem C (2010) Infection, inflammation, and chronic diseases: consequences of a modern lifestyle. Trends Immunol 31:184–190
Gold R, Linington C, Lassmann H (2006) Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain J Neurol 129:1953–1971
Haanstra KG, Hofman SO, Lopes Estevao DM, Blezer EL, Bauer J, Yang LL, Wyant T, Csizmadia V, t Hart BA, Fedyk ER (2013) Antagonizing the alpha4beta1 integrin, but not alpha4beta7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J Immunol 190:1961–1973
Hohlfeld R, Wekerle H (2004) Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: from pipe dreams to (therapeutic) pipelines. Proc Natl Acad Sci U S A 101(Suppl 2):14599–14606
Jagessar SA, Kap YS, Heijmans N, van Driel N, van Straalen L, Bajramovic JJ, Brok HP, Blezer EL, Bauer J, Laman JD, 't Hart BA (2010) Induction of progressive demyelinating autoimmune encephalomyelitis in common marmoset monkeys using MOG34-56 peptide in incomplete freund adjuvant. J Neuropathol Exp Neurol 69:372–385
Kap YS, Smith P, Jagessar SA, Remarque E, Blezer E, Strijkers GJ, Laman JD, Hintzen RQ, Bauer J, Brok HP, 't Hart BA (2008) Fast progression of recombinant human myelin/oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis in marmosets is associated with the activation of MOG34-56-specific cytotoxic T cells. J Immunol 180:1326–1337
Kerlero de Rosbo N, Hoffman M, Mendel I, Yust I, Kaye J, Bakimer R, Flechter S, Abramsky O, Milo R, Karni A, Ben-Nun A (1997) Predominance of the autoimmune response to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis: reactivity to the extracellular domain of MOG is directed against three main regions. Eur J Immunol 27:3059–3069
Kerlero de Rosbo N, Brok HP, Bauer J, Kaye JF, 't Hart BA, Ben-Nun A (2000) Rhesus monkeys are highly susceptible to experimental autoimmune encephalomyelitis induced by myelin oligodendrocyte glycoprotein: characterisation of immunodominant T- and B-cell epitopes. J Neuroimmunol 110:83–96
Lalive PH, Menge T, Delarasse C, Della Gaspera B, Pham-Dinh D, Villoslada P, von Budingen HC, Genain CP (2006) Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc Natl Acad Sci U S A 103:2280–2285
Lenz DC, Wolf NA, Swanborg RH (1999) Strain variation in autoimmunity: attempted tolerization of DA rats results in the induction of experimental autoimmune encephalomyelitis. J Immunol 163:1763–1768
Lorentzen JC, Issazadeh S, Storch M, Mustafa MI, Lassman H, Linington C, Klareskog L, Olsson T (1995) Protracted, relapsing and demyelinating experimental autoimmune encephalomyelitis in DA rats immunized with syngeneic spinal cord and incomplete Freund's adjuvant. J Neuroimmunol 63:193–205
Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, Bruck W, Parisi JE, Scheithauer BW, Giannini C, Weigand SD, Mandrekar J, Ransohoff RM (2011) Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 365:2188–2197
Ma A, Xiong Z, Hu Y, Qi S, Song L, Dun H, Zhang L, Lou D, Yang P, Zhao Z, Wang X, Zhang D, Daloze P, Chen H (2009) Dysfunction of IL-10-producing type 1 regulatory T cells and CD4(+)CD25(+) regulatory T cells in a mimic model of human multiple sclerosis in Cynomolgus monkeys. Int Immunopharmacol 9:599–608
Mader S, Gredler V, Schanda K, Rostasy K, Dujmovic I, Pfaller K, Lutterotti A, Jarius S, Di Pauli F, Kuenz B, Ehling R, Hegen H, Deisenhammer F, Aboul-Enein F, Storch MK, Koson P, Drulovic J, Kristoferitsch W, Berger T, Reindl M (2011) Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflamm 8:184
Magalhaes I, Vudattu NK, Ahmed RK, Kuhlmann-Berenzon S, Ngo Y, Sizemore DR, Wehlin L, Weichold F, Andersson J, Skeiky YA, Sadoff J, Gaines H, Thorstensson R, Spangberg M, Maeurer MJ (2010) High content cellular immune profiling reveals differences between rhesus monkeys and men. Immunology 131:128–140
McLaughlin KA et al (2009) Age-dependent B cell autoimmunity to a myelin surface antigen in pediatric multiple sclerosis. J Immunol 183:4067–4076
Menge T, von Budingen HC, Lalive PH, Genain CP (2007) Relevant antibody subsets against MOG recognize conformational epitopes exclusively exposed in solid-phase ELISA. Eur J Immunol 37:3229–3239
