
Abstract
Human immunodeficiency virus type 1 (HIV) infection presently affects more that 40 million people worldwide, and is associated with central nervous system (CNS) disruption in at least 30% of infected individuals. The use of highly active antiretroviral therapy has lessened the incidence, but not the prevalence of mild impairment of higher cognitive and cortical functions (HIV-associated neurocognitive disorders) as well as substantially reduced a more severe form dementia (HIV-associated dementia). Furthermore, improving neurological outcomes will require novel, adjunctive therapies that are targeted towards mechanisms of HIV-induced neurodegeneration. Identifying such molecular and pharmacological targets requires an understanding of the events preceding irreversible neuronal damage in the CNS, such as actions of neurotoxins (HIV proteins and cellular factors), disruption of ion channel properties, synaptic damage, and loss of adult neurogenesis. By considering the specific mechanisms and consequences of HIV neuropathogenesis, unified approaches for neuroprotection will likely emerge using a tailored, combined, and non-invasive approach.
Similar content being viewed by others

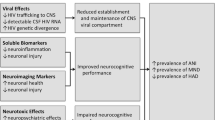
Introduction
Over 40 million people worldwide are infected by human immunodeficiency virus-1 (HIV) (UNAIDS/WHO), and, while HIV is most well known for its devastating effects on the immune system and the resulting acquired immunodeficiency syndrome (AIDS), it can also cause several neurological disorders, collectively known as HIV-associated neurocognitive disorders (HAND). HAND syndromes (minor cognitive motor disorder, HIV-associated mild neurocognitive disorder, and asymptomatic neurocognitive impairment) are characterized by cognitive, motor, and behavioral abnormalities (Kaul et al. 2005) and are classified according to patient performance in areas of neurological and behavioral functioning and neuropsychological testing (Sacktor et al. 2001; Sacktor 2002; McArthur 2004; Antinori et al. 2007). Prior to the widespread use of highly active antiretroviral therapy (HAART), 20–30% of individuals with advanced HIV infection displayed symptoms of the most severe HAND disorder, HIV-associated dementia (HAD; Gonzalez-Scarano and Martin-Garcia 2005; Kaul et al. 2005). Since the widespread use of HAART, the incidence of HAD has dramatically decreased; however, as many as 40% of HIV-positive patients continue to suffer from HAND (Sacktor et al. 2001; Sacktor 2002; McArthur 2004; Antinori et al. 2007). Furthermore, although the incidence of HAD has decreased, its prevalence is actually increasing, due in part to the longer life expectancy for individuals with HIV and to patient resistance to HAART drugs (Gonzalez-Scarano and Martin-Garcia 2005). The persistently high prevalence of milder forms of HAND (Sacktor et al. 2004) suggests a shift in the relative distribution of HAND subtypes in the post-HAART era. In patients presenting with even the milder forms of HAND, quality of life is greatly affected, with these individuals suffering from disruptions in ability to perform activities of daily living, perhaps most importantly, that of adherence to the HAART regiment (MacArthur 2004).
HAD manifests as a subcortical dementia characterized by psychomotor slowing, changes in mood and anxiety levels and deficits in memory, abstraction, information processing, verbal fluency, decision-making, and attention (Navia et al. 1986; Grant et al. 1987; Price et al. 1988; Portegies et al. 1993). Surprisingly, deficits in olfaction are common in HAD, and the severity, onset, and progression of the disease can reliably be characterized using well-established olfactory tests (Graham et al. 1995; Razani et al. 1996; Westervelt et al. 1997; Hornung et al. 1998; Mueller et al. 2002; Zucco and Ingegneri 2004; Vance 2007). As this collection of symptoms would suggest, the brain regions most commonly damaged in HAD are the basal ganglia, deep white matter, hippocampus, and cerebral cortex (Gorry et al. 2003). Pathological features of HAD include pervasive reactive astrocytosis, myelin pallor, activated resident microglia, infiltration by circulating monocytic cells, perivascular inflammation, microglial nodules, multinucleated giant cells, dendritic simplification, and cell death, both astrocytic and neuronal (Gorry et al. 2003; Kaul et al. 2005). Interestingly, rather than closely correlating with viral load in the central nervous system (CNS), clinical signs of HAD more closely associated with increased numbers of microglia, evidence of excitotoxins, and selective neuronal damage and loss (Kaul et al. 2005). These findings suggest that, although HIV invasion into the brain may be a necessary initial step in HAD progression, other mechanisms more directly lead to the functional losses associated with this disease.
HIV infection in the central nervous system
HIV primarily infects cells of the immune system, and untreated infection ultimately results in AIDS and eventually death in the vast majority of cases. Approximately 2–3 weeks after the primary infection, a patient with HIV infection enters the acute stage of infection, which presents most commonly as fever, skin rash, oral ulcers, and lymphadenopathy (Pope and Haase 2003). HIV specifically infects and kills CD4+ T cells, monocytes, and macrophages, and at this early stage of infection, the virus establishes a reservoir within the lymphoid tissue and enters into the CNS. In lymphoid tissue, productively infected, activated CD4+ T cells produce and release billions of virions daily, accounting for the systemic viral load (VL) detected by patient plasma sampling. Within the CNS the virus establishes its reservoir within the perivascular and perhaps the parenchymal macrophage populations (Morris et al. 1999). Virus detected through sampling cerebrospinal fluid (CSF) likely reflects contributions from both the systemic and CNS reservoirs, with the CNS reservoir contributing more to CSF VL in advanced infection (Ellis et al. 2000; Ritola et al. 2005).
Neuropathogenesis of HAND: overview
While neuronal cell damage and death are clearly associated with the development of HAD/HAND symptoms, HIV rarely, if ever, infects neurons themselves (McArthur et al. 2003; Gonzalez-Scarano and Martin-Garcia 2005; Kaul et al. 2005). Rather, productive HIV infection in the brain occurs in cells of the macrophage lineage, including monocyte-derived perivascular macrophages and microglia (Gorry et al. 2003; Kaul et al. 2005). According to the most widely accepted model, HIV invades the brain through a “Trojan Horse” method (Liu et al. 2002), crossing the blood–brain barrier (BBB) through infected monocytes that later differentiate into macrophages (Albright et al. 2003). This can occur within 1–2 weeks after virus enters into the systemic circulation (Davis et al. 1992; Gray et al. 1993). Subsequent infection and activation of neighboring cells occurs via direct contact with infected cells (Gonzalez-Scarano and Martin-Garcia 2005). Cells directly contacted by infected ‘Trojan horse’ cells include perivascular macrophages, astrocytes, and microglia (Williams and Hickey 2002; Gonzalez-Scarano and Martin-Garcia 2005). Importantly, while astrocytes do appear susceptible to HIV infection, they do not develop productive infection, and thus, perivascular macrophages and microglia are the only resident CNS cells capable of increasing HIV infection in the brain (Gorry et al. 2003; Gonzalez-Scarano and Martin-Garcia 2005).
Considering that HAND symptoms are closely associated with neuronal damage and loss, and the observation that HIV is unable to infect neurons, mechanisms other than direct infection must mediate the neuropathogenesis of HIV infection. Currently, two major models account for neurodegeneration and development of neurological symptoms in HAND: the direct model and the indirect model. Each of these models requires the initial productive infection of perivascular macrophages and microglia. The direct model proposes that viral proteins released from infected monocyte-derived cells cause neuronal death through direct interaction of viral proteins with neurons (Gonzalez-Scarano and Martin-Garcia 2005; Kaul et al. 2005). The indirect, or ‘bystander,’ model proposes that neuronal death is mediated by the inflammatory response mounted by infected and uninfected non-neuronal cells against HIV infection and against HIV proteins released by directly infected cells (Gonzalez-Scarano and Martin-Garcia 2005; Kaul et al. 2005). Clearly, these two models are not mutually exclusive.
The indirect model of neurodegeneration in HAND centers upon soluble factors released by non-neuronal cells as part of an inflammatory response to viral particles. When activated through direct HIV infection or through exposure to viral particles, macrophages and microglia release numerous soluble molecules, including the viral proteins, gp120, Tat, and Vpr (only released from infected cells), quinolinic and arachidonic acids, nitric oxide (NO), platelet activating factor, superoxide anions, matrix metalloproteases, chemokines, growth factors, and proinflammatory cytokines, including tumor-necrosis factor (TNF; Albright et al. 2003; Gonzalez-Scarano and Martin-Garcia 2005). While some of these molecules, such as growth factors (i.e., brain-derived neurotrophic factor) and some β-chemokines (i.e. regulated upon activation-normal T-cell expressed and secreted (RANTES)), are believed to play a neuroprotective role, a number of the other non-viral factors have proven to be neurotoxic (Kaul and Lipton 1999; Klein et al. 1999; Gonzalez-Scarano and Martin-Garcia 2005; Kaul et al. 2005). Additionally, NO and TNF impair the neuroprotective functions of astrocytes, including maintenance of the BBB and glutamate reuptake, while also increasing rates of astrocytic apoptosis (Gorry et al. 2003; Gonzalez-Scarano and Martin-Garcia 2005). The concomitant release of excessive excitatory amino acids and other N-methyl-d-aspartate glutamate receptor (NMDAR) agonists, and a reduction in glutamate reuptake can create an excitotoxic environment that results in excessive activation of NMDAR. Consequently, intraneuronal Ca2+ concentrations reach toxic levels, which results in production of free radicals (including reactive oxygen species (ROS) and NO) and in neuronal death (Gonzalez-Scarano and Martin-Garcia 2005; Kaul et al. 2005). Oxidative processes and the resulting stresses are capable of inducing cell death, even in the absence of viral infection, as suggested by the role of oxidative stress in a number of other neurodegenerative diseases (Mariani et al. 2005).
