
Abstract
There is current debate on whether genetically-manipulated plants might contain unexpected, potentially undesirable, changes in overall metabolite composition relative to that of the progenitor genotype. However, appropriate analytical technology and acceptable metrics of compositional similarity require development, particularly to allow data integration from different laboratories and different harvests. For an initial comprehensive overview of compositional similarity, we explored the use of a rapid and relatively non-selective fingerprinting technique based on flow injection electrospray ionisation mass spectrometry (FIE-MS). Six conventionally-bred potato cultivars and six experimental bioengineered potato genotypes were produced in four field blocks during two growing seasons and analysed on two different analytical instruments (LCT, Micromass in 2001 and LTQ, Thermo Finnigan in 2003). Field effects and overall process variability was found to be negligible when compared to inherited genotype variance. The data derived separately for experiments using tubers from individual harvest years were compared to assess the generalisability of models for the comparison of GM and non-GM potato tubers under investigation. This procedure proved appropriate for not only rapid assessment of similarities between plant genotypes but also to predict the identity of metabolite signals that could explain differences between genotype classes irrespective of the instrument used for analysis. Importantly, despite differences in ionisation and data acquisition properties of the two instruments the generalisation of models could be confirmed after correlation analysis of explanatory variables correctly identified the molecular origin of differences between genotypes. We conclude that FIE-MS metabolomics fingerprinting technology coupled to machine learning data analysis has great potential as a robust tool for first-pass metabolic phenotyping and, therefore, initial assessments of compositional similarities prior to use of more targeted hyphenated gas or liquid chromatography-mass spectrometry techniques.
Similar content being viewed by others

Introduction
Genetic engineering of crop plants has raised concerns that the process of gene transfer itself or unexpected regulatory or enzymatic activities of a transgene product in a new genetic background might cause crop plants to be substantially different from traditionally bred varieties, for example by the production of potentially harmful metabolites (OECD, 2001; Kok and Kuiper, 2003). As we enter the post-genomic era, ‘genome-wide’ expression profiling methods at the level of the transcriptome, the proteome and the metabolome have come to the fore, such that it is clear that for the first time we may seek to make comprehensive measurements of the working parts of biological systems at these different levels of organisation (Bino et al., 2004). We know from the theory underlying Metabolic Control Analysis (MCA), as well as from experimentation, that while changes in the levels of individual enzymes may be expected to have little effect on metabolic fluxes, they can and do have significant effects on the concentrations of a variety of individual metabolites (Fell, 1992, 1998, 2005). Thus, a global analysis at the level of the metabolome represents the optimal methodology for assessing the compositional consequences of genetic modification, whether through transgenesis, conventional breeding, irradiation or mutagenesis. This proposition is based on a number of factors:
-
as the ‘downstream’ result of gene expression, changes in the metabolome are amplified relative to changes in the transcriptome and the proteome;
-
metabolomics technology does not demand that a whole genome sequence or large EST databases be available for each crop species;
-
metabolite profiling is often cheaper and much more high throughput than proteomics and transcriptomics, making it feasible to examine large numbers of samples from plants grown under a wide range of conditions. This potential increase in sample size affords an attendant increase in confidence with regard to any conclusions reached in relation to ‘substantial equivalence’;
-
the technology is generic as a given metabolite – unlike a transcript or protein – is the same in every organism.
If GM plants are to be deemed ‘substantially equivalent’ (OECD, 2001; Kuiper et al., 2002; Kok and Kuiper, 2003;) to non-GM progenitor genotypes in their metabolism, then GM and non-GM cultivar metabolic profiles must be experimentally determined and compared. Looking for unanticipated changes in plant composition requires a non-biased assessment of ‘global’ metabolite content, providing a considerable challenge for metabolomics technology (Fiehn et al., 2000; Fiehn, 2002; Kuiper et al., 2002, 2003; Sumner et al., 2003; Bino et al., 2004; Schauer et al., 2006; Shepherd et al., 2006). Established methods for metabolite quantification include gas or high-pressure-liquid chromatography (GC and HPLC) or capillary electrophoresis (CE), usually linked to mass spectrometers (Dunn et al., 2005). Such ‘hyphenated’ mass spectrometry (MS) approaches result in detailed knowledge relating to a wide variety of individual compounds, but each only on a sub-set of metabolites. For example, by GC-time-of-flight mass spectrometry (GC-TOF-MS), the relative levels of many hundreds of compounds can be determined but it has a cut-off threshold for thermolabile or large compounds exceeding about 500 Da and has a cycle time of about 45 min/sample for data acquisition and processing. The generation of good quality GC- or LC-MS chromatograms requires highly experienced research personnel especially for data pre-processing and peak table curation. It is common for more than 50% of resolved peaks in such chromatograms to represent structurally uncharacterised chemistry (e.g. Catchpole et al., 2005). Thus, in addition to issues of equipment resolution and sensitivity, ‘hyphenated’ MS data from different instruments are difficult to align due to differences in both data pre-processing software (peak finding, spectrum deconvolution, peak annotation) and operator preferences (Buchholz et al., 2002; Jonsson et al., 2004; Hagan et al., 2007). Against this background there is a requirement not only for analytical methods that can provide a sensitive assessment of ‘global’ chemical composition but also a need for approaches that can generate data which can be aligned when produced on different instruments or in different laboratories (Kopka, 2006).
