
Abstract
Apoptosis is a major cause of cell death in the nervous system. It plays a role in embryonic and early postnatal brain development and contributes to the pathology of neurodegenerative diseases. Here, we report that activation of the P2X7 nucleotide receptor (P2X7R) in rat primary cortical neurons (rPCNs) causes biochemical (i.e., caspase activation) and morphological (i.e., nuclear condensation and DNA fragmentation) changes characteristic of apoptotic cell death. Caspase-3 activation and DNA fragmentation in rPCNs induced by the P2X7R agonist BzATP were inhibited by the P2X7R antagonist oxidized ATP (oATP) or by pre-treatment of cells with P2X7R antisense oligonucleotide indicating a direct involvement of the P2X7R in nucleotide-induced neuronal cell death. Moreover, Z-DEVD-FMK, a specific and irreversible cell permeable inhibitor of caspase-3, prevented BzATP-induced apoptosis in rPCNs. In addition, a specific caspase-8 inhibitor, Ac-IETD-CHO, significantly attenuated BzATP-induced caspase-9 and caspase-3 activation, suggesting that P2X7R-mediated apoptosis in rPCNs occurs primarily through an intrinsic caspase-8/9/3 activation pathway. BzATP also induced the activation of C-jun N-terminal kinase 1 (JNK1) and extracellular signal-regulated kinases (ERK1/2) in rPCNs, and pharmacological inhibition of either JNK1 or ERK1/2 significantly reduced caspase activation by BzATP. Taken together, these data indicate that extracellular nucleotides mediate neuronal apoptosis through activation of P2X7Rs and their downstream signaling pathways involving JNK1, ERK and caspases 8/9/3.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
ATP is an important extracellular messenger generated from various sources including release from presynaptic vesicles upon nerve stimulation in the peripheral and central nervous systems [1]. Extracellular ATP functions as a neuromodulator by activation of cell surface P2 nucleotide receptors that are widely expressed in the nervous system [2, 3]. Based on their distinct structure, P2 nucleotide receptors are classified into two families: G protein-coupled P2Y receptors (P2YR1,2,4,6,11–14; 4) and ligand-gated ion channels (P2XR1–7; 5). Functional responses to activation of these P2 receptor subtypes in cells of the nervous system under normal and pathological conditions include cell apoptosis, proliferation and migration, inflammation, ion transport, and neurotransmission [5–9]. Therefore, P2 receptors in the nervous system may prove to be useful pharmaceutical targets in the treatment of neurological disorders.
The seven subtypes of the ionotropic ATP-gated P2XR family, ranging from 379 to 595 amino acids in length, regulate the intracellular calcium concentration through ligand-stimulated increases in the cell membrane permeability to extracellular Ca2+ ions [10, 11]. In contrast to other P2XRs, the P2X7R subtype functions exclusively as a homomeric receptor [12, 13]. The P2X7R is a 595-amino acid polypeptide containing two membrane-spanning domains, a large extracellular loop, and intracellular N- and C-terminal domains including a C-terminal tail that is the longest among the cloned P2XRs [14, 15]. P2X7Rs have been reported in microglial cells [16] where they play a role in cytokine release [17]. These receptors are also found in cultured astrocytes and Schwann cells [18, 19], spinal cord neurons and in other regions of the central nervous system [20, 21]. Brief stimulation of the P2X7R with ATP results in formation of a non-selective cationic channel that promotes the influx of Ca2+ and the equilibration of the transmembrane sodium and potassium gradients leading to membrane depolarization [14, 15, 22, 23]. Subsequent responses to P2X7R activation include the formation of a nonselective pore for molecules up to 900 Da [22, 23], extensive membrane blebbing, and eventual cell death [23]. Activation of the P2X7R was reported to elicit Ca2+ influx in cerebrocortical nerve terminals [24] and hippocampal glutamate release [25], which has been confirmed by studies with P2X7R knockout mice [26]. In an ex vivo model of organotypic hippocampal cultures, P2X7R activation was shown to directly participate in cell damage induced by oxygen/glucose deprivation [27]. It has been shown that a Pro-451 to Leu polymorphism within the C-terminal tail of the P2X7R interferes with the ability of the receptor to induce cell death in murine thymocytes [28]. The P2X7R also has been shown to mediate ATP-induced cell death in human embryonic kidney cells [29] and human cervical epithelial cells [30]. A recent study demonstrated that spinal cord injury was associated with prolonged P2X7R activation of spinal cord neurons by extracellular ATP, which resulted in high-frequency spiking, increases in cytosolic calcium levels and neuronal degeneration, responses that were reversed by P2X7R inhibition [31].