Neuropathogenesis of HAND: mechanisms of inflammation-induced neuronal insults
Both past and recent studies have provided a compelling argument for a role for inflammation in triggering events leading to neurodegeneration in HIV infection. Pulliam et al. (1997) first demonstrated that individuals with HAD express higher levels of circulating activated CD14+ monocytes that co-expressed CD69 than non-HAD individuals, and hypothesized that these activated monocytes enter the brain and subsequently initiate neurotoxin production. Ryan et al. (2001) subsequently confirmed an association between plasma soluble CD14 (sCD14) and cognitive dysfunction in HIV infection. More recently, Ancuta et al. (2008) showed that elevated sCD14 and lipopolysachharide (LPS) levels are associated with HAD. These observations support an independent study which demonstrated that elevated systemic LPS levels and immune activation in chronic HIV infection resulting from microbial translocation are associated with HIV-induced depletion of gut-associated lymphoid tissue (Brenchley et al. 2006). These studies clearly support the hypothesis that systemic immune activation/inflammation triggered by HIV-induced transmicrobial translocation and monocyte activation increases the risk for development of HAND through trafficking of activated monocytes into the CNS.
The association between the abundance of activated macrophages/microglia in the CNS, neuronal damage, and cognitive dysfunction suggests that neuroinflammation resulting from systemic immune activation and/or inflammation triggers the neurodegeneration observed in HAND (Glass et al. 1995). Numerous in vitro and in vivo studies link HIV-induced inflammation-associated neurodegeneration with macrophage proinflammatory cytokine/chemokine production, excitotoxic neuronal injury, and oxidative stress (Kaul et al. 2001; Gonzalez-Scarano and Martin-Garcia). We will discuss each below.
-
a)
Chemokine/cytokine effects. In response to HIV invasion of the CNS, microglia and macrophages within the brain mount an immune response that includes the release of both α- and β-chemokines. Since neurons express chemokine receptors (Horuk et al. 1997; Lavi et al. 1997; Rottman et al. 1997; Miller and Meucci 1999; Coughlan et al. 2000; Meucci et al. 2000), this HIV-induced inflammatory response may play a critical role in the effects of HIV infection on the CNS. Studies have found elevated levels of the α-chemokines, CXCL10/P-1o and CXCL12/SDF-1α, in the brains and CSF of HAD patients (Rostasy et al. 2003; Cinque et al. 2005). α-chemokines, which are expressed in many types of CNS cells even under normal conditions, bind CXCR chemokine receptors, thereby signaling a Gi protein-dependent increase in intracellular calcium. α-chemokines can have both neuroprotective and neurotoxic effects (Kaul and Lipton 1999; Zheng et al. 1999; Khan et al. 2008). Specifically, depending on conditions, CXCL12 can act to either enhance synaptic transmission or to activate caspase-3. When cleaved by matrix metallic proteinases, CXCL12 changes its receptor specificity from CXCR4 to CXCR3, which enhances the neurotoxic functions of this chemokine (Kaul and Lipton 1999; Zheng et al. 1999; Zhang et al. 2003; Vergote et al. 2006). Similarly, CXCL10, which acts through CXCR3 without prior proteolytic cleavage, induces increase in intracellular calcium and activation of caspase-3 upon binding to its receptor (Sui et al. 2004, 2006).
In contrast to α-chemokines, β-chemokines are only expressed at relatively low levels in the brain under normal conditions. However, several β-chemokines are found at increased concentrations in the CNS following HIV infection: CCL2, MIP-1α, MIP-1β, and RANTES/CCL5 (Kelder et al. 1998). β-chemokines act through CCR receptors, and, as with α-chemokines, they are capable of both neuroprotective and neurotoxic functions in the brain (Schmidtmayerova et al. 1996). CCL5, MIP-1α, and MIP-1β all provide protection against gp120-induced neurotoxicity in vitro (Meucci et al. 1998; Kaul and Lipton 1999). Contrarily, CCL2 is associated with an increased risk of HAND, which may be due to the role of this chemokine in the brain as a monocyte chemoattractant. Furthermore, microglia activated by interferons and astrocytes activated by IL-1β and TNF-α express CCL2, which could also contribute to the positive correlation between CCL2 levels and increased risk of HAND (Kelder et al. 1998; Sevigny et al. 2004, 2007; Ragin et al. 2006). Thus, β-chemokines may contribute to neuronal toxicity via existing pathways that are overstimulated by higher than normal concentrations of these factors.
In addition to α- and β-chemokines, elevated levels of the unique chemokine, fractalkine/CX3CL1, have also been observed in the CSF of HAND patients (Pereira et al. 2001). CX3CL1, a member of the CX3C family of chemokines, binds to endothelial cells and mediates monocyte attachment, potentially increasing monocyte migration across the blood–brain barrier and into the CNS, further increasing inflammation in the brains of patients with HIV infection (Ancuta et al. 2003; Geissmann et al. 2003; Maslin et al. 2005). However, as with α- and β-chemokines, CX3CL1 appears to have both positive and negative effects in the brain and, specifically, has been shown to provide a degree of protection against neurotoxicity (Mizuno et al. 2003; Deiva et al. 2004; Limatola et al. 2005). Thus, a delicate balance of neuroprotective and neurotoxic roles of the major players in this response likely determine the effect on neurons of an HIV-induced inflammatory response in the CNS. However, it seems that much of the protective function of chemokines is a counteraction to the negative effects of inflammation, such as toxicity induced by increased intracellular calcium, and that it may, therefore, be beneficial to treat HIV patients with drugs that protect the brain from HIV-induced inflammation.
-
b)
Excitotoxicity. Excitotoxicity is the process by which excess levels of an excitatory neurotransmitter or other agent evokes prolonged periods of neuronal membrane depolarization, thereby increasing calcium (Ca2+) levels, and consequently activating proteases, endonucleases, and other enzymes which damage cellular components. The most common form of excitotoxicity in the CNS is glutamate excitotoxicity, which is mediated by the NMDAR, a voltage and ligand-gated calcium ion channel that generates excitatory postsynaptic currents through calcium influx into the neuron. The subunit composition of NMDAR varies throughout neuronal development, and, to some degree, within different brain regions (Lynch and Guttmann 2001, 2002). Thus, different brain regions can respond differently to excess glutamate and other NMDAR agonists, possibly accounting for region specific damage in diseases in which excitotoxicity plays a role, such as HAND.
Glutamate, the major excitatory neurotransmitter in the CNS, must be maintained at physiological levels within the synapse to prevent sustained, toxic calcium influx (Rothman 1984; Hyrc et al. 1997). In the HIV-infected brain, activated and infected macrophages release excitotoxic molecules that act upon the NMDAR, including released glutamate, QUIN, and the neurotoxic amine, N-Tox, and therefore, may evoke damaging periods of NMDAR activation (Giulian et al. 1990; Jiang et al. 2001; O'Donnell et al. 2006). Furthermore, activated macrophages release factors that act in a paracrine fashion to stimulate reactive CNS cells, most importantly the astrocytes and microglia. Astrocytes, in particular, play a critical role in the regulation of extracellular glutamate concentrations within the brain (Gegelashvili and Schousboe 1997; Kanai 1997; Vandenberg 1998), and when activated this normal maintenance function is altered. Therefore, altered glutamate release and uptake is thought to be a major pathway of neurodegeneration in inflammatory brain diseases such as HIV infection (Kaul et al. 2001). Notably, Ferrarese et al. (2001) reported that glutamate is elevated in the CSF of HIV-infected individuals, further supporting a role for glutamate-mediated excitotoxicity in the pathogenesis of HAND.
Although inward NMDAR-dependent currents evoked by glutamate derived from astrocytes and infected macrophages/microglia are the most commonly cited contributor to excitotoxic death in neurons of patients with HAND, other ionic currents may also play a role in excitotoxicity and neuronal dysfunction. Outwardly rectifying currents shape the action potential, interspike interval, and after hyperpolarization, and act to determine overall membrane excitability. Prolonged exposure to glutamate evokes NMDAR-mediated excitotoxicity, but it is not fully understood how outwardly rectifying channels are modulated in response to such stimuli. Compensatory mechanisms are likely initiated in order to attempt to restore and maintain the resting membrane potential. These changes in ion channel biophysics are ultimately damaging to the neuron in the long-term because equilibrium likely favors a more depolarized voltage, thus depleting energy stores and maintaining continual activation of ion channels and calcium-dependent enzymes.