As it is clear that no single metabolite profiling methodology can be totally comprehensive (Kopka et al., 2004) we have proposed previously that a hierarchical approach be utilised in which metabolite content is examined preliminarily using a chemical ‘fingerprinting’ technique to assess overall compositional similarities between sample classes (Catchpole et al., 2005). A deeper analysis can subsequently be undertaken incorporating a chromatography step in instances where significant differences in metabolome status are predicted. Metabolome fingerprinting techniques that do not incorporate a chromatographic step have a much shorter analysis cycle time and allow for a rapid pre-screen of a large number of samples which improves confidence in data model statistics. With many unknown metabolites found in crude extracts of plants and food raw materials, alignment and interpretation of data produced on different instruments should theoretically be much more straightforward using metabolome fingerprinting techniques. Spectroscopic and spectrometric techniques, such as Fourier transform infra red (FT-IR) and nuclear magnetic resonance (NMR) generate global chemical ‘fingerprints’, however, they generally require a further level of directed analysis to link any differences in wavenumber (FT-IR) or chemical shifts (NMR) to specific chemistry (Reo, 2002; Defernez and Colquhoun, 2003; Viant, 2003; Harrigan et al., 2004). In contrast, ‘fingerprinting’ techniques based on mass spectrometry (MS) offer the advantage that the measured ‘variables’ (mass to charge ratios, m/z) can more directly be linked to an individual metabolite by the additional information of atomic mass with the caveat that in some cases even accurate m/z is not sufficient to link to “individual” metabolites but only to “groups” of metabolites. Procedures utilising flow injection electrospray ionisation mass spectrometry (FIE-MS) have recently been used with success for genotyping using both plant and microbial samples (Goodacre et al., 2002; Allen et al., 2003; Hansen and Smedsgaard, 2004; Johnson et al., 2004; Catchpole et al., 2005). Such fingerprints can be regarded as simplified images of total sample composition in that the measured variables (m/z) are compiled by integrating the levels of more than one metabolite with a similar nominal mass.
Metabolite profiling of plants have been limited generally to ‘batch’ experiments performed on a single instrument over a relatively short period of time using plants grown under controlled conditions (e.g. Fiehn et al., 2000; Roessner et al., 2000). To be of use in plant breeding programmes and particularly to contribute to substantial equivalence assessment, it is essential that an experimental strategy be developed that is able to cope with the expected degree of biological and instrument variance that would be present in large scale trials performed over several growing seasons. We concentrated our effort on field-grown potatoes with 12 different genotypes cultivated in four separate blocks in field sites in Germany during two growing seasons (2001 and 2003). Two classes of experimental transgenic lines (three independent lines of each) developed in the cultivar Désirée were used to provide related examples of directed genotypic alterations in the same biochemical pathway (Hellwege et al., 2000). The first transgene coding for the enzyme sucrose:sucrose transferase (SST) produces the tri-saccharide kestose from sucrose as well as oligo-fructans up to 5 degrees of polymerisation (DP) (Hellwege et al., 1997). The second transgene was fructosyl:fructose transferase (FFT) which utilises kestose (and other larger oligo-fructans) to build insoluble, long chain inulin carbohydrate polymers. All transgenic lines were morphologically indistinguishable from the progenitor cultivar Désirée, even under field conditions. Further representation of the potato gene pool was provided by samples of four other conventional cultivars (Agria, Granola, Linda and Solara). Additionally, a separately propagated source of the Désirée cultivar (De2) was included in the analysis to provide an appropriate reference for data modelling in which metabolome differences are acknowledged to be insignificant.
When undertaking a compositional interpretation of FIE-MS fingerprints, the approaches to data analysis should reflect the types of problem under consideration. In general, both fundamental and applied research share common goals in relation to data modelling that may be reduced down to several basic questions:
-
How similar is one set of samples to another?
-
Does a sample belong to a previously described class?
-
Which variables (metabolite signals) are responsible for sample discrimination or sample clustering?
However, answering these questions can become increasingly complicated in a metabolomics context. In addition to inherent high levels of variance in metabolome data, data mining algorithms have to cope with the fact that ‘omics’ data sets often suffer from the twin problems of data dimensionality (too many variables) and data set sparcity (too few samples) (Goodacre, 2005; Broadhurst and Kell, 2006). Consequently, specific and robust strategies that go beyond traditional univariate techniques must be implemented to reduce the possibilities of deriving spurious conclusions due to the ‘curse’ of dimensionality and considerable levels of irrelevant environmental and experimental variance. Similarity is a qualitative human concept and thus it has no precise mathematical definition. Measuring ‘similarity’ relies on determining quantitative indices devised in the context of the study so that they should fulfil both mathematical coherence and biological relevance (see linked note by Enot and Draper for a detailed discussion). From a substantial equivalence point of view, it is easier and more sensible to test for similarity by understanding dissimilarities in the data rather than looking for regular patterns that do not change (Singh, 2003). In our experience (Catchpole et al., 2005; Enot et al., 2006b), supervised machine learning techniques in which data labels are used in the modelling process provide a better assessment of the degree of similarity or differences between classes of samples: the ability to discriminate classes is clearly linked to the underlying similarity between classes. However, with data of such high dimensionality supervised techniques may easily produce overoptimistic models using spurious patterns and variance unrelated to the problem (confounding effects) even if careful quality control practices have been implemented (Broadhurst and Kell, 2006). For this reason, understanding the signal behaviour responsible for class discrimination or sample clustering is as important as any assessment of the overall model robustness. Generalisability of an experiment and the biological significance of the results can only be guaranteed if the relationship between explanatory variables is investigated. In that respect, suitable modelling techniques must not only provide adequate predictive abilities but also valuable insights into the structure of the data (Breiman, 2001; Broadhurst and Kell, 2006). Many multivariate techniques may fulfil the first goal but direct interpretation of the underlying mathematical model cannot be assured unless further parsimonious models are formed to explicitly describe relationships between features (Goodacre, 2005; Jarvis and Goodacre, 2005). In contrast with many data mining strategies, the decision tree based approach Random Forest (RF) is a “two-eyed” machine learning algorithm according to Leo Breiman (Breiman, 2001, 2003) as it combines competitive predictive abilities with features useful to scientists for interpreting their results (Izmirlian, 2004; Enot et al., 2006a, 2006b).
By using plant material derived from eight separate field blocks, harvested in two separate growing seasons and analysed on two different mass spectrometers (ToF-MS: LCT, Micromass and linear ion trap, LTQ, Thermo Finnigan), a realistic assessment of FIE-MS fingerprinting technology for high throughput screening of GM genotypes can be achieved. Central to this strategy is the refinement of data analysis procedures that not only identify specific discriminatory metabolites and produce useful quantitative metrics for comparison, but which are also capable of dealing effectively with high levels of data variance of metabolomics data (Kell et al., 2001; Goodacre et al., 2004). We show that FIE-MS metabolomics fingerprinting technology coupled to powerful data analysis resources is appropriate for rapid assessment of similarities between plant genotypes. The methodology described here provides a rational approach for the comparison and rapid interpretation of FIE-MS fingerprints in order to predict the identity of metabolite signals explanatory of differences between genotype classes, irrespective of the instrument used for analysis.
