
Abstract
Warfarin is a commonly prescribed oral anticoagulant with narrow therapeutic index. It achieves anti-coagulating effects by interfering with the vitamin K cycle. Warfarin has two enantiomers, S(−) and R(+) and undergoes stereoselective metabolism, with the S(−) enantiomer being more effective. We reported the intracellular metabolic profile in HepG2 cells incubated with S(−) and R(+) warfarin by GCMS. Chemometric method PCA was applied to analyze the individual samples. A total of 80 metabolites which belong to different categories were identified. Two batches of experiments (with and without the presence of vitamin K) were designed. In samples incubated with S(−) and R(+) warfarin, glucuronic acid showed significantly decreased in cells incubated with R(+) warfarin but not in those incubated with S(−) warfarin. It may partially explain the lower bio-activity of R(+) warfarin. And arachidonic acid showed increased in cells incubated with S(−) warfarin but not in those incubated with R(+) warfarin. In addition, a number of small molecules involved in γ-glutamyl cycle displayed ratio variations. Intracellular glutathione detection further validated the results. Taken together, our findings provided molecular evidence on a comprehensive metabolic profile on warfarin-cell interaction which may shed new lights on future improvement of warfarin therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Metabolomics is the systematic study of small-molecule metabolite profiles in specific cellular processes (Daviss 2005). Small molecules, which belong to diverse categories such as organic acids, amino acids, fatty acids, carbohydrates, nucleosides and the conjugates, ketones and aldehydes, are involved in biochemical processes and provide a great deal of information on the status and functioning of a living system. They might work together with certain enzymes, or interact with other proteins or metabolites to influence cellular pathways and form metabolic networkings. Study of metabolite profile directly reflects the phenotypic alterations in response to genetic or environment changes, such as drug stimulations.
Warfarin is a commonly prescribed oral anticoagulant in patients with prosthetic valve disease, venous thromboembolism and stroke (Hirsh et al. 2001). This drug decreases blood coagulation by inhibiting vitamin K cycle, which is an essential process for the maturation of several blood coagulating factors, including prothrombin and factor VII, IX, X. In the liver, vitamin K is first reduced to its hydroquinone, which acts as a mediator to activate the enzyme γ-glutamyl carboxylase (GGCX). Several coagulating factors undergo vitamin K-dependent posttranslational modification by GGCX. Those Glutamic acid (Glu) residues on those coagulating factors are modified into γ-carboxyglutamic acid (Gla), resulting in the maturation of coagulating factors. Gla residue will mediate those coagulating factors to bind to Ca2+ in the blood stream. The binding is needed for activation of platelets as well as other downstream coagulation factors and pathways (Wu et al. 1991; Hirsh et al. 2001). Once completed the γ-carboxylation reaction, vitamin K hydroquinone is oxidized to vitamin K epoxide form. Another enzyme, vitamin K 2,3 epoxide reductase (VKOR), reduces vitamin K epoxide to its reduced form, and the cycle repeats. Warfarin inhibits vitamin K cycle through interfering with VKOR activity (Wajih et al. 2007; Tie and Stafford 2008), thus inactivate the coagulation process.
Warfarin is a racemic drug consisting of S(−) and R(+) enantiomers, with S(−) form being more effective (3–5 times) in vitro. However, this drug has a very wide therapeutic range and patients can be categorized into low-dose and high-dose groups. Many factors have been proved to affect warfarin dosage. Enantiomer drugs interaction with body has been found to response for dosage variation to some extent. Global study of cellular metabolic profile reveals the differences in biological responses between the enantiomer drugs. The findings may contribute to the improvement of warfarin therapy in terms of optimal dosage as well as giving references to new coumarin-derived drug development. To date, there are very few of studies on cellular metabolite profiles which directly reflect the biological responses from cells. Most investigations are on drug metabolism and metabolites (Wsol et al. 2001; Nassar et al. 2006; Wharfe et al. 2010). So far, a comparative study on complete metabolite profiles generated from S(−) and R(+) enantiomers of warfarin hasn’t been reported yet.