Any changes in ion channel properties, as well as membrane excitability, ultimately affect the manner in which a neuron operates and processes incoming synaptic signals. With respect to ion channels, there are several ways that HIV-related molecules can alter biophysical properties. Channel mean open probability, open duration, unitary conductance, or channel inactivation and deactivation all can be affected by various factors released by HIV-infected and/or HIV-activated immune cells in the CNS. TNF-α evokes an increase in A-type K+ currents in cultured embryonic rat cerebral cortex neurons (Houzen et al. 1997), and immune-activated macrophage-conditioned media also increases transient A-type (IA), as well as delayed rectifier (IK) K+ current in cultured rat hippocampal neurons (Hu et al. 2009). Glutamate produces a Ca2+-dependent dephosphorylation of Kv2.1 channels, which disrupts channel clusters and increases mean open channel probability and conductance (Murakoshi et al. 1997; Misonou et al. 2004). Further support for Kv channelopathy in HAD is observed in gene expression profiling studies demonstrating a markedly decreased expression of two voltage-dependent K+ channels, which could likely cause substantially decreased Kv currents (Gelman et al. 2004). Kv channels can regulate back-propagating action potentials by influencing the postsynaptic NMDAR voltage-dependent Mg2+ blockade, thereby decreasing overall membrane excitability (Paulsen and Sejnowski 2000; Johnston et al. 2003; Birnbaum et al. 2004). Hence, modulation of Kv channels by some of these compounds could significantly affect NMDAR activation, membrane excitability, and action potential propagation. Given the numerous mechanisms by which HIV infection in the brain can induce excitotoxicity, from increases in extracellular glutamate to alterations of the channels that mediate the electrochemical functioning of neurons, therapeutics directed against this particular etiology of HAND are promising candidates for treatment.
-
c)
Oxidative stress. In addition to inflammation and excitotxocity, much evidence supports a significant role for oxidative stress in the pathogenesis of HAND. In fact, oxidative stress is a well-characterized down-stream effect of both inflammation and excitotoxicity. Changes in cellular lipid metabolism that occur as a result of oxidative stress, produce characteristic molecules, such as ceramide, sphingomyelin, and hydroxynoneal, all of which are found in individuals displaying HAND (Kaul et al. 2005; Sacktor et al. 2004). Additionally, oxidized proteins were detected in the CSF of patients with HAND, further supporting a role for oxidative stress in the development of this disease (Turchan et al. 2003). Treating rat cortical neuroglial cultures with gp120 results in glial production of ROS and neuronal death, both of which are blocked when the cultures are pre-treated with an antioxidant (Wakabayashi et al. 2003). HIV-proteins may directly increase oxidative stress to neurons by inducing mitochondrial dysfunction and through interactions with membrane or cytosolic bound proteins. This finding implicates oxidative stress as an important mode of neuronal death in the indirect model of HAND neurodegeneration. Additional studies have demonstrated neuroprotective capabilities of antioxidants in vitro (Turchan et al. 2003), and therapeutic properties of antioxidants in vivo in HIV patients, showing both inhibition of mental deterioration and improvement of general health (Shor-Posner et al. 2002). These findings demonstrate a significant pathogenic role for oxidative stress in HAND, and suggest that the endogenous cellular antioxidant response may serve as a potential therapeutic target in this disease.
Neuropathogenesis of HAND: consequences of chronic neuroinflammation
The pathways of neuronal damage described above are often considered the "classical" or "central" pathophysiologies of HAD. However, new and often neglected studies have highlighted other consequences of neuroinflammation in HIV-infected and HAD individuals, that when observed in the light of physiological mechanism, become increasingly important in the study of disease progression.
-
a)
Synaptic disruption. In addition to neuronal death, HAND is associated with neuronal damage, particularly synaptic disruption. The etiologies described in this review, specifically inflammation and excitotoxicity, provide mechanisms by which this HIV-associated synaptic damage may occur. Activation of calcium-dependent proteases that disrupt the postsynaptic density (PSD) is a likely mechanism by which synapses may be altered in the HIV-infected CNS. The smooth ER, which extends into the dendritic spine, contains IP3 receptors that are tethered to mGluR and NMDARs by a complex of adaptor proteins, including Shank, GCAP, Homer, and PSD-95 (Tu et al. 1999; Sheng and Kim 2000, Sheng and Sala 2001; Sheng and Hoogenraad 2007). Secondary IP3-mediated calcium influxes are thought to play a role in LTP, however, prolonged synaptic depolarization and IP3-mediated signaling can also activate calpain proteases that can cleave PSD-95 which releases it from NMDAR (Lu et al. 2000). This could cause a large-scale decoupling of the postsynaptic complex from IP3 receptors. Interestingly, PSD-95 loss is also a hallmark sign of neurodegeneration (Gardoni 2008; Gardoni et al. 2009); hence, uncoupling or disruption of the PSD may be an important step in synaptic dysfunction and damage. Thus, inhibition of IP3-mediated calcium currents may prevent calpain or other protease activation, as well as block kinase enzymes from phosphorylating and modulating Kv channels. A summary of synaptic/cellular neurotoxic pathways and synaptic damage is presented in Figs. 1 and 2 to recapitulate the major findings discussed in this section and to illustrate the complexity of the pathological mechanisms evoked by CNS HIV infection.
-
b)
Impairment of neurogenesis. The dogma of a fixed and static CNS has been replaced by an understanding of the brain as a plastic, complex, and highly dynamic environment where new neurons are continually generated in adult animals through the process of adult neurogenesis (ANG). ANG is not confined to rodents, as recent work has demonstrated the occurrence of ANG in humans and other primates (Eriksson et al. 1998; Gage 2000; Pencea et al. 2001; Kam et al. 2009). In fact, recent studies have demonstrated that disruption of ANG is significantly involved in HAND and other neurodegenerative diseases (ND; Haughey et al. 2002; Donovan et al. 2006; Kelleher-Andersson 2006; Chen et al. 2008; Galvan and Bredesen 2007; Zhang et al. 2007; Rodriguez et al. 2008; Taupin 2009). Furthermore, substantial evidence demonstrates that ANG is involved in learning and memory, olfaction, and anxiety-related behaviors (Revest et al. 2009), all of which are functions that are disrupted in HAD and ND.

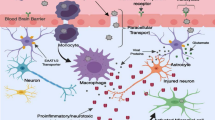
Toxicity pathways induced by HIV-associated soluble factors. Inflammatory molecules released from microglia/macrophages and astrocytes evoke NMDAR activation, as well as activation of metabotropic glutamate receptors (mGluR), receptor tyrosine kinases (RTK), voltage-gated potassium channels (Kv), other G-protein-coupled receptors (GPCR), and potentially major histocompatability complex subtype 1 receptors (MHC I). Excess calcium influx, as well as release of intracellular calcium via IP3 receptors, leads to activation of calpains and other calcium-dependent proteases, which are known to cleave postsynaptic density proteins such as PSD-95 leading to synaptic dysfunction and disassembly

Diagram detailing the numerous processes evoked by HIV infection in the brain that affect neuronal function and survival. Excess glutamate from the extracellular fluid and released from astrocytes causes excitotoxic mechanisms, such as dendritic beading, sustained NMDAR activation, increased calcium influx, and increased intracellular release of calcium. Ultimately, these processes lead to disruption of the postsynaptic density and loss of synapses. Viral proteins such, as gp120 and TAT, activate chemokine receptors, CXCR4 and CCR5, and can increase voltage-gated calcium (Cav) channels and potassium channels (Kv), leading to activation of cellular death pathways that result in mitochondrial depolarization, cytochrome p450 (Cp450) release, and ultimately DNA fragmentation associated with apoptosis. Viral proteins can also evoke increased Na+/H+ exchange, thereby increasing the pH inside astrocytes which promotes increased glutamate release and decreased glutamate uptake, thereby furthering excitotoxic damage
The two regions of the brain that contain the highest amount of newborn neurons are the hippocampus and the OB (Altman and Das 1965; Altman 1969; Lois and Alvarez-Buylla 1993; Lois and Alvarez-Buylla 1994; Eriksson et al. 1998; Gage 2000). Neural progenitor cells (NPCs) arise in the subventricular zone (SVZ) or the dentate subgranular zone (SGZ). SVZ NPCs migrate laterally and superiorly eventually terminating in the subependymal zone of the OB and SGZ NPCs migrate superiorly into the dentate gyrus of the hippocampus. After reaching their target destination, NPCs then begin to integrate into existing circuits, often forming inhibitory interneurons (Xiong et al. 2008). NPCs migrate from the SVZ to the olfactory bulb via the rostral migratory stream, which is lined with glial cells which provide trophic support to the migrating NPCs. As NPCs get closer to their destination, they begin to differentiate, and form immature neurons. Immature neurons then begin to synaptically integrate with already existing mature neurons.
HIV infection induces several processes by which ANG could be interrupted. HIV-induced alteration of general astrocyte function, including the trophic support these cells provide for both mature and immature neurons, may impair the proliferation and migration of NPCs and immature neurons along their migratory route (Fig. 3). Under this hypothesis, the OB, which is the most distant structure along the RMS, would be affected first in the earliest stages of the disease, which in fact is the case in most ND where olfaction is perturbed, such as HAND (Graham et al. 1995; Razani et al. 1996; Westervelt et al. 1997; Hornung et al. 1998; Mueller et al. 2002; Zucco and Ingegneri 2004; Vance 2007). Not surprisingly, cell cycle machinery play a role in regulating the fate of NPC (Herrup and Yang 2007). Interestingly, cell cycle proteins, such as the transcription factor, E2F1, and its regulator, the retinoblastoma gene product, exhibit increased levels and altered expression patterns in Alzheimer’s Disease (AD), PD, and HAND postmortem tissue (Jordan-Sciutto et al. 2002a, b; Hoglinger et al. 2007). Consistent with a role for these proteins in altered neurogenesis (Lee et al. 1992), mice carrying a gene-targeted deletion of E2F1 display substantially reduced ANG in the OB and hippocampus (Cooper-Kuhn et al. 2002). Furthermore, doublecortin (DCX), a microtubule protein expressed in immature neurons (Takacs et al. 2007), has several promoter sites that are regulated by cell cycle proteins, including the E2F consensus site (Karl et al. 2005). Thus, altered E2F1 function and a consequent disruption of DCX in HAND may cause an interruption in ANG, thereby contributing to the pathogenesis of these HIV-associated disorders. The CNS relies heavily on plasticity, hence, the disruption of ANG and its molecular regulation could yield devastating consequences for circuits and brain regions assaulted by HIV-induced toxicity.