Materials and methods
Biological samples
Three single transgenic (SST: S18, S20, S36) and three double transgenic (SST/FFT: SF19, SF30, SF34) lines were derived from the progenitor cultivar Désirée (Hellwege et al., 2000). Potato plants were grown under field conditions in a block design for the 2001 and 2003 growing season together with the conventional cultivars Agria, Linda, Granola, Solara and two Désirée lines (one propagated through tissue culture, the other Désirée line obtained from tuber propagation, respectively De2 and De1). The field trials were carried out by the BBA (Biologische Bundesanstalt für Land- und Forstwirtschaft) in Dahnsdorf, Germany. At harvest, approximately 48 (2001) or 40 (2003) tubers were selected at random for analysis from each of four randomly arranged field blocks. Two replicate slices of central medulla tissue (1 cm diameter, 3 mm thickness) were isolated from each tuber, snap-frozen using liquid nitrogen and stored at −80 °C in 2 × 24 well plates prior to extraction.
Tissue extraction and storage of extracts
Tuber slice homogenization was carried out in 2 ml Eppendorf tubes containing a 5 mm diameter stainless steel ball using a mixer mill (type MM200, Retsch, Germany) concomitant with metabolite extraction using 1 ml of a pre-chilled mixture of water:methanol:chloroform (2:5:2, v/v/v). Without phase fractionation, samples were shaken for 5 min at 4 °C before centrifugation to remove cell debris and storage at minus 80 °C.
Flow injection FIE-MS analysis
Potato extracts from both harvests were analysed in a randomised, but structured, run order to ensure that the full representative set of field block (×4) and genotype (×12) sample combinations were analysed each day. One set of replicates consisted of 48 biological replicates and was analysed alongside two control samples containing the extraction solvent only. A total of 12 sets of replicates of the 2001 harvest had been analysed previously in positive and negative ionisation mode (Catchpole et al., 2005) on six separate days over 4 months in batches of 96 extracts. FIE-MS was performed using an LCT mass spectrometer (Micromass Ltd., Manchester, UK). Extracts were diluted 1:50 in water/methanol (60:40 v/v) and 150 μL were dispensed in autosampler vials with polypropylene inserts. Aliquots of 40 μL were injected into a flow of 100 μL·min−1 water/methanol (60:40 v/v) using a Waters Alliance 2690 liquid chromatography (LC) system. The flow was split before the ion source to maintain a flow of 50 μL × min−1. Data were collected in positive mode and negative mode every second (0.9 s scan time, 0.1 s interscan delay) for 2 min per sample from m/z 65 to 1000. Ionisation conditions were set to 3000 V capillary voltage, 80 °C source temperature, 120 °C desolvation temperature, 100 V RF lens, 30 V sample cone voltage and 10 V extraction cone voltage.
A total of 10 sets of replicates of potato extracts from the 2003 harvest were analysed in batches of 48 extracts (=1 set of replicates) within 2 weeks. FIE-MS in positive and negative ionisation mode was performed on a LTQ linear ion-trap mass spectrometer (Thermo Finnigan). Aliquots of 20 μL crude solvent extract were injected into a flow of 60 μL·min−1 water/methanol (50:50 v/v) using a Surveyor (Thermo Finnigan) liquid chromatography (LC) system. Instrumental conditions are as follows: sheath gas (nitrogen) pressure: 40 arbitrary units, Auxiliary gas (nitrogen) pressure 5 arbitrary units, helium as collision gas (incoming pressure: 40 psi), 4.5 kV (ESI+) and 4.0 kV (ESI-) spray voltage and +15 V (ESI+) and −13 V (ESI-) capillary voltage, temperature of the heated transfer capillary set to 380 °C. Data were collected in positive mode and negative mode for 5 min per sample from m/z 50 to 2000. The raw ion intensity data were binned to nominal mass as preliminary studies indicated that binning to 0.1 u did not improve data quality (data not shown).
LC-MS profiling
The LC-MS system consisted of a Surveyor HPLC comprising autosampler and quaternary pump with integrated degasser and an LTQ linear ion trap mass spectrometer (ThermoFinnigan, San Jose, CA) running Xcalibur software (version 1.4 SR1, ThermoFinnigan). Nitrogen was used as sheath (set: 40) and auxiliary gas (set: 5) and helium as collision gas (incoming pressure: 40 psi). Potato tuber sample extracts (10 μL injection volume) were analysed by hydrophilic interaction chromatography (HILIC) on TSK Gel Amide 80 (250 mm × 2.0 mm, 5 μm particle size, TosoHaas, Montgomeryville, PA) using solvents (A) Acetonitrile and (B) 6.5 mM ammonium acetate (adjusted to pH 5.5 with acetic acid) at ambient temperature. The elution profile was as follows: at 150 μL /min flow rate 100% A for 5 min isocratic, a gradient to 20% B was concluded at 10 min, followed by a gradient finished by 60% B at 45 min, and than back to 100% B within 1 min and subsequently isocratic for 15 min. Spray voltage was set to 4.5 kV and transfer capillary maintained at 380 °C.
Data analysis
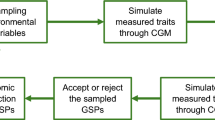
The data analysis strategy designed for the study of genetically modified organisms by FIE-MS is described in more detail in the linked note by Enot and Draper. Briefly, the overall workflow can be divided in three main steps: evaluation of main sources of variability in the experiments; development of similarity metrics using supervised machine learning techniques and inspection of the relationships between discriminatory signals.