Mass spectrometry has occupied a major position in determining the metabolic status, as it provides a better way to identify metabolites effectively, both quantitative and qualitative. Gas chromatography and mass spectrometry (GC–MS) was applied in the metabolites identification in our study. This high resolution GC separation interfaces with sensitive detection of MS is able to yield both qualitative and quantitative information on metabolites (Dunn and Ellis 2005). Samples were chemical derivatized to be more volatile before analysis and spectra generated were analyzed by chemometric methods. The established metabolomic patterns can be applied to investigate on S(−) and R(+) warfarin cellular responses.
In this study, we used GC–MS to semi-quantify the differences on metabolite levels between untreated human liver cells and those incubated by S(−) and R(+) warfarin, respectively. To better reflect in vivo condition, similar analysis was also carried out in cells incubated with warfarin in the presence of vitamin K. The results have classified the generated metabolites and identified the changes, respectively. The expressions of ten molecules including glycine, 5-oxoproline in drug-incubated groups were significantly varied compared to control groups. Several molecules including arachidonic acid and glucuronic acid expressed differentially between S(−) and R(+) warfarin. In addition, results from cellular glutathione assay further supported our results.
2 Materials and methods
2.1 Cell cultures
HepG2 cells were purchased from American Type Culture Collection and cultured in minimum essential medium (MEM, supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 U/ml)). All culture media and media supplements were purchased from Life Technologies. Then cells were maintained at 37°C in an atmosphere of 5% CO2. After cell seeding and reaching 80% confluence (10 cm tissue culture dish), in the first set, the cells were incubated with either S(−) or R (+) warfarin (commercially available in Sigma–Aldrich, Product Num.:UC213 and UC 214) at a concentration of 20 μM, respectively, for 24 h in the absence of serum. For the second set, cells were supplied with physiological concentration of vitamin K (Sigma–Aldrich) (0.5 μM) for 12 h, then again 20 μM of either S(−) or R(+) enantiomer of warfarin was used to incubate the cells separately for another 24 h, in the absence of FBS. To minimize the error, before harvest, cells were washed by cold PBS for 3 times and the numbers were counted and normalized. The same amount of cells (10 million) for each sample was harvested by trypsin in 15 ml plastic tubes by low-speed centrifugation. To minimize the factors which may contribute to metabolites’ instability and to maintain the integrity of metabolite profiles, the cells were immediately stored in liquid nitrogen before lysis.
2.2 Cell lysis, metabolite extraction and derivatization
Metabolites were extracted using mono-phasic mixture of chloroform/methanol/water, according to the generic protocols described in previous articles (Atherton et al. 2006; Mainak et al. 2009), with minor modification. Basically, 1 ml of chloroform/methanol/water mixture in the ratio of 20:50:20 was added to each sample, in a chemical fume hood since chloroform is a carcinogen. Then every sample was transferred to individual 2 ml glass tubes, as chloroform may react with plastic. Next, cells with extraction reagents were undergone ultra-sonication at room temperature in a water bath sonicator for 100 min and votexed for 2 min. Subsequently, samples were transferred to 1.5 ml centrifugation tubes and centrifuged at 4°C at 18000×g for 20 min. Then for each sample, 0.8 ml supernatant was collected. The supernatant was dried completely by centrifugal evaporator (Jouan) overnight.
For GC–MS analysis, a two-stage chemical derivatization was performed on the extracted metabolites. Firstly, methoxymation was conducted to convert enol forms of aldehydes and ketones in the metabolites to oximes or alkyloximes (Halket and Zaikin 2003). The dry samples were each dissolved in 100 μl of methoxylamine hydrochloride (20 mg/ml in pyridine, Sigma–Aldrich), and votexed for 1 min, and incubated at room temperature for 1 h. Secondly, silylation was performed by MSTFA (N-methyl-N-(trimethylsilyl) trifluoroacetamide, Sigma–Aldrich) to make the metabolites less polar and more volatile. Basically, 100ul MSTFA with 1% TMCS (Trimethylchlorosilane, Sigma–Aldrich) was applied to each sample. Samples were incubated at 70°C for 30 min, and then transferred to vials for GC–MS analysis.
