
Abstract
Purpose
The consequences of permanent inundation and storage of flood plain soils and aquatic sediments on metal mobility were studied. The main goal was to quantify kinetic mobilisation processes in order to pinpoint the conditions that pose emission risks to groundwater and surface waters.
Materials and methods
Anaerobic pore water compositions of six existing depots, containing sediments from either aquatic or terrestrial origin, were measured to obtain reliable field references. Reduction experiments were performed with SOFIE cells, in which time-dynamic measurements of reducing pore waters were carried out at the reigning redox conditions. A parallel experiment tested the possibility if sulfide-deficiencies could be compensated by the reduction of added gypsum, thus increasing the available pool of reactive S. Model calculations were performed to distinguish between the thermodynamic and kinetic processes.
Results and discussion
Reduction of flood plain soils showed that dissolved organic matter (DOM) concentrations increased up to 7-fold over time, as a result of nutrient-mediated metabolic boost of organic matter degrading microorganisms. The association of metals to DOM increased significantly due to the loss of reactive Mn-oxide and Fe-oxyhydroxide sorption phases during reduction. DOM released metals only slowly, therefore kinetically hindering the phase shifting to stable metal-sulfide precipitates. The observed effects lasted at least 10 months. In aquatic sediments, reduction rates of sulphates were six times faster, and the release of DOM occurred in lower amounts than in soils from terrestrial origin. It is shown that the addition of gypsum stimulates the formation of sulfides, thereby decreasing dissolved concentrations of most metals. On the short term however, the addition of gypsum leads to elevated concentrations of Zn and Cd due to increased electrolyte strength and subsequent Ca-metal-competition for sorption sites. This effect may be eliminated by not only adding an electron acceptor to the sediment, but also an effective donor, e.g., degradable organic matter.
Conclusions
For depots containing flood plain soils, the same kinetic mechanisms were observed as in the reduction experiments (elevated production of DOM and associated metals). It was concluded that reduction of flood plain soils may not necessarily result in efficient metal trapping. When storage is considered, a distinction should be made between sediments of terrestrial and aquatic origin.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Along the main Dutch rivers, numerous restoration and redevelopment projects are carried out to improve safety in cases of high discharges and flooding. Often, large amounts of contaminated sediments are stored under water in depots or sandpits. This practice is based on the assumption that anaerobic conditions will develop over a short time period, and cause heavy metals to immobilize through sulfide precipitation. In this paper, we show that the generality of this assumption may be seriously questioned.
Dredging activities often lead to dumping of anaerobic sediments on land, leading to chemical and physical oxidation and changes in availability of metals. Many authors have reported on these oxidation mechanisms (Holmes 1999; Buykx et al. 2000; Mayer and Jarrell 2000; Simpson et al. 2000; Xiang et al. 2000). Since legislation has become aware of the potential environmental risks involved (i.e., elevated exposure of heavy metals to humans, cattle and ecosystems), the application on land is in many EU countries restricted. As a result of this, contaminated soils and sediments are stored in waterlogged depots or pits, often without truly knowing the effects on the short or long term.
The (time-dynamic) measurement of redox-sensitive elements in anaerobic pore water is a specialized and elaborate undertaking. Only few studies report on the effects of reduction with respect to metal mobility and emissions to surrounding groundwater systems. There are some useful studies that include field or laboratory measurements (e.g., Han et al. 2001; Burton et al. 2008; Du Laing et al. 2009), and also modelling studies that use a thermodynamic approach have been reported (e.g., De Rooij and Gerrits 1995; Griffioen et al. 2006). However, the outcomes of the measurements in the field and modelling studies are not always in agreement, indicating that the uncertainty around soil inundation or sediment storage is still large. Relatively little is known about both the qualitative (mechanisms) and quantitative (concentrations and rates) aspects of this practice.
The classical concept of reduction reactions is that they proceed sequentially and from the highest towards the lowest energy yield (Berner 1981; Stumm 1984; Lovley and Goodwin 1988). However, there are sufficient indications to question whether redox processes are, in a mechanistic sense, reversible and lead to end phases that are similar to previous oxidation or reduction events. Postma and Jakobsen (1996) showed that the reduction of Mn, Fe, sulphate, and methanogenesis could be well explained by a partial equilibrium approach where the fermentive step (i.e., microbial reduction of organic matter) is rate limiting. Cooper et al. (2000) reported on the impact of competition between nitrate and iron reduction on metal geochemistry, which may eventually block the processes that immobilize zinc during reduction.
In this study, we collected samples from six large sediment depots and storage sites in The Netherlands. Reduction experiments were conducted with soils from river flood plains to mimic permanent inundation or storage. This was done by long-term inundation of aerobic soils originating from the banks of the rivers Meuse and Rhine in The Netherlands. Pore water compositions were monitored at the prevailing redox conditions for up to 10 months.
The effect of gypsum amendment on sulfide formation was studied, both experimentally and via modelling exercises. The primary goal of this paper is to give quantitative insight in the consequences of the practice mentioned above. This is the first report to propose a mechanism that explains the observed metal kinetics, involving microbial action, during reduction.