Data were log transformed and normalised to Total Ion Count before in-depth statistical analysis. Prior to any treatment, each dataset was partitioned into a training set and a test set. FIE-MS data were collected in analytical batches, each of which contained a representative selection of samples with coverage of all genotypes. It was thus possible to use specific independent data batches to form the training and test sets. The ratio of the number of training and test samples per class for each matrix was as follows: 32/16 (Potato 2001), 24/16 (Potato 2003). Training data were used to build the models, to calculate bootstrap statistical measures, to generate variable ranking lists and to produce correlation plots, whereas test data were only employed to validate model predictive abilities. All calculations were carried out in the R environment on a PowerPC G5 (dual 1.8 GHz, 2GB SDRAM):
-
Principal components analysis was carried out with the prcomp R function on the mean centered matrix and univariate analysis of the variance with the aov R function. ANOVA post-hoc tests were performed using Tukey’s Honest Significant Differences method (function TukeyHSD) (Zar, 1984). Multivariate analysis of the variance was performed with the ffmanova R package (Langsrud, 2002).
-
Linear Discriminant Analysis was implemented in R following the procedure described in (Thomaz et al., 2004). Initial PCA was carried out on the correlation matrix using the R function prcomp.
-
Random Forest (RF) models were computed with the additional R package randomForest (4.5–16). For each model 2000 trees were used. In RF prediction of new samples is performed by determining the winner class from the votes on the overall ensemble of models. Therefore, confidence in attributing a sample to a designated class can be deduced from the difference between the score (averaged number of votes) for the true class and the largest score of the rest of the classes. This is defined as the sample margin and measures the extent to which the average number of votes for the right class exceeds the average vote for any other class (i.e. the most probable misclassification). The larger the margin is, the higher is the confidence that an example belongs to the actual class.
-
Model validation was carried out initially by permutation analysis to obtain an estimate of the reliability of both RF margin and LDA eigenvalue using 2000 permutations of the class label (Good, 2000). ‘Leave-one-factor-out’ cross validation (CV) accuracies were calculated as follows: a Random Forest (RF) model is constructed using all training samples from 3 or 6 (block/batch) factor levels and then used to evaluate the predictions of the samples from the remaining level. A random partitioning was performed 50 times with identical data stratification to test the null hypothesis that the batch/block based CV accuracies is lower than the average performance accuracy obtained by random CV approximating that CV accuracies comes from a normal distribution (Dietterich, 1998). The R-package ROCR (1.0–1) was used to perform receiver operating characteristic (ROC) analysis (Sing et al., 2005). Receiver operating characteristic (ROC) curves can be used as an alternative measure of the predictive abilities of any binary classifier (Sing et al., 2005). ROC curves display the relationship between sensitivity (true-positive rate) and specificity (false-positive rate) across all possible threshold values that define the decision boundary. The most common way to summarise the ROC curve is to compute the area under the curve (AUC). As a single value measure, the AUC specifies the probability that the decision boundary assigns a higher value to a positive sample than a negative one, both chosen randomly. Bootstrap is also a data ‘resampling’ method used for estimating generalization error and is used widely to assess statistical accuracy in data mining; however the bootstrap accuracy does not always provide a good estimate of model generalisibility. In the simplest form of bootstrapping, instead of repeatedly analysing subsets of the data, you repeatedly analyse subsamples of the data. For estimating generalization error in classification problems, the .632+ bootstrap is one of the currently favoured methods that has the advantage of performing well even when there is severe overfitting (Efron and Tibshirani, 1997). A number of 30 bootstraps were employed to compute the B632+ accuracy and area under curve on the training data; note that identical partitioning was executed to allow direct comparisons between RF and LDA statistics.
-
A ranked list of explanatory variables was obtained from the RF importance score defined as the mean decrease of accuracy over all classes. Additional significance testing by permutation was performed with 2000 repetitions. Selected variables entered the hierarchical agglomerative clustering. Dissimilarity measure is defined as “one minus the absolute value of the Pearson correlation coefficient between two signals” (R-function cor). Complete agglomerative clustering is performed with the R-function hclust.
Results and discussion
Experimental design considerations
Metabolome fingerprinting by FIE-MS attempts to detect and measure total ionisable chemistry by quantifying signals at all m/z within the mass range and resolution of any particular instrument. Increases in mass resolution are concomitant with a proportional increase in data dimensionality which in turn effects experimental design with regards to the numbers of replicates required to achieve statistical robustness. In m/z fingerprinting it is commonplace to integrate all signals to the nearest nominal mass to constrain data dimensionality (Hansen and Smedsgaard, 2004). This strategy also avoids any problems with false positive or false negative signals that can result from drifts in instrument mass accuracy over time. From a pragmatic perspective nominal mass FIE-MS fingerprinting also has the advantage that any instrument in any laboratory should be able to replicate any measurements thus possibly providing extend scope for future data integration.
Although nominal mass binning can help to avoid problems associated with missing values in the data matrix, other factors can be sources of unwanted regular variance in FIE-MS data which confound multivariate analysis. Instrument ion source characteristics (both source contamination over time and molecular fragmentation/charge features) and detector sensitivity over the total mass range can differ, both between instruments and during the course of a long series of injections. Such factors can cause regular variance in signal intensity (instrument ‘drift’), which is particularly noticeable in large scale or long-term experiments. Thus to be used effectively it is important to ensure that sources of unwanted variability are either avoided or compensated in any fingerprinting strategy.