2.3 GC–MS analysis
The separation and detection of metabolites was performed by a Shimadzu QP2010plus GC–MS system (Shimadzu, Kyoto, Japan), with a 30 m × 0.25 mm i.d. DB5 capillary column (122-5033 Agilent Technologies), in the splitless mode. Each 1.0 μl aliquot of the derivatized sample was injected by Shimadzu AOC-20i+s auto-sampler (Shimadzu, Kyoto, Japan) and rinsed the needle with acetone each five times for both pre- and post-run to avoid sample cross-contamination. The solvent cutoff time was set to 3.5 min. Helium was used as carrier gas with the column flow 1.1 ml/min. The injection temperature was set to 280°C and ion source temperature was set to 200°C. The initial oven temperature was kept at 100°C for 4 min, increased at 4°C/min to 320°C. At 320°C, it held for 1.56 min. The mode for mass spectrometry was electron impact (EI) ionization (70 eV) and data acquisition was in the full scan mode (m/z from 35 to 600). Between each sample, acetone was run as blank with the same method to avoid cross-contamination. Before analysis, standard alkane series mixture (C-10 to C-40) was used to correct the retention time before analysis. This correction was performed by AART (automatic adjustment of Retention time) from Shimadzu GCMSsolution (version 2.5) software package.
2.4 Metabolites detection and quantification
The total ion chromatogram and deconvolution was acquired by Shimadzu GCMSsolution software package. The chromatogram of all the samples analyzed was under noise reduction and baseline correction before peak integration. Interesting peaks, with width at half-height being more than 2 s, were selected and subjected to library search. It is the standard to distinguish between noises and peaks. Other parameters, such as automatic assignments of unique fragment ions for each metabolite, and sensitivity of peak detection, were built-in functions in the software. In this study we used two libraries, National Institute of Standards and Technology (NIST) Mass Spectra Library and Shimadzu’s GC–MS Metabolite Mass Spectra Database to search for candidate compounds. Peaks with similarity index (SI) being more than 70% were selected and named for the compounds. If more than one compounds fulfilled the SI requirement, then Shimadzu’s GC–MS Metabolite Mass Spectra Database equipped with retention indices was used to give a more specific and reliable identification for candidate compounds. After data normalization, the large amount of metabolite information was analyzed with PCA by software SIMCA-P (version 12.0; Umetrics, Umeå, Sweden). This method is an unsupervised clustering method, which looks for linear combinations of variables that explain those biggest differences between individual samples. Relative quantification of selected compounds was performed by normalizing and comparing the relative chromatograph peak areas.
2.5 Intracellular glutathione (GSH) detection
Glutathione (GSH) plays a central role in protecting cells from free radical damages. It exists in two forms: reduced form GSH and oxidized form GSSG. The decrease of GSH/GSSG ratio reflects cellular ROS (Reactive Oxygen Species) increasing. To determine the intracellular GSH level and verify warfarin’s effect on γ-glutamyl cycle, ThiolTracker™ Violet dye (Invitrogen) was applied. This dye is a thiol reactive dye to label cells, and can be efficiently excited for imaging with fluorescence microscopes. Basically, 2 × 104 HepG2 cells were seeded to each well of 24-well plates. We still designed two sets of experiment (without and with vitamin K) as described above. When the cell reached 70% confluence, for the first set, 20 μM of S(−) enantiomer, R(+)enantiomer of warfarin were added, respectively, and incubated for 24 h. For the second set, each well was incubated with 0.5 μM vitamin K for 12 h, then again 20 μM of S(−) enantiomer, R(+) enantiomer of warfarin were added and incubated for another 24 h. Then media were removed and cells were rinsed twice by PBS. 0.5 ml PBS with final concentration 20 μM ThiolTracker Violet dye was applied to incubate the cells in each vial at 37°C for 30 min. Then the dye was removed and replaced by PBS. Cells were observed under fluorescent microscope (Olympus). The ThiolTracker Violet has excitation/emission maxima of approximately 404/526 nm and can be observed by standard fluorescent filter sets.
3 Results
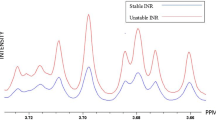
To establish a comprehensive cellular metabolite profile in response to warfarin incubation, metabolites were extracted from cells incubated with individual enantiomers of warfarin and subjected to GC–MS analysis. The experiment was performed with three independent times. Figure 1 was a representative for the overlay of chromatography from HepG2 incubated with Control and S(−) or R(+) Warfarin. A total of 80 metabolites were identified (Supplementary material provided the list of the 80 metabolites, with their names, retention times and categories). These metabolites were separated into ten categories based on their chemical compositions. These included amino acids, hydroxy acids, carbohydrates, nucleosides and nucleoside conjugates, fatty acids, alcohols, carboxylic acids, ketones, aldehydes and other organic acids which do not belong to any categories above. The results indicated that a large number of metabolites detected in both sets (with and without vitamin K) were amino acids, hydroxyl acids, carbohydrates. It was shown that amino acids eluted first, then followed by carbohydrates (mainly monosaccharides), with the fatty acids came last.