In the present study, we demonstrate that the P2X7R agonists BzATP or ATP induce nuclear condensation and DNA fragmentation in rat primary cortical neurons (rPCNs). These apoptotic responses in rPCNs were dependent on the functional expression of P2X7Rs that mediate the ERK1/2- and JNK1-dependent activation of caspases-8/9/3.
Materials and methods
Materials
Fetal bovine serum (FBS) was obtained from Hyclone (Logan, UT, USA). Dulbecco's Modified Eagle's medium (DMEM), Minimum Essential Medium (MEM), Neurobasal medium, penicillin (100 units/ml), streptomycin (100 units/ml) and B27-AO (B27 without cortex antioxidants) were obtained from Gibco-BRL (Carlsbad, CA, USA). Rabbit anti-rat ERK1/2, horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG, and HRP-conjugated goat anti-mouse IgG antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Goat anti-rat P2X7R antibody was obtained from Alomone (Jerusalem, Israel). Rabbit anti-rat caspase-8 antibody was obtained from Biovision Inc. (Mountain View, CA, USA). Mouse anti-NeuN antibody was obtained from Chemicon International Inc. (Temecula, CA, USA). Alexa Fluor TM 488 goat anti-mouse IgG antibody, Alexa Fluor TM 594 goat anti-rabbit IgG antibody, YO-PRO-1, TO-PRO-3 and the Prolong® anti-fade kit were obtained from Molecular Probes Inc. (Eugene, OR, USA). All other antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). Caspase-3 and caspase-9 inhibitors were obtained from R&D (Minneapolis, MN, USA). Caspase-8 inhibitor was obtained from BioSource International Inc. (Camarillo, CA, USA). The Precision Plus Protein Standards and nitrocellulose membranes (0.45 µm) were obtained from Bio-Rad (Hercules, CA, USA). LumiGLO chemiluminescent substrates were obtained from New England Biolabs (Beverly, MA, USA). The RNeasy Mini Kit was obtained from Qiagen (Chatsworth, CA, USA). The Apoptotic DNA-ladder kit, the First Strand cDNA Synthesis kit and the TUNEL In Situ Cell Death Detection kit were obtained from Roche (Indianapolis, IN, USA). Nucleotides and all other biochemicals were obtained from Sigma Chemical Co. (St. Louis, MO, USA).
Primary cell culture of cortical neurons
Experimental procedures for cell culture of rat primary cortical neurons (rPCNs) were carried out essentially as described by Suen et al. [32]. Briefly, cerebral cortices from 18-day-old embryos of Sprague-Dawley rats were removed and the meninges discarded. The brain tissue was mechanically dissociated in HGGMEM comprised of 1.78 g glucose, 100 IU/ml penicillin, 100 µg/ml streptomycin, 7.5 µg/ml fungizone, 2 mM glutamine in 88 ml of MEM and 10% (v/v) horse serum. The tissue clumps were dispersed with a 10 ml pipette and suspended in 6 ml of 0.25% (w/v) trypsin at 37 °C for 10 min. After incubation, 2 ml of heat-inactivated horse serum were added to block trypsin activity. Cells were centrifuged at 4000 g for 10 min at 4 °C and the pellet was suspended in ice-cold HGGMEM with 10% horse serum. The cell suspension was filtered through a sterilized 85 µm cell strainer (nylon membrane; Becton Dickinson, Franklin Lakes, NJ) and cells were counted and seeded into plastic culture plates that were precoated with poly-d-lysine. After 16–18 h and every 2–3 days thereafter, the medium was replaced with B27-AO Neurobasal medium (2 mM glutamine, 8.9 g glucose, 100 IU/ml penicillin, 100 µg/ml streptomycin, 7.5 mg/ml fungizone, 10 ml of B27 minus AO, and Neurobasal medium (Gibco-BRL) to 500 ml) and the neurons were ready for use after 7 days in culture.