Diagram detailing the various checkpoints of adult neurogenesis that can be affected by HIV infection in the brain. Neural progenitor cells and their supporting astrocytes can be damaged by inflammation associated with HIV infection. Multinucleated giant cells and microglia, especially near the rostral migratory stream, could also contribute to toxicity and damage to NPCs and immature neurons. Differentiating NPCs, as well as their integration into existing synaptic circuits, can also be affected
Another mechanism by which HIV could impair ANG is the perturbation of metabolism and associated insulin signaling pathways. Insulin in the brain enhances working memory, promotes neuronal survival, and regulates reproduction via the hypothalamic–pituitary axis (Brüning et al. 2000) This hormone acts as a neuromodulator by affecting synaptic plasticity and neurotransmitter release (Plum et al. 2005). Importantly, several lines of evidence suggest that insulin and insulin-like growth factor (IGF) are very important in ANG. Hippocampal neural progenitor cells express the insulin receptor (IR) and IGF-1 receptors, and insulin and IGF-1 are known to stimulate ANG in the dentate gyrus. ANG, synaptic plasticity and learning potential are significantly compromised in the rodent model of type 1 diabetes (Stranahan et al. 2008; Zhang et al. 2008), suggesting that both endocrine and brain insulin play a substantial role in the generation of new neurons (Kamal et al. 2000). Lang et al. (2009) also show that ANG is severely impaired in the adult type-2 diabetic GK rats, demonstrating that aberrant insulin signaling or insulin resistance can disrupt ANG. IGF-1 also has received considerable attention in recent years as a potential modulator of ANG (Aberg et al. 2000, 2003; Lichtenwalner et al. 2001; Trejo et al. 2004; Anderson et al. 2002; Perez-Martin et al. 2003a, b). In adults, peripheral IGF-1 mediates an exercise-induced enhancement of neurogenesis in the hippocampus (Trejo et al. 2001) and also has a direct proliferative effect on adult hippocampal progenitor cells in culture (Aberg et al. 2003). Intracerebroventricular infusion of IGF-1also eliminates the modest decrease in ANG that occurs in advanced geriatric animals (Lichtenwalner et al. 2001).
HIV infection alters insulin signaling, glucose homeostasis, lipid distribution, and metabolism in patients with or without HAART therapy (Grinspoon and Bilezikian 1992; Sellmeyer and Grunfeld 1996; Grinspoon et al. 1998; Kotler et al. 1999). Mechanisms of metabolic disruption in HIV-infected patients remain unclear, but it is hypothesized that peripheral chemokine signaling is at least partially involved in the alteration of insulin and glucose homeostasis (Hardy et al. 2001; Carper et al. 2008). In addition, patients on HAART therapy experience more pronounced metabolic disturbance, leading to aberrant lipid distribution (Safrin and Grunfeld 1999). HAART and some medications used to treat behavioral perturbations in HAD are known to cause a prolonged form of insulin resistance (Grinspoon and Bilezikian 1992; Hardy et al. 2001; Hughes et al. 2005; Carper et al. 2008). Some of these medications can bind the insulin receptor kinase domain and inhibit downstream phosphorylation of targets, or reduce voltage-gated K+ channels and other anion currents (Neye et al. 2006), which are known to affect glucose sensitivity and insulin response signaling (Xu et al. 2004; Desir 2005; Li et al. 2006; Tschritter et al. 2006). Thus, altered signaling from the IR could potentially cause a form of insulin resistance, and may be one of the factors by which HAND continues to progress in patients on HAART.
Therapeutics for HAND; current trials and considerations
Recent and past clinical therapeutic trials for the treatment of HAND have focused on drugs as adjuncts to current HAART, and although only modest success with adjunctive therapies has been achieved the need for more effective protection against HAND has clearly been recognized (Clifford et al. 2009). In addition more effective utilization of HAART drugs, based upon individual drug CNS penetration and efficacy represents an immediate approach, and more effective delivery of HAART drugs through nanoparticle delivery offers exciting possibilities for future consideration (Dou et al. 2009). Some adjunctive therapies to HAART studied thus far include NMDAR antagonists, calcium channel blockers, antioxidants, and anti-inflammatory drugs that either specifically or non-specifically target suspected key pathways in HIV-induced neuronal injury. Several recent, large-scale trials conducted through the AIDS Clinical Trials Group (ACTG) focus on three of these drugs: minocycline, memantine, and selegiline.
Minocycline: anti-inflammatory
Minocycline is a second-generation tetracycline antibiotic derivative developed in the 1960s, and has become an attractive drug candidate to treat HAND and other neurodegenerative diseases. In addition to the antibiotic properties of this molecule, it also potentially has protective and anti-inflammatory effects in the CNS. Minocycline is able to cross the blood–brain barrier at a substantial rate (Colovic and Caccia 2003), thus allowing delivery at much lower concentrations than other medications. Minocycline has neuroprotective effects in quite a few models of neurodegenerative diseases, as well as in traumatic and ischemic brain injury (Chen et al. 2000; Du et al. 2001; Sanchez Mejia et al. 2001; Arvin et al. 2002; Van Den Bosch et al. 2002; Wu et al. 2002; Metz et al. 2004). Minocycline has the advantages of being inexpensive to produce, readily available in the pharmaceutical market, and safe to administer over a long treatment time course.
The mechanisms by which minocycline could potentially provide protection against HAND are numerous. Minocycline suppresses JNK activation and lowers nitric oxide levels in the brain in SIV models of infection (Follstaedt et al. 2008), and, in parallel with this finding, minocycline suppresses NO-induced activation of p38 and JNK in vitro. Moreover, minocycline can inhibit ASK1 activation and disrupts the cyclical inflammation/virus replication that is often the precursor to the encephalitic state (Zink et al. 2005). Neuroprotective properties of minocycline are also linked to suppressed activation of p38 mitogen-activated protein kinase, a key physiopathology of SIV encephalitis (Barber et al. 2004). In addition, minocycline can also protect the brain by inhibiting the immune cell infiltration and activation of microglia that lead to encephalitis (Tikka and Koistinaho 2001; Tikka et al. 2001; Zink et al. 2005). Finally, minocycline significantly inhibits HIV and SIV replication in vitro (Zink et al. 2005), making this drug an excellent candidate for limiting HIV replication within the brain, which has remained a difficult anatomical compartment for therapeutic interventions for HIV due to the lack of BBB permeability of many such drugs. A multicenter study of minocycline for the treatment of HAND (ACTG5235) is currently underway (http://clinicaltrials.gov/ct2/results?term=HIV+dementia).
Memantine: anti-excitotoxicity
Memantine is a voltage-dependent, open channel NMDAR blocker that decreases prolonged conductance of calcium via a simple uncompetitive bimolecular reaction with the receptor that does not appear to interfere with physiological function (Chen and Lipton 1997; Rammes et al. 2008). Memantine is effective in gp120 transgenic mice and HIVE SCID mice. Furthermore, it appears to have some efficacy in clinical trials in patients with Alzheimer’s Disease, another neurodegenerative disease associated with excitotoxicity (Raber et al. 1996; Anderson et al. 2004; Lipton and Chen 2003; Tariot et al. 2004), and is, in fact, currently FDA-approved for AD treatment.
The neuroprotective potential for NMDAR antagonists such as memantine, in cases of HAND has recently been investigated in a therapeutic multicenter trial of Namenda (memantine; Schifitto et al. 2007b). However, no clinically significant benefit in neuropsychological and behavioral testing performance was observed during the 16-week treatment phase of the trials, though there were potential neuroprotective effects demonstrated by magnetic resonance spectroscopy in the frontal white matter and parietal cortex in treated individuals. Further trials of pharmaceuticals that block excitotoxic pathways, in particular glutamate and QUIN production, and trial designs of longer duration are clearly needed and are likely to follow in the future (Bandaru et al. 2007; Brew et al. 2007; Clifford 2008; Evans et al. 2007).
Selegiline: antioxidant
Selegiline is a monoamine oxidase type B inhibitor (MAOIB) that has recently been used in clinical trials for the treatment of HAD (Sacktor et al. 2000; Schifitto et al. 2007a, 2009). There is evidence that Selegiline has a low-dose trophic effect on neurons, and also can reduce oxygenated free radicals. Transdermal administration of Selegiline results in less monoamine peripheral metabolites than the oral route, suggesting that certain toxicities may be avoided with higher transdermal doses of this pharmaceutical (Schifitto et al. 2007a, 2009). However, the most recent study with the transdermal application system did not yield expected results (Schifitto et al. 2009). This system demonstrated no effect on either magnetic resonance spectroscopy metabolites or protein carbonyl concentration from CSF, which can be a measure of oxidative stress. The authors hypothesize that changes in oxidative stress may occur before neural imaging performed in this study; hence, selegiline may yet prove to be an effective treatment as more advanced clinical analyses become available.