2 Methods
2.1 Measurements in storage sites
Sediment cores were collected from six large sediment depots: Slufter (51°55′31 N, 4°00′15E), Amerikahaven (52°42′23 N, 4°46′35E), IJsseloog (52°35′56 N, 5°44′34E), Moorlag (51°58′26 N, 5°51′43E), Asselt (51°14′19 N, 6°00′28E), and Meers (51°49′13 N, 5°38′22E). Data on depot history and sediment origin were provided by the Dutch Ministry of Transport and Public Works (Rijkswaterstaat, unpublished). These depots receive their sediments from the local area. The first three depots contain sediments that originate from the aquatic environment, e.g., from dredging activities. The last three depots contain soils from flood plains that were sanitised and redeveloped. According to Dutch environmental quality standards, the soils and sediments in these depots are considered moderately to heavily contaminated with heavy metals.
From each depot, cores were collected from a ship at a depth of 3 m or more below the water-sediment interface, using a Vibro corer operated from a crane. These cores were 70 cm in length, with a diameter of 20 cm. The core ends were sealed on site, and stored in bags that were flushed with nitrogen gas from portable cylinders. In the laboratory, the columns were brought in a grade 6 N2-gas glove box at 15°C in a climate control chamber. The column walls were perforated with a 1-mm drill, and SOFIE® probes (see section 2.2) were inserted on two depths around the core centre. In this way, pore water compositions were measured at the occurring redox status in duplicate.
2.2 Floodplain soil reduction
To simulate the reduction of aerobe terrestrial soils, reduction experiments were conducted with three flood plain soils. These were collected from: “Meuse-north”, (51°15′55 N, 6°01′27E) near the city of Beesel; “Meuse-south” (51°13′27 N, 5°59′16E) near Asselt; and “Rhine” (51°58′15 N, 5°51′46E) near Arnhem. All soils were classified as Fluvisols (FAO 1998). These metal-contaminated pastures were selected because they were eligible for future restoration works and subsequent large-scale storage in deep groundwater pits.
The top 0.05–0.35 m was sampled in an undisturbed manner using SOFIE® cells (Sediment or Fauna Incubation Experiment), described in detail by Vink (2002). These cells enable undisturbed sampling, and accurate probing of pore water over time, while the redox status maintains intact. Up to 16 probes can be inserted at desired depths via air-tight connectors in the cell wall. A probe consists of a polyethersulfone semi-permeable polymer, which retains and transports pore water without disrupting equilibrium conditions. The polymer discriminates colloidal fractions >0.1 μm. This mesh size is impermeable to bacteria, so pore water that passes the polymer is sterile. This is a large advantage, since microbial processes (e.g., redox transformations) do no longer occur in the extracted pore water sample.
After sampling, cells were placed in a climate-controlled room (15 ± 2°C). The room was darkened to prevent the production of CO2 by algae in the cells. The cells were inundated with river water via an upward flow in the column by connecting the deepest probe connector to a water reservoir via a siphon construction. The soils became waterlogged after 8 till 14 h, and 0.1 m of water was maintained on the soil surface. Oxygen was removed from the system by flushing the headspace in the cell with N2-gas (grade 6 purity) during the course of the experiments, which varied from 225 to 283 days. Pore water was sampled periodically in the centre of the core by using the membrane probes described earlier and a slow rotation peristaltic pump. Samples were taken in duplicate, and Eh and pH were measured directly in the pore water. Pore water samples for metal analyses were acidified with ultrapure HNO3. All samples were analysed within 30 min after sampling. At the end of the experiments, the sediment was sampled for characteristics and total metal contents by aqua regia (HNO3/HCl, 3:1 microwaved) destruction procedure NEN 6961.
2.3 Gypsum amendment
A parallel experiment was conducted to test whether sulfide-deficiencies could be compensated for by the reduction of CaSO4.2H2O (gypsum), thus increasing the available pool of reactive S. Gypsum is a raw produce that is cheap and readily available, and could therefore be applied on a large scale. Although the application of gypsum to soils is a much-applied practice to increase cation adsorption (e.g., Singh 1984; Fahrenhorst et al. 1999), the effects of reduction of gypsum on the geochemical composition and subsequent mobility of metals has never been reported in this context.
The soil from “Meuse-north” was selected for these experiments. This site is scheduled for redevelopment works, and large amounts of soil are destined to be stored in former sand pits. Former studies from this site included detailed sampling for characterisation and modelling studies (Vink 1999). The mean content of S was 17.8 ± 9.1 (SD) mmole.kg−1 (n = 18), and it was concluded earlier that this amount may be insufficient to bind all available metals in the sediment when stored under anaerobic conditions. Therefore, two doses of S were applied to the sediment: 50 and 500 mmole.kg−1, by using ultrapure gypsumdihydrate, CaSO4.2H2O. These amounts were based on theoretical S-deficiencies of the soil for the sum of heavy metals, assuming that only the reactive Fe-phase (as defined by Postma and Jakobsen 1996) will initially associate with S. In this case, dosages correspond to approximately 0.2 and 2 times the theoretical S-deficiency of 260 mmole.kg−1 calculated from data from Tables 1 and 2. The gypsumdihydrate was mixed homogeneously with the soil, prior to inundation. Dissolved sulfides were measured with a Ag/S solid-state ion selective electrode (ISE; Cole Parmer; l.o.d.: 1 μg.l−1). A five-point calibration curve was made from stock solutions, which was prepared by dissolving crystals of Na2S.9H2O in deoxygenated millipore water. The ISE was placed at 5 cm depth into the sediment via an air-tight connector in the lid of the cell over the course of the experiment. Physical sediment properties were determined via standard analytical NEN protocols.