RT-PCR analysis of P2XR mRNA expression
Total RNA was isolated from rPCNs using the RNeasy Mini Kit (Qiagen). cDNA was synthesized from the purified RNA using the First Strand cDNA Synthesis Kit for RT-PCR (AMV) (Roche). Five percent of the synthesized cDNA was used as a template in PCR reactions with the Expand High Fidelity PCR System (Roche). Specific 338 Q. Kong et al. oligonucleotide primers were designed to selectively amplify cDNA for individual P2XR subtypes, as shown in Table 1. The amplification was performed using 35 cycles of denaturation at 95 °C for 30 s, with annealing at 60 °C for 30 s and extension at 72 °C for 1 min. The resulting PCR products were resolved on a 1% (w/v) agarose gel containing 10 µg/ml ethidium bromide and photographed under UV illumination.
Single cell calcium assay
The [Ca2+]i was quantified in single cells with the Ca2+-sensitive fluorescent dye fura-2, using an InCyt Dual-Wavelength Fluorescence Imaging System (Intracellular Imaging, Cincinnati, OH). Rat PCNs cultured on a poly-d-lysine-coated coverslip were incubated with 2.5 µM fura-2-acetoxymethylester at 37 °C for 30 min in physiological salt solution (PSS) containing (mM) NaCl 138, KCl 5, CaCl2 2, MgCl2 1, HEPES 10, glucose 10, pH 7.4, and washed with PSS. The coverslip with fura-2-loaded cells was positioned on the stage of an inverted epifluorescence microscope (Nikon; model TMD) and stimulated with agonists at 37 °C, as described in the figure legends. Cells were exposed to 340/380 nm light and fluorescence emission at 505 nm was converted to [Ca2+]i using a standard curve created with solutions containing known concentrations of Ca2+. Increases in [Ca2+]i were measured by subtracting basal [Ca2+]i from the maximum agonist-induced increase in [Ca2+]i. The percentage of cells that responded to ATP/BzATP was also determined. Viable cells were identified by responsiveness to carbachol and non-responding cells were eliminated.
Detection of nuclear condensation
Rat PCNs were cultured in B27-AO Neurobasal medium on poly-d-lysine-coated coverslips in 12-well plates until 50–70% confluence. After nucleotide treatments, medium was removed and cells were washed once and incubated with DAPI-methanol staining solution (1 µg/ml) for 15 min at 37 °C. The staining solution was removed and the cells were washed with methanol. The inverted coverslip was placed onto a microscope slide using glycerol as the mounting agent. Nuclear morphology was observed under a fluorescence microscope (Nikon, Eclipse TE300) with a 340/380 nm excitation filter and a LP 430 nm barrier filter. DAPI has an absorbance maximum at 340 nm and an emission maximum at 488 nm in aqueous solution [33].
DNA laddering assay
Rat PCNs were cultured in B27-AO Neurobasal medium in 100 mm dishes to 70% confluence, and incubated in serumfree medium overnight with or without BzATP. Then, rPCNs were treated for 2 h with 500 µM H2O2 and DNA was isolated using the Apoptotic DNA-ladder kit (Roche, Indianapolis, IN) according to the manufacturer's instructions. DNA also was isolated from lyophilized apoptotic U937 cell lysate (provided with the DNA-ladder assay kit) for use as a positive control. Briefly, the medium was replaced with 200 µl of PBS and 200 µl of binding buffer (6 M guanidine-HCl, 10 mM urea, 10 mM TrisYHCl, pH 4.4, 20% (v/v) Triton X-100). After 10 min at 15–25 °C, 100 µl of isopropanol was added and the samples were transferred to a filter tube and centrifuged for 1 min at 10,000 g (Spectrafuge 18 M, Labnet, Edison, NJ). The fluid in the collection tube was discarded and the upper filter column was washed with 500 µl of washing buffer (20 mM NaCl and 2 mM TrisYHCl, pH 7.5, 40% (v/v) ethanol) followed by centrifugation twice for 1 min at 13,000 g. After the residual washing buffer was removed, the DNA was eluted with 200 µl of pre-warmed (70 °C) elution buffer (10 mM Tris, pH 8.5). The concentration and purity of the eluted DNA was determined by measuring the optical density at 260 and 280 nm. DNA was electrophoresed in a 2% (w/v) agarose gel at 80 V for 1 h in TBE buffer (89 mM Tris base, 89 mM boric acid, 2 mM EDTANa2-salt, pH 8.3) and visualized under UV light.