Characteristics of FIE-MS fingerprint data in large scale experiments
Instrument ‘drift’ is often a significant problem in mass spectrometry experiments generating complex metabolite ‘fingerprints’ of crude tissue extracts, as data cannot easily be calibrated against purified standard chemicals. An assessment was therefore made of the signal variance in FIE-MS fingerprints comparing traditional cultivars with two classes of genetically modified potato genotypes (Roessner et al., 2001; Catchpole et al., 2005; Kopka et al. 2005; Shepherd et al., 2006). FIE-MS data was generated in six injection batches over a 4-month period (600 injections = 576 samples plus controls) to check that instrument drift was not a major concern. Nominal mass FIE-MS fingerprinting was performed using two different instruments (Thermo LTQ linear ion trap and Micromass LCT). The latter instrument has been used in an effective mass range of up to m/z 1000 whereas the linear ion trap proved to be accurate for fructans of up to m/z 2000. For comparability reasons, only the mass range in common between the two platforms (m/z = 110–997) was used for data modelling. The typical LTQ-FIE-MS fingerprint (+ve ion mode) illustrated in figure 1 shows that the majority of strong signals in the potato tuber extracts were below m/z 1000. Signal strengths at selected m/z-values are illustrated in the inset of figure 1. Although considerable variation in signal intensities is apparent, general intensity differences between different sample meta-classes (i.e. non-GM cultivars < SST and SST/FFT GM lines) are evident and signals potentially relating to molecular isotopes were found in expected ratios. This large number of samples derived from independent field sites over a 2-year period provided plenty of scope to examine the effect of environmental variance and analytical variance on large-scale metabolomics experiments. Initially, a multivariate analysis of the variance adapted from Langsrud, 2002, was conducted on a subset of the original data to determine the impact of two known experimental factors (field block and analytical batch) using the two near isogenic lines Désirée De1 and De2. Field block is the most significant experimental factor reported (p < 10−7). However, this effect is of the same magnitude as for the comparison of near isogenic lines effect one (p < 10−6), illustrating that both, field block and analytical batch do not seem to have any noticeable impact. In contrast, genotype differences are clearly dominating the experiment. Typically, one-way ANOVA conducted on each PCA dimension (PCs) to test genotype differences clearly showed highly significant effects on the first 20 principal components (F(11,564)>10, p< < 10−10). Post-hoc comparisons were performed on each PC to identify the origins of the significant ANOVA F using Tukey’s ‘Honest Significant Differences’ method. Effects observed on PC1 and, to a lesser extent PC5, are linked to the genetic modification whereas the remaining PCs are related to features associated with cultivar compositional differences (Catchpole et al., 2005). In addition to the relative estimation of the influence of the experimental factors, the impact of both block and batch effects on the predictive abilities of models discriminating GM to non-GM lines was assessed by means of ‘leave-one-factor-out’ cross validation (CV). This approach is a special case of classical K-fold cross validation where samples constituting a fold also share an identical factor level (for e.g. same batch or field block). The resulting accuracy is then tested against the classifier performance computed with a similar but random partitioning. Only a significant loss of accuracy while discriminating De1 and De2 between the block based CV (accuracy = 57%) and random 4-fold CV (accuracy: mean=72 ± 3%) is detected at p < 10−5. For the comparisons involving the other genotypes, block and batch constrained CV fell neatly into the limits of their corresponding random process. It can be concluded from this set of analyses that field effect and overall process variability is negligible compared to between genotype differences.

Characteristics of FIE-MS data. Representative LTQ FIE-MS +ve spectrum of a double transgenic potato tuber sample. Inset: nominal ion intensities of masses characteristic for a 4DP-fructan (4 degrees of polymerization) shown for SST/FFT double transgenic lines, SST single transgenic lines and all other cultivars.
Evaluating phenotypic distances in potato genotypes
Previous publications (Somorjai et al., 2003; Wu et al., 2003; Catchpole et al., 2005; Broadhurst and Kell, 2006; Enot et al., 2006b) have stressed the importance of performing several data analysis approaches to minimise false discoveries in metabolome modelling. In this respect, our strategy relies on a succession of data mining approaches to assess the similarly between FIE-MS fingerprints of each potato genotype. Initially, Linear Discriminant Analysis (LDA) performed on the training data using all 12 genotypes provides a useful ‘first pass check’ to confirm that the potato genotype meta-classes could be adequately discriminated in tubers produced in two separate growth seasons (figure 2). Accuracies on an independent test set were respectively 77 and 73% for the 2001 and 2003 harvest models with over 90% of miss-classification reported only within the GM meta-classes SST and SST/FFT as well as between the Désirée lines (Catchpole et al., 2005). In a second stage, pair-wise comparisons of individual classes were conducted to delineate the more understandable assessment of similarities between the GM potato lines and the progenitor cultivar (Désirée). Table 1 gathers quantitative measures of phenotypic ‘distances’ and predictive abilities of several modelling techniques, which were provided by calculation of classification accuracy (bootstrap error calculated on the training data) and area under curve (AUC), together with two model estimates provided as the margin in RF and eigenvalue in LDA. Due to the popularity of PCA in the metabolomics community, ANOVA statistics (F and corresponding p-value) calculated on the first PC are reported for comparison. In all binary comparisons GM potato classes can be discriminated from the progenitor with good accuracy and AUC values approaching 1 in both harvest years. Additional similarity measures provide a more sensitive estimation of group differences than accuracy or AUC that have reached their maximal possible value. As illustrated in Table 1, production of additional novel fructans by the double transgenic lines is translated into higher RF model margins and greater LDA eigenvalues. This is corroborated by the analysis of the variance on the first principal component where the effect of the genetic modification is significant for SST lines (p < 10−10) and even more for the SST/FFT lines (p < 10−20).

Projection of the training samples on the first two discriminant functions for the 2001 (left) and 2003 (right) FIE-MS +ve datasets using linear discriminant analysis (LDA).
To determine the significance of such results, model properties must be examined using a null hypothesis stating that the statistical measure under study is not relevant to the biological problem. As a result, significance is usually presented by a p-value and/or interval of confidence to accept or reject the model. An alternative approach exploits characteristics of models that do not contain much relevant information such as one involving two near isogenic lines of Désirée (De1 and De2) where expected (non significant) differences should be related to tissue culture propagation or experimental variance. Identical simulations as for the progenitor-GM line comparisons are reported in Table 1. Model margins and LDA eigenvalues in the De1–De2 comparisons are in the same order as the significance level p < 0.001 determined by permutation testing (c.a. 0.15 and 1.5 for RF margin and LDA eigenvalue respectively) and the AUC is consistent with a generally accepted 0.8 threshold for model significance in the literature (Broadhurst and Kell, 2006; Enot et al., 2006b). By contrast, statistical measures derived from the progenitor-GM models are much larger, implying that potentially strong genuine compositional differences exist between Désirée 1 and the genetically modified lines.