2.4 Pore water analyses
In the inundation experiments (section 2.2 and 2.3), pore water was sampled on a weekly basis in duplicate from the core centre at −0.15 m using the SOFIE probes. Sampling intensity was adjusted according to the rate at which concentration shifts of the studied compounds occurred. Pore water samples were analyzed for As, Cd, Cu, Cr, Ni, Pb, Zn, Fe, Mn, Na, Ca, and Mg using high-resolution inductively coupled plasma mass spectrometry (Thermo Electron Element2 field sector, dual mode secondary electron multiplier with linear dynamic range of six orders; limit of detection (lod) min/max: 0.003 μg.l−1 for Cd to 6.0 μg.l−1 for Zn). Internal standardization of elements was checked with In115/Bi209/Li6. External reference standards and internal quality samples were used for quality control. Analytical lod are 3× standard deviations in blanks.
NO −2 , NO −3 , PO 2−4 , SO 2−4 , and Cl−were analyzed with an ion chromatograph using a High Capacity analytical column AS9-HC (Dionex), based on conductivity detection with chemical suppression (lod min/max: 0.025 mg.l−1 for NO −2 to 0.05 mg.l−1 for PO 2−4 ). All anions were thus measured sequentially under equivalent conditions. NH +4 was measured with a Skalar Segmented Flow analyzer SAN + 6250 matrix photometer (lod: 0.022 mg.l−1). Dissolved organic carbon was determined with a Shimadzu 5000 TOC analyzer (lod: 0.45 mg.l−1). All measurements in this study were performed with 6-point calibration curves from standard reference stock solutions.
2.5 Speciation modelling
As explained earlier, the soil from “Meuse-north” was expected to have insufficient sulfide content after full reduction, which would consequently result in insufficient metal trapping.
The outcome of the reduction experiments is the overall result of chemical, thermodynamic processes and microbial processes. To distinguish between the thermodynamic part and the kinetic part that dictate the overall results, thermodynamic speciation calculations were performed for the “Meuse-north” soil. These calculations were performed with the chemical speciation model BIOCHEM-ORCHESTRA (Vink and Meeussen 2007). This model is based on the chemical module BIOCHEM (Schröder et al. 2005; Vink and Schröder 2006) which was modified with the NICA-DONNAN concept for complexation of metals to DOM (Benedetti et al. 1995), and coupled to the ORCHESTRA framework (Meeussen 2003). Unlike most chemical speciation models, BIOCHEM-ORCHESTRA is capable of simulating redox changes over time by relating water saturation and duration to reduction potential for each layer. The model contains modules for precipitation and dissolution of solids, based on an extended database of mineral components with up-to-date thermodynamic constants for the ferro/ferric and sulphur/sulfide minerals. Model schematization started at oxic steady state, followed by water saturation, thus simulating inundation and subsequent reduction. We assumed that DOM consisted of equal amounts of fulvic and humic components. Starting values for the model schematization were adapted from Tables 1 and 2. Pore water compositions from both oxic and reduced layers in soil cores of the same site from an earlier campaign (Vink 1999) were used to validate the model for this purpose.
3 Results and discussion
3.1 Measurements in storage sites
Characteristics of the cores taken from the depots are shown in Table 1 (solid phase) and 2 (pore water). Based on measurements of redox potential (Eh), [NO2/NO −3 ], [NH +4 ] and [SO 2−4 ], all sediments are considered to have sulphate reducing conditions.
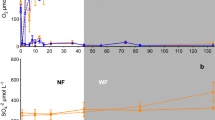
Figure 1 shows DOC and Zn concentrations in pore water collected from the six depots. The results immediately show that in situ concentrations of DOC in the depots that contain floodplain sediments have significant elevated values (up to a factor 5) compared with the depots with dredged sediments from aquatic environments. Depots Asselt and Meers have DOC concentrations of respectively 165 and 215 mg/l, which is in the same order as the maximum concentrations that we observed in the reduction experiments. Also, Zn concentrations are a factor of 10 higher (266 and 204 μg/l, respectively) in the “terrestrial” depots. This is strong evidence that the proposed mechanisms in Fig. 2 do indeed occur in the field. This is an important finding, not only from a chemical, but also from a management point of view. In risk assessments for metal emissions, the origin of sediment does matter, since dredged sediments and floodplain sediments do behave differently in terms of metal mobility following storage, inundation and reduction.

Concentrations of DOC and metals in pore water, collected from six depots (error bars are standard deviations). Depots on the right (Moorlag, Asselt, Meers) contain soils from terrestrial environments and have higher concentrations of DOC and metals in pore waters than depots on the left (Slufter, Amerika haven, IJsseloog) that contain sediments from aquatic environments

Schematization of reduction mechanisms in time. Reductive production of Mn2+ increases the metabolic organic matter fermentation and the production of DOM, which prolongs the retention of metals in solution (DOM dissolved organic matter, M metal, S sulfide)
3.2 Floodplain soil reduction
Sediment and pore water characteristics are listed in Table 3. Compared with the Rhine location, the two Meuse locations are generally more clayey and organic, and have a higher pH. All soils are rich in iron. The carbon to sulphur ratio in Meuse-north is twice as high as in Meuse-south (C/S = 161 and 86, respectively), which is higher than commonly observed for pastures (Nguyen and Goh (1994) report a mean value of 90). Net-loss of S in this flood plain soil is not very likely, since surface water provides a significant S-source during floods (40–70 mg.l−1 SO 2−4 (Waterbase 1999–2008)). It is possible that the Meuse-north site may have a lower organic matter turnover rate than the Meuse-south location, but this was not further investigated.