Overlay of representative GC/MS chromatography of HepG2 cells incubated by warfarin S(−) and R(+) enantiomers
For those peaks detected, the peak area was calculated and normalized using internal standard ribitol. Relative quantification assessment was performed by comparing the peak areas of individual samples. Results were summarized in Table 1 for cells incubated with the individual enantiomers of warfarin, and for cells incubated with the individual enantiomers of warfarin in the presence of vitamin K.
A total of ten metabolites showed changes in their level in Table 1. Most metabolites displayed similar changes in cells incubated with either S(−) or R(+) warfarin. However, some of them showed changes to one but not all enantiomers of warfarin.
For example, arachidonic acid, whose function is involved in coagulation process, was up-regulated for around 40% in cells incubated with S(−) warfarin but not in those incubated with R(+) warfarin, while glucuronic acid showed down-regulated in R(+) warfarin but not that much in S(−) warfarin. In addition, glycine, 5-oxoproline, l-glutamine, l-valine and l-threonine showed up-regulated in both enantiomers with similar ratio; while l-serine, l-glutamate and l-lactic acid showed down-regulated. The other set (vitamin K enhanced) showed the same results.
Subsequently, PCA results provided more information on multi-variables. Separation was according to the principle components values on untreated, S(−) warfarin and R(+) warfarin incubated samples. Each of the samples incubated by control, S(−) warfarin and R(+) warfarin in the three independent experiments was analyzed. The results could be found in Fig. 2. In the score plot (Fig. 2, Panel 1, a), different groups had been clearly separated according to the scores. The further the distance in the plot showed, the more different the samples were. It can be concluded that S(−) warfarin has greater variance than R(+) warfarin from the control group. In the loading plot (Fig. 2, Panel 1, b), the relative peak areas of individual metabolites contributed to the loading values. Those metabolites with larger variation between drug-incubated and non-incubated samples were dispensed further from the origin. In addition, the results from vitamin K enhanced set (Fig. 2, Panel 2, c and d), provided further validation.


PCA results for relative peak intensities between different samples. Panel 1 a Score plot and b loading plot for HepG2 cells incubated by warfarin enantiomers only. In the score plot, C1–C3 were cells without any treatment; S1–S3 were cells incubated by S(−) enantiomer of warfarin; R1–R3 were cells incubated by R(+) enantiomer of warfarin. In the loading plot, three PCs were selected to represent a spatial distribution of different metabolites. Panel 2, c Score plot and d loading plot for HepG2 cells incubated by warfarin enantiomers, in the presence of vitamin K. In the score plot, V1–V3 were cells incubated by vitamin K only; S1–S3 were cells incubated by vitamin K then S(−) enantiomer of warfarin; R1–R3 were cells incubated by vitamin K then R(+) enantiomer of warfarin. In the loading plot, three PCs were selected to represent a spatial distribution of different metabolites
Several amino acids, including glycine, 5-oxoproline, l-glutamate were involved in glutathione (GSH) generation. To detected the intracellular glutathione level for the validation of changes on above amino acids by S(−) and R(+) warfarin, glutathione level was measured and results were shown in Fig. 3, while a were the results for samples incubated with warfarin alone and b are the results for samples incubated with warfarin in the presence of vitamin K. In the first set, glutathione level showed increased in S(−) and R(+) warfarin incubated samples, while the level was increased more in HepG2 cells incubated by S(−) warfarin than R(+) warfarin. In the second set, the glutathione level was enhanced in the S(−) and R(+) warfarin, which was consistent with the results from 1st set. It indicated that warfarin enantiomers have effects on enhancing the intracellular glutathione levels.