Identification of explanatory variables that reflect compositional differences
While estimating the probability that any model based on FIE-MS data could be useful, it is crucial for the concept of substantial equivalence to identify individual or subsets of features responsible for sample discrimination. In the present case, transgenic potatoes have been engineered to express novel enzymes associated with fructan metabolism and thus it is expected that phenotypic differences with regard to progenitor genotype will be found in relation to fructan metabolism. An illustration of the output from RF analysis is given in figure 3. The ranking of ions in LCT data for discrimination between conventional cultivars, to separate cultivars from both classes of transgenic potatoes or to distinguish both transgenic classes from each other, is aligned to the rank order of ions discriminating all potato genotypes. A distinct hierarchy can be seen in the rank of important ions. Six ions used to discriminate GM lines and conventional cultivars were in the top ten of the ranked list of variables used to classify all genotypes. The strongest features used to classify all genotypes are related to those m/z signals that distinguish GM lines from cultivars, confirming PCA observations where the genetic modification is the dominant factor on the first principal component. There was hardly any overlap between ions ranked within the top ∼40 for discrimination between cultivars and the ∼20 top-ranked variables discriminating the GM-lines from conventional cultivars which were all predicted to represent fructan molecules (see Catchpole et al., 2005 for details of signal annotations).

Ranking of all discriminatory variables (m/z signals) according to the Random Forest Importance Score in models derived from the LCT-FIE-MS analysis of 2001 field harvest using all genotypes (12 lines), all cultivars (6 lines), cultivars vs. GM lines (2 classes) and all STT vs. all SST/FFT (2 classes). The number in boxes indicates the rank of the variable in each model. The far right column lists the predicted metabolites (×DP: number of degrees of polymerization, fructans).
Model generalisability and interpretation
In most published metabolomics studies, experiments can be considered as “static” as they usually deal with a given problem, with a set of given treatments at a given time. However, the relevance of a model and its derived knowledge can only be validated and shown to be generalisable if the same results are obtained across experiments repeated at different time points, using different growth batches of materials or even different instruments/laboratories, but aiming to address the same biological hypothesis. With two sets of GM potatoes produced alongside common cultivars in two growth seasons analysed using two very different mass spectrometers it is possible to investigate whether explanatory signals identified in separate fingerprinting experiments result in identical models. Additionally, it can be determined whether the explanatory signals are associated with discrete chemistry that makes biological sense in terms of the lines under study.
Sensu stricto superposition of two metabolomics datasets generated from different experiments remains a challenging task. In the present study, discrimination between GM to non-GM potatoes from one harvest, using the second harvest to construct the model proved to be difficult by both machine learning approaches. This is partly due to a combination of instrument differences related to dynamic ranges, ionization characteristics and data acquisition techniques. To approach this problem, Random Forest models were constructed by comparing De1 fingerprints with data from either all SST lines or all SST/FFT genotypes in both harvest years. Figure 4a presents a scatter plot of explanatory m/z signals identified in RF models representing data of two different mass spectrometers and tubers harvested in 2 years, respectively using an Importance Score significance threshold of 0.003 (Enot et al., 2006b). Each of the De1_SST comparisons show the common signals with greatest importance scores on the diagonal; however, a group of signals (Zone I in figure 4a) appear to be relatively much more important in the 2003 sample (LTQ data). A similar phenomenon is observed in the comparison of the De1_SST/FFT models where one group of signals (Zone I in figure 4a) seems to be more important in the 2001 samples (LCT data). As predicted from the weaknesses of their respective model characteristics, examination of the top ranked signals in the De1_De2 comparisons did not highlight any common features between the two harvest years (data not shown).

Scatterplot of explanatory variables reflecting GM phenotype in two separate harvest years. The data points are represented respectively as either (a) nominal m/z value, (b) correlation analysis clade or (c) Degrees of fructan polymerisation of predicted progenitor metabolite. Zone I contains mainly high molecular weight signals (> 800 m/z) that have good representation in LTQ data (2003). Zone II contains almost entirely signals relating to double charged fructan oligomers of 5DP or greater that have good representation in LCT data (2001).
To further investigate this phenomenon, a threshold value based on either the actual importance score or its associated p-value was applied to reduce the number of variables in the list for subsequent analysis (Enot et al., 2006b). A correlation analysis on this sub-set of variables allowed for the identification of m/z signals associations in order to simplify the attribution of putative chemical identity in deeper analyses. Figure 5 illustrates a typical output derived from all signals at p < 0.001 in any comparison of De1 with either SST or SST/FFT genotypes using the 2001 data. Explanatory signals are grouped in specific correlation clusters that are characteristic for each GM class. In three of the clades (A, B, & C) several of the tightly correlated m/z-numbers differ by 1 mass unit, suggesting that these represent isotopes of the same molecular species. Furthermore, pairs of signals differ by 16 mass units suggesting the presence of correlated sodium and potassium salt adducts of the same analytes. Interestingly, the majority of these signals are highly ranked in models derived from both LCT and LTQ data (see figure 4a and 4b). Three further groupings of correlated signals are discernible (D–F) and in many cases the selected variables are half the mass of those found in clades A–C, indicating that such signals may represent doubled charged fructan ions (m/z with z = 2).

Complete hierarchical agglomerative clustering to visualise correlated and interpreted identity of explanatory signals discriminating De1 and each GM potato line.
The identity of fructan signals was further investigated by looking at the relationship of correlated signals in HILIC LC-MS profiles of the same potato extracts (see figure 6). Signals derived from potential isotopes, salt adducts and double charged ions of individual fructans were located as predicted in discrete peaks representing different degrees of polymerisation (DP). In figure 4c, the explanatory variables are labelled by fructan type. In all four data sets the majority of selected features are characteristic for fructan signals of different degrees of polymerization. In De1_SST/FFT models the explanatory signals with high significance only in the 2001 data are mainly double charged fructan species that are much more abundant in LCT-FIE-MS analysis. Conversely, the majority of the explanatory signals with higher significance in the potato 2003 LTQ-FIE-MS data are high molecular weight fructans (predominantly single-charged) which are weakly represented in the LCT data from 2001 (figure 1). These findings are presumably closely related to differences in ion source geometry and charge transfer. Although highlighted signal lists differ per se between instruments, deeper investigation revealed that both models represent the same metabolome differences.