Inundation effectively lowered redox potentials in pore water (Fig. 3). Dissolved oxygen concentrations in inundation water dropped from 10 ± 2 to 3.5 ± 1 mg.l−1 over the first 25 days. Along reduction, pH decreased gradually in the Meuse sediments. In the Rhine sediment, pH increased temporarily by almost a full unit, due to alkalinity production. This was most probably caused by anaerobic microbial respiration of organic matter (raising pCO2) and simultaneous calcite dissolution. After approximately 5 months, pH stabilizes and a net increase of half a pH-unit compared with the initial aerobic state is maintained. The final pH of the three soils differs by only 0.4 unit, indicating that the pH of the dissolved phase is regulated by the dissolution of carbonates.

Pore water concentrations during reduction in three inundated soils
While oxygen is metabolized, nitrites and nitrates act as alternative electron acceptors, and are at least partially converted to ammonium. Denitrification rates varied around 30 ± 3 μg.ml−1.d−1 for the three soils, which is quite uniform, but are somewhat lower than Siegrist et al. (1999) found in activated sludge (70–100 μg.ml−1.d−1).
Further reduction lead to the dissolution of Mn-oxides and Fe-oxyhydroxides (Patrick and Verloo 1998; Van Cappellen et al. 1998). Dissolved concentrations of Mn(II) and Fe(II) increased by a factor 20 within 7 days. Cooper et al. (2000), who studied the formation of mineral phases during reduction, stated that microbial reduction of Fe(III) was inhibited when NO −3 was present as an alternative electron acceptor. In this study, denitrification and Mn(IV) and Fe(III) reduction occurred simultaneously, and even simultaneous Fe(III) and SO 2−4 reduction was reported earlier (Postma and Jakobsen 1996; Vink 2002). Francis and Dodge (1990) showed that the release of metals from Fe-oxyhydroxides was facilitated by microbial action, and that dissolved Mn(II) facilitated this mechanism (Guha et al. 2001).
Reduction rates were fast in the first 3 weeks after inundation. However, the reduction of sulphate did not start until approximately 2 months. Dissolved sulfides were not detected until approximately 80 days.
[PO4] in pore water increased due to two main mechanisms: (1) dissolution of inorganic phases such as strengite, FePO4 (Simpson et al. 2000), and (2) P-release via decomposition of particulate organic matter, which is indicated by the increase of [NH +4 ] (Lopes, 2004). The effect of P-release from the solid state to solution is enhanced by what was suggested by Siegrist et al. (1999). They found a 6-fold P-release in sludge that became anaerobic, which they attributed to a reduced phosphate uptake by microorganisms. Due to the reduction of Fe(III) to Fe(II), large amounts of [Fe2+] occurs in solution, and the released phosphate precipitates to form vivianite minerals (Fe3(PO4)2). This subsequently results in P-immobilization. This up-and-down trend of PO4− concentrations during reduction is clearly recognized in Fig. 3, and is most pronounced for the Rhine and Meuse-south soils.
While organic matter undergoes incomplete mineralization, degradation products are formed, resulting in a 6- to 7-fold increase of DOC in all three soils. The sheer magnitude of this increase is remarkable, since it is generally assumed that the metabolic activity of microorganisms is low at sub-oxic or anaerobic conditions. Evidently, the mineralization of organic matter is not inhibited due to the flooding, at least not for a short-term period after inundation.
The observed trends give a strong indication that the release of phosphates may boost the mineralization of organic matter, followed by the production of DOM. The dissolution of Mn-oxides may also play a role, since Mn2+ is an essential micronutrient in many biochemical reactions (Tietz 1957; Wakil et al. 1957). In aerobic conditions, oxides like Mn are poorly soluble, resulting in low concentrations in pore water and hence low bioavailability (Vink et al. 1999). This effect should be further investigated, using abiotic blanks and cell counts to identify the role of microbial degradation of organic matter, in order to quantify its significance in relation to the chemical processes that occur during reduction of soils and sediments.
The dissolution reactions of phosphates and subsequent association with other cations in solution are very sensitive to small changes in pH. Assuming [CO 2−3 ] ∼ pCO2 and with [Ca2+] = 1.10−3 mol.l−1, we find that Mn2+ concentrations may drop 50% when pH decreases only from 7.2 to 7.0. However, in PO 2−4 dominated systems, 95% of total Mn will still be present as relatively soluble MnHPO4 at this pH, and Mn will therefore continue to stay in solution for a prolonged period. Moreover, Mn2+ will not form insoluble sulfide phases (MnS) easily when Fe2+ is present (Morse and Luther 1999), which is almost always the case.
These results show that during reduction, favourable conditions occur that facilitate the transformation of particulate organic matter to DOM, as a result of the release of essential elements. This effect is temporal, since further reduction creates, obviously, unfavourable respiratory conditions due to complete oxygen depletion. However, during this temporal boost, a high turnover of organic matter occurs, and hence the production of DOM. The left hand side of Fig. 2 schematizes the mechanisms mentioned above.