Therapeutics for HAND; future considerations for adjunctive therapies
In addition to current treatment approaches (HAART drug delivery, NMDAR antagonists, antioxidants, and anti-inflammatory drugs), novel agents and routes of delivery are also under consideration. Recent successes in the CNS delivery of trophic factors (insulin, IGF-1, and neurotrophin) have provoked interest in these agents as neuroprotectants against HIV. The development of intranasal delivery (IND) of neurotrophin, insulin, and IGF-1 has emerged as a potential therapeutic technique to reduce damage associated with stroke and memory degeneration associated with HAND, AD, or diabetes (Kern et al. 1999; Thorne and Frey 2001; Benedict et al. 2004; Thorne et al. 2004, 2008; Ross et al. 2004; Reger et al. 2008a; Reger et al. 2008b; Hanson and Frey 2007; Reagan 2007; Ross et al. 2008). IND allows rapid delivery from the nose to the CNS via an extracellular route, which does not require selective hormone binding or axonal transport mechanisms (Dhanda et al. 2005; Hanson and Frey 2007). One of the most interesting aspects of IND is that after molecule administration, there is virtually no rise in blood concentration of that particular molecule. Insulin, IGF-1, FGF-2, and HB-EGF have all been tested intranasally and were not elevated in the serum of the test subjects/animals nor were peripheral glucose levels affected (Jin et al. 2003; Thorne et al. 2004; Reger et al. 2006; Hanson and Frey 2007, 2008; Reger et al. 2008a, b; Marks et al. 2009). Delivery time and clearance rates are also quite remarkable for IND, where some studies show anywhere from 6 to 20 min of peak concentration (Thorne et al. 1995, 2004) and 4–9 h of clearance from the CSF (Thorne et al. 2004; Hanson and Frey 2007). Delivery systems for IND are fairly simple, mimicking commercially available nasal spray mechanical design, or for murine models, a standard P10-20 pipetteman and associated sized plastic tip (Thorne et al. 2004; Benedict et al. 2007; Hanson and Frey 2007; Francis et al. 2008; Marks et al. 2009). There is also no associated damage to the olfactory epithelium, olfactory bulb, or disruption of olfactory receptor neuron axonal targeting (Marks et al. 2009). Thus, for drug delivery to the CNS, IND appears to be a non-invasive, fast, safe, and cost-effective solution with little, if any, of the peripheral side effects that are often associated with oral and site-injection medications.
IND of insulin and other molecules has been demonstrated to mitigate many of the behavioral and pathological deficits associated with HAND and other ND. Specifically, Marks et al. (2009) demonstrated that chronic intranasal insulin delivery enhances odor discrimination and short- and long-term memory, as well as reduces anxiety. Reger et al. (2006, 2008a, b) also demonstrated that acute intranasal insulin delivery enhances memory in AD patients. IND also has rapid and continual effects where subjects receiving acute and/or chronic IND insulin have reported extended periods of enhanced mood, reduced anger, and enhanced self-confidence in clinical trials (Benedict et al. 2004). In a recent comprehensive review of HAND pathophysiology, the authors state "Now more than ever, patients are in need of therapy in combination with ART in order to alleviate the potentially greater neuropsychological decline over a longer lifespan" (Ferris et al. 2008). Thus, a combined HAART and intranasal therapy intended to address multiple etiologies of HIV-induced damage in the CNS could be a promising regimen for persons at high risk for HAND.
Conclusion
The therapeutic targets for HAND neuroprotection discussed in this review, as well as others, clearly need further consideration if effective treatment regimens for HAND are to be identified and developed. We believe that effective control of CNS viral replication through HAART is the essential primary approach, but that it should be complemented with adjunctive CNS-directed therapeutics. HIV-induced inflammation and its consequences, expressed both systemically and within the CNS, are attractive targets that could be approached through multiple pharmacologic agents. Minocycline is a promising candidate for treating the negative effects of this inflammatory response. Excitotoxicity is another mechanism of neural damage evoked by HIV and HIV-related molecules in the brain. The open-channel NMDAR blocker, memantine, has proven beneficial in animal models of HAND, but the recently published short-term clinical trial results are disappointing, and it is thus unclear how successful this agent will be in the long-term. The possibilities for antioxidant therapies to reduce oxidative stress are quite numerous. Selegiline is one of many antioxidants, but it also yielded disappointing results in a recent short-term clinical trial. It is clear that the possibilities for future HAND treatments abound and that novel classes of potential neuroprotectant agents as well as increased duration of clinical trial testing and novel routes of administration should be strongly considered in future trials.
References
Aberg MA, Aberg ND, Hedbacker H, Oscarsson J, Eriksson PS (2000) Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci 20:2896–2903
Aberg MA, Aberg ND, Palmer TD, Alborn AM, Carlsson-Skwirut C, Bang P, Rosengren LE, Olsson T, Gage FH, Eriksson PS (2003) IGF-I has a direct proliferative effect in adult hippocampal progenitor cells. Mol Cell Neurosci 24:23–40
Albright AV, Soldan SS, Gonzalez-Scarano F (2003) Pathogenesis of human immunodeficiency virus-induced neurological disease. J Neurovirol 9:222–227
Altman J (1969) Autoradiographic and histological studies of postnatal neurogenesis. 3. Dating the time of production and onset of differentiation of cerebellar microneurons in rats. J Comp Neurol 136:269–293
Altman J, Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124:319–335
Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D (2003) Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med 197
Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D (2008) Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS ONE 3:e2516
Anderson ER, Gendelman HE, Xiong H (2004) Memantine protects hippocampal neuronal function in murine human immunodeficiency virus type 1 encephalitis. J Neurosci 24:7194–7198
Anderson MF, Aberg MA, Nilsson M, Eriksson PS (2002) Insulin-like growth factor-I and neurogenesis in the adult mammalian brain. Brain Res Dev Brain Res 134:115–122
Antinori A, Trotta MP, Lorenzini P, Torti C, Gianotti N, Maggiolo F, Ceccherini-Silberstein F, Nasto P, Castagna A, De LA, Mussini C, Andreoni M, Perno CF (2007) Virological response to salvage therapy in HIV-infected persons carrying the reverse transcriptase K65R mutation. Antivir Ther 12:1175–1183
Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM (2002) Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol 52:54–61
Bandaru VV, McArthur JC, Sacktor N, Cutler RG, Knapp EL, Mattson MP, Haughey NJ (2007) Associative and predictive biomarkers of dementia in HIV-1-infected patients. Neurology 68
Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC (2004) Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol 164:355–362
Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, Kern W (2004) Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29:1326–1334
Benedict C, Hallschmid M, Schultes B, Born J, Kern W (2007) Intranasal insulin to improve memory function in humans. Neuroendocrinology 86:136–142
Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA (2004) Structure and function of Kv4-family transient potassium channels. Physiol Rev 84:803–833
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC (2006) Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371
Brew BJ, Halman M, Catalan J, Sacktor N, Price RW, Brown S, Atkinson H, Clifford DB, Simpson D, Torres G, Hall C, Power C, Marder K, Mc Arthur JC, Symonds W, Romero C (2007) Factors in AIDS dementia complex trial design: results and lessons from the abacavir trial. PLoS Clin Trials 2
Brüning JC, Burks EJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn R (2000) Role of brain insulin receptor in control of body weight and reproduction. Science 289:2122–2125
Carper MJ, Cade WT, Cam M, Zhang S, Shalev A, Yarasheski KE, Ramanadham S (2008) HIV-protease inhibitors induce expression of suppressor of cytokine signaling-1 in insulin-sensitive tissues and promote insulin resistance and type 2 diabetes mellitus. Am J Physiol Endocrinol Metab 294:E558–E567
Chen HS, Lipton SA (1997) Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol 499(Pt 1):27–46
Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6:797–801
Chen Q, Nakajima A, Choi SH, Xiong X, Sisodia SS, Tang YP (2008) Adult neurogenesis is functionally associated with AD-like neurodegeneration. Neurobiol Dis 29:316–326
Cinque P, Bestetti A, Marenzi R, Sala S, Gisslen M, Hagberg L, Price RW (2005) Cerebrospinal fluid interferon-gamma-inducible protein 10 (IP-10, CXCL10) in HIV-1 infection. J Neuroimmunol 168
Clifford DB (2008) HIV-associated neurocognitive disease continues in the antiretroviral era. Top HIV Med 16
Clifford DB, Fagan AM, Holtzman DM, Morris JC, Teshome M, Shah AR, Kauwe JS (2009) CSF biomarkers of Alzheimer disease in HIV-associated neurologic disease. Neurology
Colovic M, Caccia S (2003) Liquid chromatographic determination of minocycline in brain-to-plasma distribution studies in the rat. J Chromatogr B 791:337–343