TUNEL assay
After treatment with nucleotides or H2O2, rPCNs were fixed with fresh 4% (v/v) paraformaldehyde in PBS and treated with 3% (v/v) H2O2 in methanol to inhibit endogenous peroxidase activity. Then, DNA strand breaks in fixed cells were detected with a TUNEL In Situ Cell Death Detection kit (Roche) according to the manufacturer's instructions. The number of TUNEL-positive cells and the total cell number were determined by fluorescence (Nikon, Eclipse TE300) using an excitation wavelength in the range of 450–500 nm and a detection wavelength in the range of 515–565 nm (green) and light microscopy with cell counting, respectively, at 200× magnification.
Application of P2X7 antisense oligonucleotide
Antisense oligonucleotide treatment was performed as previously described [65]. Phosphorothioate-modified oligonucleotides to rat P2X 7 were synthesized and purified by Integrated DNA Technologies (ADT, Coralville, IA, USA). Sequences were designed as follows: sense 5′-AGAGCGT GAATTACGGCACATCAA-3′, antisense 5′-TTGATGGT GCCGTAATTCACGCTCT-3′. Sequences were checked for uniqueness with the National Center for Biotechnology Information's Basic Local Alignment Search Tool [34]. The rPCNs were incubated with 0.1 or 1 µmol/L P2X7 sense or antisense S-oligonucleotide for 6 h in serum-free medium containing 1.4% (v/v) DOTAP liposomal reagent (Roche Diagnostics). Fresh serum-free medium was added with or without BzATP, and cells were cultured for an additional 18 h.
Western blot analysis
Rat PCNs cultured in 6-well plates were treated with 300 µM BzATP in the presence or absence of inhibitors as indicated. The cells were washed twice with ice-cold PBS, and lysed for 5 min in 200 µl of ice-cold lysis buffer (20 mM TrisYHCl, pH 7.5, 2% (w/v) sodium dodecyl sulfate (SDS), and 1 mM sodium orthovanadate). Lysate (30 µg of protein) was added to Laemmli sample buffer (187.5 mM TrisYHCl, pH 6.8, 6% (w/v) SDS, 1.8% (v/v) b-mercaptoethanol and 0.003% (w/v) bromophenol blue). Samples were heated for 5 min at 96–100 °C, and subjected to 7.5% (w/v) SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes for protein immunoblotting. After overnight blocking at 4 °C with 5% (w/v) fat-free milk in TBS-T (10 mM TrisYHCl, pH 7.4, 120 mM NaCl, and 0.1% (v/v) Tween-20), membranes were incubated with either 1:1000 dilution of anti-ERK1/2, anti-phospho-ERK1/2, anti-phospho-JNK, or anti-P2X7R antibodies for 2 h at room temperature, or 1:1000 dilution of anti-cleaved caspase-3, anti-caspase-3, anti-cleaved caspase-9, anti-caspase-9, or anti-caspase-8 antibodies overnight at 4 °C followed by incubation with HRP-conjugated anti-rabbit or anti-mouse IgG antibodies (1:1000 dilution in TBS-T containing 5% (w/v) fat-free milk) for 1 h at room temperature. For normalization of protein loading, the membranes were stripped of antibodies by incubation for 30 min at 60 °C in stripping buffer (62.5 mM TrisYHCl, pH 6.8, 100 mM 2-mercaptoethanol, and 2% (w/v) SDS), and re-probed with 1:2000 dilution of rabbit polyclonal anti-actin antibody (2 h at room temperature; Cytoskeleton, Denver, CO) followed by 1:2000 dilution of HRP-conjugated anti-rabbit IgG antibody (1 h at room temperature). Protein immunoreactivity was visualized on autoradiographic film using the LumiGlo Chemiluminescence System (New England BioLabs), according to the manufacturer's instructions. The protein bands detected on X-ray film were quantified using a computer-driven scanner and Quantity One software (Bio-Rad, Hercules, CA). The activation levels of kinases or caspases were calculated as a percentage of control (i.e., total kinase, caspase, or actin).