3.3 Heavy metal response to reduction
As is shown in Fig. 3 and discussed in section 3.2, DOM concentrations increased significantly in all three soils during reduction. The differences in DOM-formation rates between soils most likely depend on the reactivity of the local humic matter and other factors that influence microbial activity (such as pH and availability of nutrients). The mechanisms behind these local differences are not further discussed here. But it is clear that compared with the Meuse sediments, the Rhine sediment releases larger amounts of PO4, NH4, arsenic, chromium and nickel.
In most cases, metal concentrations increased during reduction as a result of association with DOM, in some cases far exceeding quality standards for groundwater and surface water (e.g., As, Cd, Cr, Ni, Pb). In fact, none of these sediments meet the Dutch target quality standards for both deep and shallow groundwater over the course of the experiments. So it becomes clear that in anaerobic environments, release (and possibly emission) of metals actually occurs.
For arsenic, increasing concentrations following reduction is a well-known phenomenon (Cherry et al. 1979; Pierce and Moore 1982; Macur et al. 2004). As(V), arsenate, is reduced to As(III), arsenite, which is very mobile and has led to elevated groundwater concentrations on many occasions. Chromium(VI) can be reduced to Cr(III) by reactions with e.g., Fe (acting as a catalyst (Eary and Rai 1988)) and organic matter (Kozuh et al. 2000). The direct relation between DOM and reduction rates of chromium was discussed by Buerge and Hug (1998), and Francis and Dodge (1990) showed that Cr was liberated by Fe-oxyhydroxides by—again—microbial action. Later, Guha et al. (2001) showed that dissolved Mn(II) facilitates this mechanism. In summary, there are sufficient indications to state that the dissolution of metals results from the formation of DOM, mediated by the availability of the essential nutrients.
Reduction and subsequent dissolution of Mn(IV–VII)-oxides and Fe(III)-oxyhydroxides initially leads to the release of metals (e.g., Dong et al. 2000). Once in solution, most metals will redistribute over the remaining sorption phases. DOM is an excellent provider of bonding sites (Sauvé et al. 1997; Christensen et al. 1999; Eyrolle and Benaim, 1999). If sulfides have not yet formed, DOM is the dominant and most accessible provider of bonding sites for metals that are released in solution. This was confirmed by the modelling results summarized in Table 4, showing the dominance of metal-DOM fractions in solution.
The increase in DOM concentrations during reduction not always leads to prolonged mobilization of metals. For example, copper shows a prolonged mobilization in the Meuse-north sediment, but not in the Rhine and Meuse-south sediments. Probably the most important reason for this is the fact that Cu, of all metals except Hg, forms the most stable sulfide complexes. The Cu–S solubility product, Ksp, is in the order of 10−36, compared with 10−19 for Ni–S (Fisher and Peters 1970). For Meuse-north, both the modelling results (section 3.5) and the dissolved-sulfide (HS−) measurements presented in Fig. 3 show a sulfide deficiency for Meuse-north. So instead of associating to the first-formed sulfides, as occurs in Rhine and Meuse-south, copper in Meuse-north predominantly binds to DOM. This observation does not contradict the classical idea that Cu always and massively binds to DOM. Indeed, DOM dominates the speciation of Cu in the aqueous phase, as we will show in section 3.5. But DOM concentrations almost always far exceed concentrations of copper (mg-range vs. μg-range), and available sorption sites may already be occupied. The formation of even more DOM in that case not necessarily leads to redistribution of Cu.
An explanation for the observed time lap between sulfide formation (see Fig. 3: after 80 days of inundation) and actual formation of metal-sulfide compounds (after 10 months) is found in the kinetic constraint of release of metals from DOM. To form complexes with sulfides, metals first need to dissociate from DOM. However, desorption rates are often much slower than sorption rates (Strawn et al. 1998), and sorption reactions of metals to DOM may even be non-reversible. Evidence for this was provided by several authors (reviewed by Sutton and Sposito 2005). In any case, desorption of metals from dissolved humic substances is slow or partly even irreversible. This consequently results in prolonged increased metal concentrations in solution. This mechanism is shown in the right hand side of Fig. 2, and completes the overall schematization of the relation between thermodynamically regulated processes and the kinetic mechanisms during reduction.
The occurrence of kinetic constraints, i.e., the fast association of metals with DOM and slow release to S-phases, was in fact observed earlier by some authors, but not explicitly explained. In experiments with anaerobic sediments, Buykx et al. (2000) and Simpson et al. (2000) found that a large portion of sediment-bound metals (73–95%) were non-sulfide phases. They concluded that reactive sulfide phases play a limited role in controlling the speciation of metals entering sulfide-rich environments. This may indeed often be the case, since sediments in lakes and rivers are mostly not at equilibrium. Given the fact that metal-sulfide complexes are among the most stable ones, dissolved metal concentrations in a sulfide-controlled sediment should therefore approach zero. This is however very rarely reported, and also our measurements in anaerobic depots do not show zero-concentrations of metals.
3.4 Gypsum amendment
Two-tailed paired variance analyses (p < 0.05) showed that the difference in metal concentrations between the two gypsum dosages (section 2.3) were not significant, except for Zn and, obviously, Ca and SO4. The smallest dose (50 mmole S.kg−1) was sufficient to eliminate the S-deficiency in the sediments. Time-dynamic examples are shown in Fig. 4. The following trends are observed:
-
1)
As, Cu, and Pb concentrations in pore water are significantly lower after gypsum amendment.