Intracellular glutathione level of HepG2 cells incubated with drugs. a glutathione level of 1st set of experiment (without vitamin K pre-incubation). The left group was fluorescent view and the right group was fluorescent view with corresponding light view. b glutathione level of 2nd set of experiment (with vitamin K pre-incubation). The left group was fluorescent view and the right group was fluorescent view with corresponding light view
4 Discussion
4.1 Influence on γ-glutamyl cycle and glutathione level by warfarin
In our results, it has been shown that several amino acids varied in their expression ratio. The elevated level of glycine, l-glutamine, 5-oxoproline and decreased level of l-glutamate may be associated with warfarin induced glutathione production thus subsequently influenced γ-glutamyl cycle and hepatic γ-glutamyl transferase. An overall change in the cellular metabolic process by warfarin was depicted in Fig. 4.

Over all changes in the cellular metabolic process by warfarin. The expression of GST, which is associated with Vitamin K Epoxide Reductase (VKOR) may be affected by warfarin incubation. The expression of glutathione may also be affected by warfarin thus influence γ-glutamyl cycle with related metabolites such as glycine, 5-oxoproline and glutamate. This cycle is a transport system for amino acids. Its regulation by warfarin may lead to increase of hepatic γ-glutamyl transferase activity. In addition, warfarin metabolism is stereo-selective. Arachidonic acid and glucuronic acid are affected by S(−) and R(+) warfarin, respectively. Expression ratio elevated molecules were highlighted in red while reduced highlighted in blue. An increased GSH-to-GSSG ratio is considered indicative of reduced ROS. GGCX γ-glutamyl carboxylase, VKOR vitamin K 2,3 epoxide reductase, NQO1 NAD(P)H quinone oxidoreductase 1, GST: Glutathione-S-tranferase, AA amino acid
It has been reported that warfarin administration increased both serum and hepatic γ-glutamyltransferase activity in rats (Lake and Grasso 1996). This protein γ-glutamyltransferase is a critical enzyme on γ-glutamyl cycle, which is a transport system for amino acids and generation of glutathione (Orlowski and Meister 1970; Meister 1974).
In γ-glutamyl cycle, glutathione is broken down to glycine, cysteine and glutamate. Four enzymes catalyze the reaction. γ-glutamyl transferase transfers γ-glutamyl moiety of glutathione to one amino acid, generation γ-glutamyl amino acid and cysteinylglycine. The cysteinylglycine is split to glycine and cysteine. γ-glutamyl cyclotransferase converts γ-glutamyl amino acid to 5-oxoproline and corresponding amino acid. Thus the process has transported the amino acid transmembranely. After that, 5-oxoproline is converted to glutamate by 5-oxoprolinase. The generated glutamate is combined with cysteine and glycine coming from previous cysteinylglycine and the glutathione synthetase to re-synthesize glutathione. Then the cycle repeats.
Glutathione is an antioxidant which helps protect cells from reactive oxygen species (ROS). It exists in reduced form (GSH) and oxidized disulfide form (GSSG), and a decreased GSH-to-GSSG ratio is considered oxidative stress generated by ROS. Warfarin increases the hepatic total glutathione level that the increase is primarily due to elevated GSH levels with very little effect on GSSG level (Lake and Grasso 1996). The results may indicate a decreased level of ROS generation on warfarin administration. In our previous study on intracellular protein profiles generated by HepG2 cells incubated with warfarin enantiomers, we have reported that protein DJ-1, which is a protein marker for intracellular ROS, was down-regulated in the cells incubated by warfarin (Bai et al. 2010). This suggested a reduced level of intracellular ROS by warfarin, which was consistent with our metabolic finding herein.
Glutathione is the substrate of Glutathione-S-tranferase (GST). As it has been proposed that GST superfamily contained a groups of warfarin-sensitive GST, which was associated with Vitamin K Epoxide Reductase (VKOR) and was a part of the trans-membrane enzyme (Cain et al. 1997). The outcome is: the elevation of glutathione may also be due to the deduced VKOR complex activity by warfarin. Thus affects γ-glutamyl cycle with several metabolites and hepatic γ-glutamyl transferase.
In addition, as γ-glutamyl cycle transports amino acids transmembranely, the elevation or decrease of other intracellular l-amino acid levels, such as l-valine, l-serine or l-threonine may be attributed to the change of amino acid transportation by γ-glutamyl cycle.
4.2 Influence on uptake of vitamin K by warfarin
Another possible reason for elevated level of intracellular glycine is: one function of glycine is to combine with cholic acid and to form bile acid (Kevresan et al. 2006). The binding will enhance the hepatic uptake of fat-soluble vitamin K (Hollander and Rim 1976). In that case, increase of unbound intracellular glycine may reflect the reduced uptake of vitamin K.