Mills KH (2011) TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol 11:807–822
Nam KH (2000) Experimental autoimmune encephalomyelitis in cynomolgus monkeys. J Vet Sci 1:127–131
Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, Axthelm MK, Picker LJ (2002) Development and homeostasis of T cell memory in rhesus macaque. J Immunol 168:29–43
Ransohoff RM (2006) EAE: pitfalls outweigh virtues of screening potential treatments for multiple sclerosis. Trends Immunol 27:167–168
Rose LM, Clark EA, Hruby S, Alvord EC Jr (1987) Fluctuations of T- and B-cell subsets in basic protein-induced experimental allergic encephalomyelitis (EAE) in long-tailed macaques. Clin Immunol Immunopathol 44:93–106
Rose LM, Richards TL, Petersen R, Peterson J, Hruby S, Alvord EC Jr (1991) Remitting-relapsing EAE in nonhuman primates: a valid model of multiple sclerosis. Clin Immunol Immunopathol 59:1–15
Shaw CM, Alvord EC Jr, Hruby S (1988) Chronic remitting-relapsing experimental allergic encephalomyelitis induced in monkeys with homologous myelin basic protein. Ann Neurol 24:738–748
Sriram S, Steiner I (2005) Experimental allergic encephalomyelitis: a misleading model of multiple sclerosis. Ann Neurol 58:939–945
't Hart BA, Amor S (2003) The use of animal models to investigate the pathogenesis of neuroinflammatory disorders of the central nervous system. Curr Opin Neurol 16:375–383
't Hart BA, Massacesi L (2009) Clinical, pathological, and immunologic aspects of the multiple sclerosis model in common marmosets (Callithrix jacchus). J Neuropathol Exp Neurol 68:341–355
't Hart BA, Bakker NP, Jonker M, Bontrop RE (1993) Resistance to collagen-induced arthritis in rats and rhesus monkeys after immunization with attenuated type II collagen. Eur J Immunol 23:1588–1594
't Hart BA, Amor S, Jonker M (2004) Evaluating the validity of animal models for research into therapies for immune-based disorders. Drug Discov today 9:517–524
't Hart BA, Bauer J, Brok HP, Amor S (2005) Non-human primate models of experimental autoimmune encephalomyelitis: variations on a theme. J Neuroimmunol 168:1–12
van Beek J, van Meurs M, 't Hart BA, Brok HP, Neal JW, Chatagner A, Harris CL, Omidvar N, Morgan BP, Laman JD, Gasque P (2005) Decay-accelerating factor (CD55) is expressed by neurons in response to chronic but not acute autoimmune central nervous system inflammation associated with complement activation. J Immunol 174:2353–2365
Visser L, Jan de Heer H, Boven LA, van Riel D, van Meurs M, Melief MJ, Zahringer U, van Strijp J, Lambrecht BN, Nieuwenhuis EE, Laman JD (2005) Proinflammatory bacterial peptidoglycan as a cofactor for the development of central nervous system autoimmune disease. J Immunol 174:808–816
von Budingen HC, Hauser SL, Ouallet JC, Tanuma N, Menge T, Genain CP (2004) Frontline: epitope recognition on the myelin/oligodendrocyte glycoprotein differentially influences disease phenotype and antibody effector functions in autoimmune demyelination. Eur J Immunol 34:2072–2083
Acknowledgements
This work was supported in France by the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Commissariat à l’Energie Atomique et aux Energies alternatives (CEA). A travel grant by the Fondation pour l’Aide à la Recherche sur la Sclérose En Plaques (ARSEP) allowed personnel exchange between France and the Netherlands. The authors thank Henk van Westbroek for help with preparing the figures.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Krista G. Haanstra, S. Anwar Jagessar and Anne-Laure Bauchet contributed equally to the study.
Che Serguera and Bert A. ’t Hart share senior authorship.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Haanstra, K.G., Jagessar, S.A., Bauchet, AL. et al. Induction of Experimental Autoimmune Encephalomyelitis With Recombinant Human Myelin Oligodendrocyte Glycoprotein in Incomplete Freund’s Adjuvant in Three Non-human Primate Species. J Neuroimmune Pharmacol 8, 1251–1264 (2013). https://doi.org/10.1007/s11481-013-9487-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-013-9487-z