-
2)
Zn and Cd concentrations in pore water rise by a factor 2; the addition of gypsum leads to increased release from the solid phase;
-
3)
Little effects were observed for Ni and Cr. Ni2+ has slower reaction kinetics than the other metals (Morse and Luther 1999; Snodgrass 1980), which indicates that those metals form sulfide phases prior to Ni. Cr is inert to reaction with sulfide.

Pore water concentrations during reduction in Meuse-north (solid line) and with two doses of gypsum (circles, 50 mmole S.kg−1, triangles, 500 mmole S.kg−1). Vertical bars represent analytical variation
Elevated concentrations of Zn and Cd occur, due to the addition of Ca. This enhances competition with Ca2+ ions for bonding sites. In addition, and perhaps more importantly, relatively large fractions of solid-phase Zn and Cd are in the oxic state associated with the carbonate phase (Smith and Martell 1976; Tessier et al. 1979), and are exchanged with Ca via the same mechanism (Warwick et al. 1998). For As, Pb, and Cu, the competition with Ca2+ for sorption sites is of lesser importance, because of mainly two reasons: (1) their capacity to form stable ligand complexes with OM (both SOM and DOM; Sauvé et al. 1997; Shi et al. 1998; Christensen et al. 1999). Pb, and Cu in particular, will remain in the OM-phase, disregarding the ionic strength of the composite solution; (2) the physical behaviour of DOM in the presence of large amounts of Ca2+. Römkens and Dolfing (1998) concluded from their experiments with organic ligands that stoichiometric changes of DOM took place when calcium was added to the solution. We indeed observed significant disappearance of DOM from pore water, as a result of flocculation and subsequent precipitation. In their experiments with 400 mg.l−1 Ca, which is comparable to concentrations in the gypsum-enriched soils of this experiment, Eyrolle and Benaim (1999) immobilized 40% to 60% of the soluble copper through this flocculation mechanism. This order of magnitude agrees very well with the observations presented in Fig. 4. By increasing [Ca2+] from 100 to 550 mg.l−1, DOC concentrations drop 38%. Accordingly, many metal concentrations decline with the same order of magnitude.
3.5 Speciation modelling; the effect of gypsum amendment
Results of the speciation calculation for the final anaerobic steady state, and the effect of gypsum addition, are shown in Table 4. Speciation in this case is dictated by the thermodynamic stability of the involved species including sorption to DOM. The stable reduction end-point was checked using Fe(III) and Fe(II) as a redox couple, which is expressed by the Nernst equation: \( {E_{\rm{h}}} = {E^0} + {\hbox{RT}}/{\hbox{nF ln}}\left[ {{\hbox{F}}{{\hbox{e}}^{{3} + }}} \right]/\left[ {{\hbox{F}}{{\hbox{e}}^{{2} + }}} \right] \). The standard potential E0 (0.77 V) was calculated from the Gibbs free energy derived from thermodynamic tables. This yields \( 0.{77} + 0.0{59}.{ \log }\left( {\left[ {{\hbox{F}}{{\hbox{e}}^{{3} + }}} \right]/\left[ {{\hbox{F}}{{\hbox{e}}^{{2} + }}} \right]} \right) = --0.{232} {\hbox{V}} \), which is in agreement with the end-point measurements, being −0.248 V after 10 months. For these calculations it was assumed that in the aerobic state all dissolved iron was present as Fe(III), and in the anaerobic state as Fe(II).
The results show that: (1) DOM plays a major part in metal speciation, and dominates the metal fraction in the dissolved phase, even when sulfides are present; (2) Free ion concentrations are relatively large for Cd2+ (36% of total dissolved), Zn2+ (14%) and Pb (13%), but concentrations decrease some orders of magnitude when gypsum is added and the reactive pool of sulfide is increased; (3) Sulfide deficiency occurs in the natural soil and limits the complete immobilization of Cd, Pb, Zn, but not Cu. This limitation is compensated by the addition of 50 mmole S in the form of gypsum.
4 Conclusions
Metal speciation during reduction cannot be attributed to thermodynamic relationships alone, but also results from differences in exchange reaction kinetics. For reducing pore waters, it was demonstrated that the association of metals with DOM plays an important role in chemical speciation, and that the production of DOM is facilitated by the release of nutrients from the solid phase. It is demonstrated that DOM releases metals only slowly, therefore kinetically hindering the phase shifting to stable metal-sulfide precipitates. The observed differences between Rhine and Meuse locations demonstrate that the significance of the described mechanisms is dictated by local conditions.
It was shown that addition of gypsum stimulates the formation of sulfides. As a result, metal concentrations decrease significantly (ranging from 24% for Cr to 64% for Pb after 6 months of inundation; p < 0.05). On the short term however, the addition of gypsum leads to elevated concentrations of Zn and Cd due to increased Ca-metal-competition for bonding sites. We conclude that not only an effective electron acceptor has to be added to the sediment, but also an effective donor, such as easily degradable organic matter.
In situ measurements in depots showed that sediments originating from flood plains follow the same mechanisms that were observed in the reduction experiments. Both DOC concentrations and associated metals increase. This mechanism did not occur in depots that contain sediments from aquatic origin. This is an important finding, and shows that when permanent inundation or sediment storage is considered, a clear distinction should be made between sediments of terrestrial and aquatic origin.