4.3 Influence on glucuronidation by warfarin stereo-selectivity
The next down-regulated metabolite is glucuronic lactone. It is the stable form of glucuronic acid, as glucuronic acid normally does not exist in free acid. Glucuronidation is a process of glucuronic acid conjugated with xenobiotic metabolism of substances such as drugs, to facility the transport and elimination of those substances (Jansing et al. 1992; Ritter 2000). The conjugation involves glycosidic bonds. It mainly occurs in the liver and is a part of phase II metabolism of drugs. Until very recently, it has been reported that glucuronidation could abolish the binding between warfarin and its targeting enzyme, VKOR, due to introduction of a bulky acidic sugar (Miller et al. 2009). In our results, R(+) warfarin incubated sample showed a more decreased level of this metabolite, compared to S(−) warfarin. It consistent with the previous finding that R(+) warfarin may be more tended to be glucuronidated. The tendency of R(+) warfarin may contribute to the lower biological activity of this enantiomer.
4.4 Influence on arachidonic acid metabolism by warfarin stereo-selectivity
Another elevated metabolite is arachidonic acid. It is a polyunsaturated omega-6 fatty acid and presents in the phospholipids of cell membranes. In addition, it is also involved in cellular signaling (Baynes et al. 2005). Warfarin undergoes stereo-selective metabolism and S(−) enantiomer are mainly metabolized by Cytochrome P450 2C9 (CYP2C9) and R(+) warfarin by CYP1A2 and CYP3A4. In the previous study, it has been addressed that CYP2C9 enzymes metabolize warfarin and arachidonic acid (Kaur-Knudsen et al. 2009). Possibly, the presence of S(−) warfarin enantiomer may affect the metabolism of arachidonic acid. Arachidonic acid is the precursor for synthesis thromboxane A2, which has prothrombotic properties (Catella et al. 1986). This thromboxane A2 enhances the liver-generated fibrinogen bindings to platelets for activation (Al Mondhiry et al. 1983). Thus it does not exclude the possibility that the presence of warfarin, by regulating arachidonic metabolism, may indirectly affect the coagulating process.
5 Concluding remarks
We reported the intracellular metabolic profile in HepG2 cells incubated with S(−) and R(+) warfarin, using GC–MS. Glucuronic acid, arachidonic acid showed variations on cells incubated with S(−) warfarin and R(+) warfarin. For other metabolites like glycine, 5-oxoproline and glutamate, the expressions each were found to be the same in cells incubated with either S(−) or R(+) warfarin. Our results indicated that the expression of those metabolites may partially explain the different bio-activities between the two enantiomers and addressed that the cycle for generation of glutathione may be interfered by warfarin. Taken together, our findings provided molecular evidence on a comprehensive metabolite profile on warfarin-cell interaction which may shed new lights on future improvement of warfarin therapy.
References
Al Mondhiry, H., Ballard, J. O., et al. (1983). Fibrinogen interaction with human platelets: Effect of other coagulation factors, prostaglandins and platelet inhibitors. Thrombosis Research, 31, 415–426.
Atherton, H. J., Bailey, N. J., et al. (2006). A combined 1H-NMR spectroscopy- and mass spectrometry-based metabolomic study of the PPAR ± null mutant mouse defines profound systemic changes in metabolism linked to the metabolic syndrome. Physiological Genomics, 27, 178–186.
Bai, J., Sadrolodabaee, L., et al. (2010). A comparative proteomic analysis of HepG2 cells incubated by S(−) and R(+) enantiomers of anti-coagulating drug warfarin. Proteomics, 10, 1463–1473.
Baynes, J., et al. (2005). Medical biochemistry. Philadelphia: Elsevier.
Cain, D., Hutson, S. M., et al. (1997). Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. Journal of Biological Chemistry, 272, 29068–29075.
Catella, F., Healy, D., et al. (1986). 11-dehydrothromboxane B2: A quantitative index of thromboxane A2 formation in the human circulation. Proceedings of the National Academy of Sciences of the United States of America, 83, 5861–5865.
Daviss, B. (2005). Growing pains for metabolomics. Scientist, 19, 25–28.
Dunn, W. B., & Ellis, D. I. (2005). Metabolomics: Current analytical platforms and methodologies. TrAC Trends in Analytical Chemistry, 24, 285–294.