Ion chromatograms of five selected fructan peaks in LTQ-HILIC-LC-MS ranging from (a) 3 degrees of polymerization (3DP) to (e) 7DP. Shown are traces of representative single and double-charged ions found to be discriminatory in ‘cultivars vs. GM lines’ and SST vs. SST/FFT’ models, in both or either of the LCT-FIE-MS and LTQ-FIE-MS analyses.
Concluding remarks
The present report represents one of the first studies to test the generalisability of metabolome models coping with variance relating to both, analytical equipment and environmental field scale effects. We show that nominal mass FIE-MS fingerprinting can accurately and rapidly detect differences between potato genotypes. Machine learning tools proved to cope well with data variance characteristics and were able to produce explicit and informative models. Although the two transgenic lines were affected in the same area of metabolism, FIE-MS fingerprinting easily reported the classes. Several similarity metrics proved to be consistent in assessing compositional differences under metabolomics fingerprinting constraints associated with a small sample size. Inspection of the characteristics of models with low information content is proposed as an alternative to hypothesis testing in order to estimate metrics significance and compare models in any new experimental system. The study conceivably illustrates the importance of interpreting the similarities between genotypes at the level of the explanatory variables. Specifically, an assessment of generalisability across analytical platforms and experimental conditions was only fully made when the relationship between explanatory signals was explored in depth.
References
Allen J., Davey H.M., Broadhurst D., Heald J.K., Rowland J.J., Oliver S.G., Kell D.B. (2003) High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 21:692–696
Bino R.J., Hall R.D., Fiehn O., Kopka J., Saito K., Draper J., Nikolau B.J., Mendes P., Roessner-Tunali U., Beale M.H., Trethewey R.N., Lange B.M., Wurtele E.S., Sumner L.W. (2004) Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 9:418–425
Breiman L. (2001) Random forests. Machine Learning 45:5–32
Breiman L. (2001) Statistical modeling: the two cultures. Statis. Sci. 16:199–215
Breiman, L. (2003). Two-eyed algorithms and problems. LECTURE NOTES IN COMPUTER SCIENCE, 9–9
Broadhurst D.I., Kell D.B. (2006) Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2:171–196
Buchholz A., Hurlebaus J., Wandrey C., Takors R. (2002) Metabolomics: quantification of intracellular metabolite dynamics. Biomol. Eng. 19:5–15
Catchpole G.S., Beckmann M., Enot D.P., Mondhe M., Zywicki B., Taylor J., Hardy N., Smith A., King R.D., Kell D.B., Fiehn O., Draper J. (2005) Hierarchical metabolomics demonstrates substantial compositional similarity between genetically modified and conventional potato crops. Proc. Natl. Acad. Sci. USA 102:14458–14462
Defernez M., Colquhoun I.J. (2003) Factors affecting the robustness of metabolite fingerprinting using 1H NMR spectra. Phytochemistry 62:1009–1017
Dietterich, T.G. (1998) Approximate statistical test for comparing supervised classification learning algorithms, MIT Press
Dunn W.B., Bailey N.J., Johnson H.E. (2005) Measuring the metabolome: current analytical technologies. Analyst 130:606–625
Efron B., Tibshirani R. (1997) Improvements on cross-validation: the. 632+ bootstrap method. J. Am. Statis. Assoc. 92:548–560
Enot D.P., Beckmann M., Draper J. (2006). On the interpretation of high throughput MS based metabolomics fingerprints with random forest. Complife 06:226–235
Enot D.P., Beckmann M., Overy D., Draper J. (2006) Predicting interpretability of metabolome models based on behaviour, putative identity, and biological relevance of explanatory signals. Proc. Natl. Acad. Sci. USA 103:14865–14870
Fell D.A. (1992) Metabolic control analysis: a survey of its theoretical and experimental development. Biochem. J. 286:3–330
Fell D.A. (1998) Increasing the flux in metabolic pathways: a metabolic control analysis perspective. Biotechnol. Bioeng. 58:121–124
Fell D.A. (2005) Enzymes, metabolites and fluxes. J. Exp. Bot. 56:267–272
Fiehn O. (2002) Metabolomics-the link between genotypes and phenotypes. Plant Mol. Biol. 48:155–171
Fiehn O., Kopka J., Dormann P., Altmann T., Trethewey R.N., Willmitzer L. (2000) Metabolite profiling for plant functional genomics. Nat. Biotechnol. 18:1157–1161
Good, P. (2000). Permutation tests: a practical guide to resampling methods for testing hypotheses. Springer series in statistics
Goodacre R. (2005) Making sense of the metabolome using evolutionary computation: seeing the wood with the trees. J. Exp. Bot. 56:245–254
Goodacre R., Vaidyanathan S., Bianchi G., Kell D.B. (2002) Metabolic profiling using direct infusion electrospray ionisation mass spectrometry for the characterisation of olive oils. Analyst 127:1457–1462
Goodacre R., Vaidyanathan S., Dunn W.B., Harrigan G.G., Kell D.B. (2004) Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 22:245–252
Hagan S.O., Dunn W.B., Knowles J.D., Broadhurst D., Williams R., Ashworth J.J., Cameron M., Kell D.B. (2007) Closed-loop, multiobjective optimization of two-dimensional gas chromatography/mass spectrometry for serum metabolomics. Anal. Chem. 79:464–476
Hansen M.E., Smedsgaard J. (2004) A new matching algorithm for high resolution mass spectra. J. Am. Soc. Mass Spectrom. 15:1173–1180
Harrigan G.G., LaPlante R.H., Cosma G.N., Cockerell G., Goodacre R., Maddox J.F., Luyendyk J.P., Ganey P.E., Roth R.A. (2004) Application of high-throughput Fourier-transform infrared spectroscopy in toxicology studies: contribution to a study on the development of an animal model for idiosyncratic toxicity. Toxicol. Lett. 146:197–205