References
Benedetti MF, Milne CJ, Kinniburgh DG, VanRiemsdijk WH, Koopal L (1995) Metal ion binding to humic substances: application of the non-ideal competitive adsorption model. Environ Sci Technol 29:446–457
Berner RA (1981) A new geochemical classification of sedimentory environments. J Sediment Petrol 51:359–365
Buerge IJ, Hug SJ (1998) Influence of organic ligands on chromium(VI) reduction by iron(II). Environ Sci Technol 32:2092–2099
Burton ED, Bush RT, Sullivan LA, Johnston SG, Hocking RK (2008) Mobility of arsenic and selected metals during re-flooding of iron and organic rich acid sulfate soil. Chem Geol 253:64–73
Buykx SEJ, Bleijenberg M, VandenHoop MAGT, Loch JPG (2000) The effect of oxidation and acidification on the speciation of heavy metals in sulfide rich freshwater sediments using a sequential extraction procedure. J Environ Monitor 1:23–28
Cherry JA, Shaikh AU, Tallman DE, Nicholson RV (1979) Arsenic species as an indicator of redox conditions in groundwater. J Hydrol 43:373–392
Christensen JB, Botma JJ, Christensen TH (1999) Complexation of Cu and Pb by DOM in polluted groundwater: a comparison of experimental data and predictions by computer speciation models (WHAM and MINTEQA2). Wat Res 15:3231–3238
Cooper DG, Picardal F, Rivera J, Talbot C (2000) Zinc immobilization and magnetite formation via ferric oxide reduction by Shewanella putrefaciens. Environ Sci Technol 34:100–106
De Rooij NM, Gerrits HJ (1995) The transport of pollutants from a sludge desposal site. European Water Poll Control 5(5):43–55
Dong Y, Ma LQ, Rhue RD (2000) Relation of enhanced Pb solubility to Fe partitioning in soils. Environ Poll 110:515–522
Du Laing G, Meers E, Dewispelaere M, Rinklebe J, Vandecasteele B, Verloo MG, Tack FMG (2009) Effect of water table level on metal mobility at different depths in wetland soils of the Scheldt estuary, Belgium. Water Air Soil Pollut 202:353–367
Eary LE, Rai D (1988) Chromate removal from aqueous wastes by reduction with ferrous iron. Environ Sci Technol 22:972–977
Eyrolle F, Benaim JY (1999) Metal available sites on colloidal organic compounds in surface waters. Wat Res 4:995–1004
Fahrenhorst C, Botschek J, Skowronek A, Ferraz J (1999) Application of gypsum and lime to increase cation adsorption of a geric ferralsol in the Brazilian Amazon region. J Plant Nutr Soil Sci 162:41–47
FAO (1998) World reference base for soil resources. Int Soc Soil Sci, Rome, ISBN 92-5-104141-5
Fisher RB, Peters DG (1970) Chemical equilibrium. Saunders College Publishing, Orlando, FL
Francis AJ, Dodge CJ (1990) Anaerobic microbial remobilization of toxic metals coprecipitated with iron oxide. Environ Sci Technol 24:373–378
Griffioen J, Heerdink R, Kuijper MJM, Roelofsen F, Rozemeijer JC, Van der Grift B (2006) Evaluation of the environmental efficacy of soil storage in depots with respect to ground water effects (in Dutch). TNO report 2006-U-R0174/A, Utrecht, The Netherlands
Guha H, Saiers JE, Brooks S, Jardine P, Jayachandran K (2001) Chromium transport, oxidation, and adsorption in manganese coated sand. J Contam Hydrol 3(4):311–334
Han FX, Banin A, Triplett GB (2001) Redistribution of heavy metals in arid-zone soils under a wetting-drying cycle soil moisture regime. Soil Sci 1:18–28
Holmes J (1999) Fate of incorporated metals during mackinawite oxidation in sea water. Appl Geochem 3:277–281
Kozuh N, Stupar J, Gorenc B (2000) Reduction and oxidation processes of chromium in soils. Environ Sci Technol 34:112–119
Lopes P (2004) Composition of porewater and benthic fluxes in the mesohaline es grau lagoon (Minorca, Spain) during spring and early summer. Wetlands 24(4):796–810
Lovley DR, Goodwin S (1988) Hydrogen concentrations as an indicator of the predominant terminal electron accepting reactions in aquatic sediments. Geochim Cosmochim Acta 52:2993–3003
Macur RE, Jackson CR, Botero LM, McDermott TR, Inskeep WP (2004) Bacterial populations associated with the oxidation and reduction of arsenic in an unsaturated soil. Environ Sci Technol 38:104–111
Mayer TD, Jarrell WM (2000) Phosphorus sorption during iron(II) oxidation in the presence of dissolved silica. Wat Res 16:3949–3956
Meeussen JCL (2003) ORCHESTRA: an object-oriented framework for implementing chemical equilibrium models. Environ Sci Technol 37:1175–1182
Morse JW, Luther GW (1999) Chemical influences on trace metal-sulfide interactions in anoxic sediments. Geochim Cosmochim Acta 19(20):3373–3378
Nguyen ML, Goh KM (1994) Sulphur cycling and its implications on sulphur fertilizer requirements of grazed grassland ecosystems. Agric Ecosyst Environ 49:173–206