Cooper-Kuhn CM, Vroemen M, Brown J, Ye H, Thompson MA, Winkler J, Kuhn HG (2002) Impaired adult neurogenesis in mice lacking the transcription factor E2F1. Mol Cell Neurosci 21:312–323
Coughlan CM, McManus CM, Sharron M, Gao Z, Murphy D, Jaffer S, Choe W, Chen W, Hesselgesser J, Gaylord H, Kalyuzhny A, Lee VM, Wolf B, Doms RW, Kolson DL (2000) Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience 97
Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, Young SA, Mills RG, Wachsman W, Wiley CA (1992) Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 42
Deiva K, Geeraerts T, Salim H, Leclerc P, Hery C, Hugel B, Freyssinet JM, Tardieu M (2004) Fractalkine reduces N-methyl-d-aspartate-induced calcium flux and apoptosis in human neurons through extracellular signal-regulated kinase activation. Eur J Neurosci 20
Desir GV (2005) Kv1.3 potassium channel blockade as an approach to insulin resistance. Expert Opin Ther Targets 9:571–579
Dhanda DS, Frey WH, Leopold D, Kompella UB (2005) Approaches for drug deposition in the human olfactory epithelium. Drug Deliv Technol 5:64–72
Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ (2006) Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer's disease. J Comp Neurol 495:70–83
Dou H, Grotepas CB, McMillan JM, Destache CJ, Chaubal M, Werling J, Kipp J, Rabinow B, Gendelman HE (2009) Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J Immunol 183:661–669
Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM (2001) Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A 98:14669–14674
Ellis RJ, Gamst AC, Capparelli E, Spector SA, Hsia K, Wolfson T, Abramson I, Grant I, McCutchan JA (2000) Cerebrospinal fluid HIV RNA originates from both local CNS and systemic sources. Neurology 54:927–936
Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4:1313–1317
Evans SR, Yeh TM, Sacktor N, Clifford DB, Simpson D, Miller EN, Ellis RJ, Valcour V, Marra CM, Millar L, Schifitto G (2007) Selegiline transdermal system (STS) for HIV-associated cognitive impairment: open-label report of ACTG 5090. HIV Clin Trials 8
Ferrarese C, Aliprandi A, Tremolizzo L, Stanzani L, De Micheli A, Dolara A, Frattola L (2001) Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 57
Ferris MJ, Mactutus CF, Booze RM (2008) Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: current status of dopamine system vulnerability in NeuroAIDS. Neurosci Biobehav Rev 32:883–909
Follstaedt SC, Barber SA, Zink MC (2008) Mechanisms of minocycline-induced suppression of simian immunodeficiency virus encephalitis: inhibition of apoptosis signal-regulating kinase 1. J Neurovirol 14:376–388
Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH, Toth C (2008) Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain 131:3311–3334
Gage FH (2000) Mammalian neural stem cells. Science 287:1433–1438
Galvan V, Bredesen DE (2007) Neurogenesis in the adult brain: implications for Alzheimer's disease. CNS Neurol Disord Drug Targets 6:303–310
Gardoni F (2008) MAGUK proteins: new targets for pharmacological intervention in the glutamatergic synapse. Eur J Pharmacol 585:147–152
Gardoni F, Marcello E, Di Luca M (2009) Postsynaptic density-membrane associated guanylate kinase proteins (PSD-MAGUKs) and their role in CNS disorders. Neuroscience 158:324–333
Gegelashvili G, Schousboe A (1997) High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol 52:6–15
Geissmann F, Jung S, Littman DR (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19
Gelman BB, Soukup VM, Schuenke KW, Keherly MJ, Holzer C III, Richey FJ, Lahart CJ (2004) Acquired neuronal channelopathies in HIV-associated dementia. J Neuroimmunol 157:111–119
Giulian D, Vaca K, Noonan CA (1990) Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 250
Glass JD, Fedor H, Wesselingh SL, McArthur JC (1995) Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol 38:755–762
Gonzalez-Scarano F, Martin-Garcia J (2005) The neuropathogenesis of AIDS. Nat Rev Immunol 5:69–81
Gorry PR, Ong C, Thorpe J, Bannwarth S, Thompson KA, Gatignol A, Vesselingh SL, Purcell DF (2003) Astrocyte infection by HIV-1: mechanisms of restricted virus replication, and role in the pathogenesis of HIV-1-associated dementia. Curr HIV Res 1
Grant RM, Wiley JA, Winkelstein W (1987) Infectivity of the human immunodeficiency virus: estimates from a prospective study of homosexual men. J Infect Dis 156:189–193
Gray F, Hurtrel M, Hurtrel B (1993) Early central nervous system changes in human immunodeficiency virus (HIV)-infection. Neuropathol Appl Neurobiol 19
Graham CS, Graham BG, Bartlett JA, Heald AE, Schiffman SS (1995) Taste and smell losses in HIV infected patients. Physiol Behav 58:287–293
Grinspoon S, Corcoran C, Miller K, Wang E, Hubbard J, Schoenfeld D, Anderson E, Basgoz N, Klibanski A (1998) Determinants of increased energy expenditure in HIV-infected women. Am J Clin Nutr 68:720–725
Grinspoon SK, Bilezikian JP (1992) HIV disease and the endocrine system. N Engl J Med 327:1360–1365
Hanson LR, Frey WH (2007) Strategies for intranasal delivery of therapeutics for the prevention and treatment of neuroAIDS. J Neuroimmune Pharmacol 2:81–86
Hanson LR, Frey WH (2008) Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci 9(Suppl 3):S5
Hardy H, Esch LD, Morse GD (2001) Glucose disorders associated with HIV and its drug therapy. Ann Pharmacother 35:343–351
Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP (2002) Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer's disease. Neuromolecular Med 1:125–135
Herrup K, Yang Y (2007) Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci 8:368–378
Hoglinger GU, Breunig JJ, Depboylu C, Rouaux C, Michel PP, Varez-Fischer D, Boutillier AL, Degregori J, Oertel WH, Rakic P, Hirsch EC, Hunot S (2007) The pRb/E2F cell-cycle pathway mediates cell death in Parkinson's disease. Proc Natl Acad Sci U S A 104:3585–3590
Hornung DE, Kurtz DB, Bradshaw CB, Seipel DM, Kent PF, Blair DC, Emko P (1998) The olfactory loss that accompanies an HIV infection. Physiol Behav 64:549–556
Horuk R, Martin AW, Wang Z, Schweitzer L, Gerassimides A, Guo H, Lu Z, Hesselgesser J, Perez HD, Kim J, Parker J, Hadley TJ, Peiper SC (1997) Expression of chemokine receptors by subsets of neurons in the central nervous system. J Immunol 158
Houzen H, Kikuchi S, Kanno M, Shinpo K, Tashiro K (1997) Tumor necrosis factor enhancement of transient outward potassium currents in cultured rat cortical neurons. J Neurosci Res 50:990–999
Hu D, Liu J, Xiong H (2009) Enhancement of neuronal outward delayed rectifier K+ current by human monocyte-derived macrophages. Glia 57:1492–1500
Hughes CA, Cashin RP, Eurich DT, Houston S (2005) Risk factors for new-onset diabetes mellitus in patients receiving protease inhibitor therapy. Can J Infect Dis Med Microbiol 16:230–232
Hyrc K, Handran SD, Rothman SM, Goldberg MP (1997) Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J Neurosci 17
Jiang ZG, Piggee C, Heyes MP, Murphy C, Quearry B, Bauer M, Zheng J, Gendelman HE, Markey SP (2001) Glutamate is a mediator of neurotoxicity in secretions of activated HIV-1-infected macrophages. J Neuroimmunol 117
Jin K, Xie L, Childs J, Sun Y, Mao XO, Logvinova A, Greenberg DA (2003) Cerebral neurogenesis is induced by intranasal administration of growth factors. Ann Neurol 53:405–409
Johnston D, Christie BR, Frick A, Gray R, Hoffman DA, Schexnayder LK, Watanabe S, Yuan LL (2003) Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci 358:667–674
Jordan-Sciutto KL, Malaiyandi LM, Bowser R (2002a) Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J Neuropathol Exp Neurol 61:358–367
Jordan-Sciutto KL, Wang G, Murphey-Corb M, Wiley CA (2002b) Cell cycle proteins exhibit altered expression patterns in lentiviral-associated encephalitis. J Neurosci 22:2185–2195
Kam M, Curtis MA, McGlashan SR, Connor B, Nannmark U, Faull RLM (2009) The cellular composition and morphological organization of the rostral migratory stream in the adult human brain. J Chem Neuroanat 37:196–205
Kamal A, Biessels GJ, Duis SE, Gispen WH (2000) Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: interaction of diabetes and ageing. Diabetologia 43:500–506
Kanai Y (1997) Family of neutral and acidic amino acid transporters: molecular biology, physiology and medical implications. Curr Opin Cell Biol 9:565–572
Karl C, Couillard-Despres S, Prang P, Munding M, Kilb W, Brigadski T, Plotz S, Mages W, Luhmann H, Winkler J, Bogdahn U, Aigner L (2005) Neuronal precursor-specific activity of a human doublecortin regulatory sequence. J Neurochem 92:264–282
Kaul M, Lipton SA (1999) Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A 96:8212–8216
Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410
Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA (2005) HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ 12 (Suppl 1)
Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE (1998) Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol 44
Kelleher-Andersson J (2006) Discovery of neurogenic, Alzheimer's disease therapeutics. Curr Alzheimer Res 3:55–62
Kern W, Born J, Schreiber H, Fehm HL (1999) Central nervous system effects of intranasally administered insulin during euglycemia in men. Diabetes 48:557–563
Khan MZ, Brandimarti R, Shimizu S, Nicolai J, Crowe E, Meucci O (2008) The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell Death Differ