Hellwege E.M., Czapla S., Jahnke A., Willmitzer L., Heyer A.G. (2000) Transgenic potato (Solanum tuberosum) tubers synthesize the full spectrum of inulin molecules naturally occurring in globe artichoke (Cynara scolymus) roots. Proc. Natl. Acad. Sci. USA 97:8699–8704
Hellwege E.M., Gritscher D., Willmitzer L., Heyer A.G. (1997) Transgenic potato tubers accumulate high levels of 1-kestose and nystose: functional identification of a sucrose sucrose 1-fructosyltransferase of artichoke (Cynara scolymus) blossom discs. Plant J. 12:1057–1065
Izmirlian G. (2004) Application of the random forest classification algorithm to a SELDI-TOF proteomics study in the setting of a cancer prevention trial. Ann. NY Acad. Sci. 1020:154–174
Jarvis R.M., Goodacre R. (2005) Genetic algorithm optimisation for pre-processing and variable selection of spectroscopic data. Bioinformatics 27:860–868
Johnson H.E., Broadhurst D., Kell D.B., Theodorou M.K., Merry R.J., Griffith G.W. (2004) High-throughput metabolic fingerprinting of legume silage fermentations via Fourier transform infrared spectroscopy and chemometrics. Appl. Environ. Microbiol. 70:1583–1592
Jonsson P., Gullberg J., Nordstrom A., Kusano M., Kowalczyk M., Sjostrom M., Moritz T. (2004) A strategy for identifying differences in large series of metabolomic samples analyzed by GC/MS. Anal. Chem. 76:1738–1745
Kell D.B., Darby R.M., Draper J. (2001) Genomic computing. Explanatory analysis of plant expression profiling data using machine learning. Plant Physiol. 126:943–951
Kok E.J., Kuiper H.A. (2003) Comparative safety assessment for biotech crops. Trends Biotechnol. 21:439–444
Kopka J. (2006) Current challenges and developments in GC-MS based metabolite profiling technology. J. Biotechnol. 124:312–322
Kopka J., Fernie A., Weckwerth W., Gibon Y., Stitt M. (2004) Metabolite profiling in plant biology: platforms and destinations. Genome Biol. 5:109
Kopka J., Schauer N., Krueger S., Birkemeyer C., Usadel B., Bergmuller E., Dormann P., Gibon Y., Stitt M., Willmitzer L., Fernie A.R., Steinhauser D. (2005) GMD@CSB.DB: the Golm metabolome database. Bioinformatics 21:1635–1638
Kuiper H.A., Kleter G.A., Noteborn H.P., Kok E.J. (2002) Substantial equivalence–an appropriate paradigm for the safety assessment of genetically modified foods? Toxicology 182:427–431
Kuiper H.A., Kok E.J., Engel K.H. (2003) Exploitation of molecular profiling techniques for GM food safety assessment. Curr. Opin. Biotechnol. 14:238–243
Langsrud O. (2002) 50–50 multivariate analysis of variance for collinear responses. J. Roy. Statis. Soc. Series D (The Statistician) 51:305–317
OECD (2001) Report of the OECD workshop on the nutritional assessment of novel foods and feeds, Organisation for Economic Co-operation and Development
Reo N.V. (2002) NMR-based metabolomics. Drug Chem. Toxicol. 25:375–382
Roessner U., Luedemann A., Brust D., Fiehn O., Linke T., Willmitzer L., Fernie A. (2001) Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 13:11–29
Roessner U., Wagner C., Kopka J., Trethewey R.N., Willmitzer L. (2000) Technical advance: simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 23:131–142
Schauer N., Semel Y., Roessner U., Gur A., Balbo I., Carrari F., Pleban T., Perez-Melis A., Bruedigam C., Kopka J., Willmitzer L., Zamir D., Fernie A.R. (2006) Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 24:447–454
Shepherd L.V., McNicol J.W., Razzo R., Taylor M.A., Davies H.V. (2006) Assessing the potential for unintended effects in genetically modified potatoes perturbed in metabolic and developmental processes. Targeted analysis of key nutrients and anti-nutrients. Trans. Res. 15:409–425
Sing T., Sander O., Beerenwinkel N., Lengauer T. (2005) ROCR: visualizing classifier performance in R. Bioinformatics 21:3940–3941
Singh S. (2003) Multiresolution estimates of classification complexity. Pattern Anal. Machine Intell., IEEE Trans. on 25:1534–1539
Somorjai R.L., Dolenko B., Baumgartner R. (2003) Class prediction and discovery using gene microarray and proteomics mass spectroscopy data: curses, caveats, cautions. Bioinformatics 19:1484–1491
Sumner L.W., Mendes P., Dixon R.A. (2003) Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochemistry 62:817–836
Thomaz, C.E., Boardman, J.P., Hill, D.L.G., Hajnal, J.V., Edwards, D.D., Rutherford, M.A., Gillies, D.F. and Rueckert, D. (2004). Using a maximum uncertainty LDA-based approach to classify and analyse mr brain images. Lect. Notes Comput. Sci. 3216:291–300
Viant M.R. (2003) Improved methods for the acquisition and interpretation of NMR metabolomic data. Biochem. Biophys. Res. Commun. 310:943–948
Wu B., Abbott T., Fishman D., McMurray W., Mor G., Stone K., Ward D., Williams K., Zhao H. (2003) Comparison of statistical methods for classification of ovarian cancer using mass spectrometry data. Bioinformatics 19:1636–1643
Zar J.H. (1984) Biostatistics. 2nd edn. Englewood Cliffs, New Jersey: Prentice-Hall
Acknowledgements
The authors wish to acknowledge the valuable contributions made to this work by Oliver Fiehn and colleagues (M.P.I., Golm and now UC, Davis) who provided the potato samples. Thanks also to Jim Heald and Robert Darby (Biological Sciences, Aberystwyth) for supporting the LCT analysis. The potato data was generated as part of a project (G02006) funded by the UK Foods Standards Agency. The University of Wales, Aberystwyth, supported M.B. and D.P.E.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Enot, D.P., Beckmann, M. & Draper, J. Detecting a difference – assessing generalisability when modelling metabolome fingerprint data in longer term studies of genetically modified plants. Metabolomics 3, 335–347 (2007). https://doi.org/10.1007/s11306-007-0064-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-007-0064-4