Patrick WH, Verloo M (1998) Distribution of soluble heavy metals between ionic and complexed forms in a saturated sediment as affected by pH and redox conditions. Wat Sci Technol 6(7):165–172
Pierce ML, Moore CB (1982) Adsorption of arsenite and arsenate on amorphous iron hydroxide. Wat Res 16:1247–1253
Postma D, Jakobsen R (1996) Redox zonation: equilibrium constraints on the Fe(III)/SO4 − reduction interface. Geochim Cosmochim Acta 17:3169–3175
Römkens PFAM, Dolfing J (1998) Effect of Ca on the solubility and molecular size distribution of DOM and Cu binding in soil solution samples. Environ Sci Technol 32:363–369
Sauvé S, McBride M, Henderschot W (1997) Soil solution speciation of Pb2+: effects of organic matter and pH. Environ Poll 2:149–155
Schröder TJ, Hiemstra T, Vink JPM, Van der Zee SEATM (2005) Modeling of the solid-solution partitioning of heavy metals and arsenic in embanked flood plain soils of the rivers Rhine and Meuse. Environ Sci Technol 18:7176–7184
Shi B, Allen HE, Grass MT, Ma H (1998) Modelling copper partitioning in surface waters. Wat Res 12:3756–3764
Siegrist H, Brunner I, Koch G, Phan LC, Le VC (1999) Reduction of biomass decay rate under anoxic and anaerobic conditions. Wat Sci Technol 1:129–137
Simpson SL, Apte SC, Batley GE (2000) Effect of short-term resuspension events on the oxidation of cadmium, lead, and zinc sulfide phases in anoxic estuarine sediments. Environ Sci Technol 34:4533–4537
Singh SS (1984) Increase in neutral salt extractable cation exchange capacity of some acid soils as affected by CaSO4 applications. Canad J Soil Sci 64:153–161
Smith RM, Martell AE (1976) Critical stability constants IV: inorganic complexes. Plenum, New York
Snodgrass WJ (1980) Distribution and behavior of nickel in the aquatic environment. In: Nriagu JO (ed) Nickel in the environment. Wiley, New York
Strawn DG, Scheidegger AM, Sparks DL (1998) Kinetics and mechanisms of Pb(II) sorption and desorption at the aluminium oxide-water interface. Environ Sci Technol 32:2596–2601
Stumm W (1984) Interpretation and measurement of redox intensity in natural waters. Schweitz Zeitschrift für Hydrologie 46:291–296
Sutton R, Sposito G (2005) Molecular structure in soil humic substances: the new view. Environ Sci Technol 23:9009–9014
Tessier A, Campbell PGC, Bisson M (1979) Sequential extraction procedure for the speciation of particulate trace metals. Anal Chem 7:844–851
Tietz A (1957) Mechanisms of fatty acid synthesis. Biochem Biophysica Acta 25:303–310
Van Cappellen P, Viollier E, Roychoudhury R, Clark L, Ingall E, Lowe K, Dichristina T (1998) Biochemical cycles of manganese and iron at the oxic-anoxic transition of a stratified marine basin, Orca basin, Gulf of Mexico. Environ Sci Technol 32:2931–2939
Vink JPM (1999) Availability of heavy metals in the Meuse floodplain and in reducing sediments (in Dutch). RIZA report 99.014, Lelystad
Vink JPM (2002) Measurement of heavy metal speciation over redox gradients in natural water-sediment interfaces and implications for uptake by benthic organisms. Environ Sci Technol 23:5130–5138
Vink JPM, Meeussen JCL (2007) BIOCHEM-ORCHESTRA: A tool for evaluating chemical speciation and ecotoxicological impacts of heavy metals in river floodplain systems. Environ Poll 148:833–841
Vink JPM, Schröder TJ (2006) Biochem-Orchestra DSS. Biochem version 2.0. Structural and functional setup. User manual V2. RIZA, Lelystad
Vink JPM, Schraa G, Van der Zee SEATM (1999) Nutrient effects on microbial transformation of pesticides in nitrifying surface waters. Environ Toxicol 14:329–338
Wakil SJ, Porter JW, Gibson DM (1957) Mechanisms of fatty acid synthesis. Biochem Biophysica Acta 24:453–461
Warwick P, Hall A, Pashley V, VanderLee J, Maes A (1998) Zinc and cadmium mobility in sand: effects of pH, speciation, CEC, humic acid and metal ions. Chemosphere 10:2283–2290
Waterbase (1999–2008). Available at: http://www.waterbase.nl (accessed June 2009)
Xiang L, Chan LC, Wong JWC (2000) Removal of heavy metals from anaerobically digested sewage sludge by isolated indigenous iron-oxidizing bacteria. Chemosphere 1(2):283–287
Acknowledgments
Part of this study was financed by the EU/KP6 programme AquaTerra, contract GOCE-CT-2003-505428 and the Strategic Research funds of Deltares.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Ian G. Droppo
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Vink, J.P.M., Harmsen, J. & Rijnaarts, H. Delayed immobilization of heavy metals in soils and sediments under reducing and anaerobic conditions; consequences for flooding and storage. J Soils Sediments 10, 1633–1645 (2010). https://doi.org/10.1007/s11368-010-0296-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-010-0296-1