Klein RS, Williams KC, Alvarez-Hernandez X, Westmoreland S, Force T, Lackner AA, Luster AD (1999) Chemokine receptor expression and signaling in macaque and human fetal neurons and astrocytes: implications for the neuropathogenesis of AIDS. J Immunol 163:1636–1646
Kotler DP, Rosenbaum K, Wang J, Pierson RN (1999) Studies of body composition and fat distribution in HIV-infected and control subjects. J Acquir Immune Defic Syndr Hum Retrovirol 20:228–237
Lang BT, Yan Y, Dempsey RJ, Vemuganti R (2009) Impaired neurogenesis in adult type-2 diabetic rats. Brain Res 1258:25–33
Lavi E, Strizki JM, Ulrich AM, Zhang W, Fu L, Wang Q, O'Connor M, Hoxie JA, Gonzalez-Scarano F (1997) CXCR-4 (Fusin), a co-receptor for the type 1 human immunodeficiency virus (HIV-1), is expressed in the human brain in a variety of cell types, including microglia and neurons. Am J Pathol 151
Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359:288–294
Li Y, Wang P, Xu J, Desir GV (2006) Voltage-gated potassium channel Kv1.3 regulates GLUT4 trafficking to the plasma membrane via a Ca2+-dependent mechanism. Am J Physiol Cell Physiol 290:C345–C351
Lichtenwalner RJ, Forbes ME, Bennett SA, Lynch CD, Sonntag WE, Riddle DR (2001) Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Neuroscience 107:603–613
Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, Di Angelantonio S, Ragozzino D, Eusebi F (2005) Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J Neuroimmunol 166
Lipton SA, Chen HS (2003) Paradigm shift in neuroprotective drug development: clinically tolerated NMDA receptor inhibition by memantine. Cell Death Differ 11:18–20
Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, Roberts J, Pushkarsky T, Bukrinsky M, Witte M, Weinand M, Fiala M (2002) Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol 76:6689–6700
Lois C, Alvarez-Buylla A (1993) Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci U S A 90:2074–2077
Lois C, Alvarez-Buylla A (1994) Long-distance neuronal migration in the adult mammalian brain. Science 264:1145–1148
Lu X, Rong Y, Baudry M (2000) Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neurosci Lett 286:149–153
Lynch DR, Guttmann RP (2001) NMDA receptor pharmacology: perspectives from molecular biology. Curr Drug Targets 2
Lynch DR, Guttmann RP (2002) Excitotoxicity: perspectives based on N-methyl-D-aspartate receptor subtypes. J Pharmacol Exp Ther 300
Mariani E, Polidori MC, Cherubini A, Mecocci P (2005) Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. J Chromatogr B Analyt Technol Biomed Life Sci 827:65–75
Marks DR, Tucker K, Cavallin MA, Mast TG, Fadool DA (2009) Awake intranasal insulin delivery modifies protein complexes and alters memory, anxiety, and olfactory behaviors. J Neurosci 29:6734–6751
Maslin CL, Kedzierska K, Webster NL, Muller WA, Crowe SM (2005) Transendothelial migration of monocytes: the underlying molecular mechanisms and consequences of HIV-1 infection. Curr HIV Res 3
McArthur JC (2004) HIV dementia: an evolving disease. J Neuroimmunol 157(1–2):3–10
McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N (2003) Human immunodeficiency virus-associated dementia: an evolving disease. J Neurovirol 9
Metz LM, Zhang Y, Yeung M, Patry DG, Bell RB, Stoian CA, Yong VW, Patten SB, Duquette P, Antel JP, Mitchell JR (2004) Minocycline reduces gadolinium-enhancing magnetic resonance imaging lesions in multiple sclerosis. Ann Neurol 55:756
Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ (1998) Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A 95
Meucci O, Fatatis A, Simen AA, Miller RJ (2000) Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A 97
Miller RJ, Meucci O (1999) AIDS and the brain: is there a chemokine connection? Trends Neurosci 22
Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS (2004) Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 7:711–718
Mizuno T, Kawanokuchi J, Numata K, Suzumura A (2003) Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res 979
Morris A, Marsden M, Halcrow K, Hughes ES, Brettle RP, Bell JE, Simmonds P (1999) Mosaic structure of the human immunodeficiency virus type 1 genome infecting lymphoid cells and the brain: evidence for frequent in vivo recombination events in the evolution of regional populations. J Virol 73:8720–8731
Mueller C, Temmel AF, Quint C, Rieger A, Hummel T (2002) Olfactory function in HIV-positive subjects. Acta Otolaryngol 122:67–71
Murakoshi H, Shi G, Scannevin RH, Trimmer JS (1997) Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol Pharmacol 52:821–828
Navia BA, Jordan BD, Price RW (1986) The AIDS dementia complex: I. Clinical features. Ann Neurol 19:517–524
Neye Y, Dufer M, Drews G, Krippeit-Drews P (2006) HIV protease inhibitors: suppression of insulin secretion by inhibition of voltage-dependent K+ currents and anion currents. J Pharmacol Exp Ther 316:106–112
O'Donnell LA, Agrawal A, Jordan-Sciutto KL, Dichter MA, Lynch DR, Kolson DL (2006) Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the NMDA receptor subtypes. J Neurosci 26
Paulsen O, Sejnowski TJ (2000) Natural patterns of activity and long-term synaptic plasticity. Curr Opin Neurobiol 10:172–179
Pencea V, Bingaman KD, Freedman LJ, Luskin MB (2001) Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol 172:1–16
Pereira CF, Middel J, Jansen G, Verhoef J, Nottet HS (2001) Enhanced expression of fractalkine in HIV-1 associated dementia. J Neuroimmunol 115
Perez-Martin M, Azcoitia I, Trejo JL, Sierra A, Garcia-Segura LM (2003a) An antagonist of estrogen receptors blocks the induction of adult neurogenesis by insulin-like growth factor-I in the dentate gyrus of adult female rat. Eur J NeuroSci 18:923–930
Perez-Martin M, Cifuentes M, Grondona JM, Bermudez-Silva FJ, Arrabal PM, Perez-Figares JM, Jimenez AJ, Garcia-Segura LM, Fernandez-Llebrez P (2003b) Neurogenesis in explants from the walls of the lateral ventricle of adult bovine brain: role of endogenous IGF-1 as a survival factor. Eur J NeuroSci 17:205–211
Plum L, Schubert M, Bruning JC (2005) The role of insulin receptor signaling in the brain. Trends Endocrinol Metab 16:59
Pope M, Haase AT (2003) Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat Med 9:847–852
Portegies P, Enting RH, De GJ, Algra PR, Derix MM, Lange JM, Goudsmit J (1993) Presentation and course of AIDS dementia complex: 10 years of follow-up in Amsterdam. AIDS, The Netherlands, pp 669–675
Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, Cleary P (1988) The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science 239:586–592
Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS (1997) Unique monocyte subset in patients with AIDS dementia. Lancet 349:692–695
Raber J, Toggas SM, Lee S, Bloom FE, Epstein CJ, Mucke L (1996) Central nervous system expression of HIV-1 Gp120 activates the hypothalamic–pituitary–adrenal axis: evidence for involvement of NMDA receptors and nitric oxide synthase. Virology 226:362–373
Ragin AB, Wu Y, Storey P, Cohen BA, Edelman RR, Epstein LG (2006) Monocyte chemoattractant protein-1 correlates with subcortical brain injury in HIV infection. Neurology 66
Rammes G, Danysz W, Parsons CG (2008) Pharmacodynamics of memantine: an update. Curr Neuropharmacol 6:55–78
Razani J, Murphy C, Davidson TM, Grant I, McCutchan A (1996) Odor sensitivity is impaired in HIV-positive cognitively impaired patients. Physiol Behav 59:877–881
Reagan LP (2007) Insulin signaling effects on memory and mood. Curr Opin Pharmacol 7:633–637
Reger MA, Watson GS, Frey Ii WH, Baker LD, Cholerton B, Keeling ML, Belongia DA, Fishel MA, Plymate SR, Schellenberg GD (2006) Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol Aging 27:451–458
Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, Plymate SR, Cherrier MM, Schellenberg GD, Frey WH, Craft S (2008a) Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis 13:323–331
Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, Fishel MA, Plymate SR, Breitner JCS, DeGroodt W, Mehta P, Craft S (2008b) Intranasal insulin improves cognition and modulates {beta}-amyloid in early AD. Neurology 70:440–448
Revest JM, Dupret D, Koehl M, Funk-Reiter C, Grosjean N, Piazza PV, Abrous DN (2009) Adult hippocampal neurogenesis is involved in anxiety-related behaviors. Mol Psychiatry
Ritola K, Robertson K, Fiscus SA, Hall C, Swanstrom R (2005) Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of HIV-1-associated dementia. J Virol 79:10830–10834
Rodriguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, Verkhratsky A (2008) Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS ONE 3:e2935
Ross TM, Martinez PM, Renner JC, Thorne RG, Hanson LR, Frey WH (2004) Intranasal administration of interferon beta bypasses the blood-brain barrier to target the central nervous system and cervical lymph nodes: a non-invasive treatment strategy for multiple sclerosis. J Neuroimmunol 151:66–77
Ross TM, Zuckermann RN, Reinhard C, Frey WH (2008) Intranasal administration delivers peptoids to the rat central nervous system. Neurosci Lett 439:30–33
Rostasy K, Egles C, Chauhan A, Kneissl M, Bahrani P, Yiannoutsos C, Hunter DD, Nath A, Hedreen JC, Navia BA (2003) SDF-1alpha is expressed in astrocytes and neurons in the AIDS dementia complex: an in vivo and in vitro study. J Neuropathol Exp Neurol 62
Rothman S (1984) Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci 4
Rottman JB, Ganley KP, Williams K, Wu L, Mackay CR, Ringler DJ (1997) Cellular localization of the chemokine receptor CCR5. Correlation to cellular targets of HIV-1 infection. Am J Pathol 151