Halket, J. M., & Zaikin, V. G. (2003). Derivatization in mass spectrometry—1. Silylation. European Journal of Mass Spectrometry, 9, 1–21.
Hirsh, J., Dalen, J. E., et al. (2001). Oral anticoagulants: Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest, 119, 8S–21S.
Hollander, D., & Rim, E. (1976). Vitamin K2 absorption by rat everted small intestinal sacs. American Journal of Physiology, 231, 415–419.
Jansing, R. L., Chao, E. S., et al. (1992). Phase II metabolism of warfarin in primary culture of adult rat hepatocytes. Molecular Pharmacology, 41, 209–215.
Kaur-Knudsen, D., Bojesen, S. E., et al. (2009). Common polymorphisms in CYP2C9, subclinical atherosclerosis and risk of ischemic vascular disease in 52000 individuals. Pharmacogenomics Journal, 9, 327–332.
Kevresan, S., Kuhajda, K., et al. (2006). Biosynthesis of bile acids in mammalian liver. European Journal of Drug Metabolism and Pharmacokinetics, 31, 145–156.
Lake, B. G., & Grasso, P. (1996). Comparison of the hepatotoxicity of coumarin in the rat, mouse, and Syrian hamster: A dose and time response study. Fundamental and Applied Toxicology, 34, 105–117.
Mainak, M., Koon, K. P., et al. (2009). Development and validation of a gas chromatography/mass spectrometry method for the metabolic profiling of human colon tissue. Rapid Communications in Mass Spectrometry, 23, 487–494.
Meister, A. (1974). The gamma glutamyl cycle. Diseases associated with specific enzyme deficiencies. Annals of Internal Medicine, 81, 247–253.
Miller, G. P., Jones, D. R., et al. (2009). Assessing cytochrome P450 and UDP-glucuronosyltransferase contributions to warfarin metabolism in humans. Chemical Research in Toxicology, 22, 1239–1245.
Nassar, A., Talaat, R. E., et al. (2006). The impact of recent innovations in the use of liquid chromatography-mass spectrometry in support of drug metabolism studies: Are we all the way there yet? Current Opinion in Drug Discovery & Development, 9, 61–74.
Orlowski, M., & Meister, A. (1970). The gamma-glutamyl cycle: A possible transport system for amino acids. Proceedings of the National Academy of Sciences of the United States of America, 67, 1248–1255.
Ritter, J. K. (2000). Roles of glucuronidation and UDP-glucuronosyltransferases in xenobiotic bioactivation reactions. Chemico-Biological Interactions, 129, 171–193.
Tie, J. K., & Stafford, D. W. (2008). Structure and function of vitamin K epoxide reductase. Vitamin K (Vol. 78, pp. 103–130). San Diego: Elsevier Academic Press Inc.
Wajih, N., Hutson, S. M., et al. (2007). Disulfide-dependent protein folding is linked to operation of the vitamin K cycle in the endoplasmic reticulum—a protein disulfide isomerase-VKORC1 redox enzyme complex appears to be responsible for vitamin K-1 2,3-epoxide reduction. Journal of Biological Chemistry, 282, 2626–2635.
Wharfe, E. S., Winder, C. L., et al. (2010). Monitoring the effects of chiral pharmaceuticals on aquatic microorganisms by metabolic fingerprinting. Applied and Environmental Microbiology, 76, 2075–2085.
Wsol, V., Kral, R., et al. (2001). Stereospecificity and stereoselectivity of flobufen metabolic profile in mate rats in vitro and in vivo: Phase I of biotransformation. Chirality, 13, 754–759.
Wu, S. M., Cheung, W. F., et al. (1991). Cloning and expression of the cDNA for human gamma-glutamyl carboxylase. Science, 254, 1634–1636.
Acknowledgments
This work was supported by Grant NMRC/EDG/0033/2008 (WN Chen). J Bai, MX Wang are recipients of graduate research scholarship from Nanyang Technological University, Singapore.
Conflict of interest
None.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bai, J., Wang, M.X., Chowbay, B. et al. Metabolic profiling of HepG2 cells incubated with S(−) and R(+) enantiomers of anti-coagulating drug warfarin. Metabolomics 7, 353–362 (2011). https://doi.org/10.1007/s11306-010-0262-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-010-0262-3