Ryan LA, Zheng J, Brester M, Bohac D, Hahn F, Anderson J, Ratanasuwan W, Gendelman HE, Swindells S (2001) Plasma levels of soluble CD14 and tumor necrosis factor-alpha type II receptor correlate with cognitive dysfunction during human immunodeficiency virus type 1 infection. J Infect Dis 184:699–706
Sacktor N, Schifitto G, McDermott MP, Marder K, McArthur JC, Kieburtz K (2000) Transdermal selegiline in HIV-associated cognitive impairment: pilot, placebo-controlled study. Neurology 54:233
Sacktor N, Tarwater PM, Skolasky RL, McArthur JC, Selnes OA, Becker J, Cohen B, Miller EN (2001) CSF antiretroviral drug penetrance and the treatment of HIV-associated psychomotor slowing. Neurology 57:542–544
Sacktor N (2002) The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol 8(Suppl 2):115–121
Sacktor N, Haughey N, Cutler R, Tamara A, Turchan J, Pardo C, Vargas D, Nath A (2004) Novel markers of oxidative stress in actively progressive HIV dementia. J Neuroimmunol 157
Safrin S, Grunfeld C (1999) Fat distribution and metabolic changes in patients with HIV infection. AIDS 13:2493–2505
Sanchez Mejia RO, Ona VO, Li M, Friedlander RM (2001) Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery 48:1393–1399
Schifitto G, Zhang J, Evans SR, Sacktor N, Simpson D, Millar LL, Hung VL, Miller EN, Smith E, Ellis RJ, Valcour V, Singer E, Marra CM, Kolson D, Weihe J, Remmel R, Katzenstein D, Clifford DB, The AA (2007a) A multicenter trial of selegiline transdermal system for HIV-associated cognitive impairment. Neurology 69:1314–1321
Schifitto G, Navia BA, Yiannoutsos CT, Marra CM, Chang L, Ernst T, Jarvik JG, Miller EN, Singer EJ, Ellis RJ, Kolson DL, Simpson D, Nath A, Berger J, Shriver SL, Millar LL, Colquhoun D, Lenkinski R, Gonzalez RG, Lipton SA (2007b) Memantine and HIV-associated cognitive impairment: a neuropsychological and proton magnetic resonance spectroscopy study. AIDS 21
Schifitto G, Yiannoutsos CT, Ernst T, Navia BA, Nath A, Sacktor N, Anderson C, Marra CM, Clifford DB (2009) Selegiline and oxidative stress in HIV-associated cognitive impairment. Neurology
Schmidtmayerova H, Sherry B, Bukrinsky M (1996) Chemokines and HIV replication. Nature 382
Sellmeyer DE, Grunfeld C (1996) Endocrine and metabolic disturbances in human immunodeficiency virus infection and the acquired immune deficiency syndrome. Endocr Rev 17:518–532
Sevigny JJ, Albert SM, McDermott MP, McArthur JC, Sacktor N, Conant K, Schifitto G, Selnes OA, Stern Y, McClernon DR, Palumbo D, Kieburtz K, Riggs G, Cohen B, Epstein LG, Marder K (2004) Evaluation of HIV RNA and markers of immune activation as predictors of HIV-associated dementia. Neurology 63
Sevigny JJ, Albert SM, McDermott MP, Schifitto G, McArthur JC, Sacktor N, Conant, K Selnes, OA Stern Y, McClernon DR, Palumbo D, Kieburtz K, Riggs G, Cohen B, Marder K, Epstein LG (2007) An evaluation of neurocognitive status and markers of immune activation as predictors of time to death in advanced HIV infection. Arch Neurol 64
Sheng M, Hoogenraad CC (2007) The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 76:823–847
Sheng M, Kim E (2000) The Shank family of scaffold proteins. J Cell Sci 113(Pt 11):1851–1856
Sheng M, Sala C (2001) PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci 24:1–29
Shor-Posner G, Lecusay R, Morales G, Campa A, Miguez-Burbano MJ (2002) Neuroprotection in HIV-positive drug users: implications for antioxidant therapy. J Acquir Immune Defic Syndr 31(Suppl 2)
Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP (2008) Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 18:1085–1088
Sui Y, Potula R, Dhillon N, Pinson D, Li S, Nath A, Anderson C, Turchan J, Kolson D, Narayan O, Buch S (2004) Neuronal apoptosis is mediated by CXCL10 overexpression in simian human immunodeficiency virus encephalitis. Am J Pathol 164
Sui Y, Stehno-Bittel L, Li S, Loganathan R, Dhillon NK, Pinson D, Nath A, Kolson D, Narayan O, Buch S (2006) CXCL10-induced cell death in neurons: role of calcium dysregulation. Eur J Neurosci 23
Takacs J, Zaninetti R, Vig J, Vastagh C, Hamori J (2007) Postnatal expression pattern of doublecortin (DCX) in some areas of the developing brain of mouse. Ideggyogy Sz 60:144–147
Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I (2004) Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 291:317–324
Taupin P (2009) Adult neurogenesis and the pathogenesis of Alzheimer's disease. Med Sci Monit 15:LE1
Thorne RG, Frey WH (2001) Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet 40:907–946
Thorne RG, Emory CR, Ala TA, Frey WH (1995) Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res 692:278–282
Thorne RG, Pronk GJ, Padmanabhan V, Frey WH (2004) Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 127:481–496
Thorne RG, Hanson LR, Ross TM, Tung D, Frey Ii WH (2008) Delivery of interferon-[beta] to the monkey nervous system following intranasal administration. Neuroscience 152:785–797
Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21:2580–2588
Tikka TM, Koistinaho JE (2001) Minocycline provides neuroprotection against N-methyl-d-aspartate neurotoxicity by inhibiting microglia. J Immunol 166:7527–7533
Trejo JL, Carro E, Torres-Aleman I (2001) Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J Neurosci 21:1628–1634
Trejo JL, Carro E, Lopez-Lopez C, Torres-Aleman I (2004) Role of serum insulin-like growth factor I in mammalian brain aging. Growth Horm IGF Res 14(Suppl A):S39–S43
Tschritter O, Machicao F, Stefan N, Schafer S, Weigert C, Staiger H, Spieth C, Haring HU, Fritsche A (2006) A new variant in the human Kv1.3 gene is associated with low insulin sensitivity and impaired glucose tolerance. J Clin Endocrinol Metab 91:654–658
Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF (1999) Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23:583–592
Turchan J, Pocernich CB, Gairola C, Chauhan A, Schifitto G, Butterfield DA, Buch S, Narayan O, Sinai A, Geiger J, Berger JR, Elford H, Nath A (2003) Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology 60
Van Den Bosch L, Tilkin P, Lemmens G, Robberecht W (2002) Minocycline delays disease onset and mortality in a transgenic model of ALS. NeuroReport 13:1067–1070
Vance DE (2007) Olfactory and psychomotor symptoms in HIV and aging: potential precursors to cognitive loss. Med Sci Monit 13:SC1–SC3
Vandenberg RJ (1998) Molecular pharmacology and physiology of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol 25:393–400
Vergote D, Butler GS, Ooms M, Cox JH, Silva C, Hollenberg MD, Jhamandas JH, Overall CM, Power C (2006) Proteolytic processing of SDF-1alpha reveals a change in receptor specificity mediating HIV-associated neurodegeneration. Proc Natl Acad Sci U S A 103:19182–19187
Visentin S, Renzi M, Levi G (2001) Altered outward-rectifying K(+) current reveals microglial activation induced by HIV-1 Tat protein. Glia 33:181–190
Walker TL, Yasuda T, Adams DJ, Bartlett PF (2007) The doublecortin-expressing population in the developing and adult brain contains multipotential precursors in addition to neuronal-lineage cells. J Neurosci 27:3734–3742
Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M (2003) Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35:238–245
Westervelt HJ, McCaffrey RJ, Cousins JP, Wagle WA, Haase RF (1997) Longitudinal analysis of olfactory deficits in HIV infection. Arch Clin Neuropsychol 12:557–565
Williams KC, Hickey WF (2002) Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu Rev Neurosci 25
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22:1763–1771
Xiong K, Luo DW, Patrylo PR, Luo XG, Struble RG, Clough RW, Yan XX (2008) Doublecortin-expressing cells are present in layer II across the adult guinea pig cerebral cortex: partial colocalization with mature interneuron markers. Exp Neurol 211:271–282
Xu J, Wang P, Li Y, Li G, Kaczmarek LK, Wu Y, Koni PA, Flavell RA, Desir GV (2004) The voltage-gated potassium channel Kv1.3 regulates peripheral insulin sensitivity. Proc Natl Acad Sci U S A 101:3112–3117
Zhang C, McNeil E, Dressler L, Siman R (2007) Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer's disease. Exp Neurol 204:77–87
Zhang K, Rana F, Silva C, Ethier J, Wehrly K, Chesebro B, Power C (2003) Human immunodeficiency virus type 1 envelope-mediated neuronal death: uncoupling of viral replication and neurotoxicity. J Virol 77
Zhang WJ, Tan YF, Yue JT, Vranic M, Wojtowicz JM (2008) Impairment of hippocampal neurogenesis in streptozotocin-treated diabetic rats. Acta Neurol Scand 117:205–210
Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng YC, Gelbard HA, Shepard RB, Swartz JM, Gendelman HE (1999) Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol 98:185–200
Zink MC, Uhrlaub J, DeWitt J, Voelker T, Bullock B, Mankowski J, Tarwater P, Clements J, Barber S (2005) Neuroprotective and anti-human immunodeficiency virus activity of minocycline. JAMA 293:2003–2011
Zucco GM, Ingegneri G (2004) Olfactory deficits in HIV-infected patients with and without AIDS dementia complex. Physiol Behav 80:669–674
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Kathryn A. Lindl and David R. Marks contributed equally to this study.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lindl, K.A., Marks, D.R., Kolson, D.L. et al. HIV-Associated Neurocognitive Disorder: Pathogenesis and Therapeutic Opportunities. J Neuroimmune Pharmacol 5, 294–309 (2010). https://doi.org/10.1007/s11481-010-9205-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-010-9205-z