Immunofluorescence
After fixation and permeabilization, rPCNs cultured on coverslips in 6-well plates were incubated in 2.5% (w/v) BSA and 2.5% (v/v) goat serum in PBS for 1 h at room temperature followed by incubation overnight with mouse anti-NeuN antibody (1:100 dilution; Chemicon) and rabbit anti-cleaved caspase-3 antibody (1:100 dilution; Cell Signaling) in 0.25% BSA and 0.25% goat serum in PBS. Cells were washed three times with PBS and then incubated with 1:200 dilution of goat anti-mouse Alexa Fluor 488- or goat anti-rabbit Alexa Fluor 594-conjugated IgG antibody (Molecular Probes, Eugene, OR) in 0.25% BSA and 0.25% goat serum in PBS for 1 h at 37 °C. Nuclei were stained with TO-PRO-3 (Molecular Probes), cells were rinsed with PBS and mounted on glass slides in ProLong antifade reagent (Molecular Probes). Images were obtained on a Bio-Rad MRC 1024 laser scanning confocal imaging system coupled to an Olympus IX70 inverted microscope. The imaging system was controlled by Laser Sharp software (version 3.2; Bio-Rad, Hercules, CA). Alexa 488-labeled slices were excited by 488 nm laser light, and images were acquired with a 522 nm emission filter with a bandwidth of 35 nm, whereas Alexa 594-labeled slices were excited by 568 nm light and images acquired with a 585 nm longpass emission filter. Settings of the confocal microscope were optimized for imaging in an initial experiment and then equivalently applied to all sections. Laser power and photomultiplier tube (PMT) gain were adjusted to ensure that even the brightest fluorescence was below saturation levels. Neurons were randomly chosen and scanned at 512 × 512 pixel resolution.
Statistical analysis
Results are expressed as the means ± S.E.M. Statistical analysis of data was performed using Graph Pad Prism version 4.0. Statistical significance was determined by oneway ANOVA between groups followed by post hoc Newman-Keuls test. Differences were considered statistically significant when P < 0.05.
Results
Purity of rat cortical neurons in primary culture
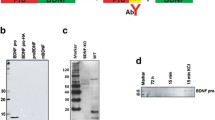
The purity of cultured rat primary neurons was determined by monitoring morphology with phase contrast microscopy and by immunofluorescence assay with the neuron-specific marker NeuN and the nuclear stain, TO-PRO-3 (Figure 1A). NeuN staining was found primarily in the nucleus of the neurons with lighter staining in the cytoplasm. About 95% of the cells were determined to be cortical neurons based on the ratio of NeuN positive cells to total cells counted by TO-PRO nuclear staining (data not shown).

Purity of rat primary cortical neurons (rPCNs) and P2X7R expression. (A) Rat PCNs were cultured on coverslips in 6-well plates and neurons were labeled with mouse NeuN monoclonal antibody (Panel a) and cell nuclei with TO-PRO-3 (Panel b); merged picture shown in Panel c. (B) RNA was isolated from rPCNs maintained in culture for 7–10 days and RT-PCR was used to amplify mRNAs to P2X1–7Rs, as described in the Materials and methods. The amplified PCR products were resolved by gel electrophoresis, and data shown are representative of results from three independent experiments. Results are shown of PCRs performed in the presence (+) or absence (−) of reverse transcriptase (RT). G3PDH primers were used to amplify G3PDH mRNA as a positive control. (C) P2X7R protein expression was detected by Western blot analysis, as described in the Materials and methods. Human 1321N1 cells expressing the recombinant human P2X7R or the empty expression vector pLXSN were used as positive (+) or negative (−) controls, respectively. Precision Plus Protein Standards are indicated as ‘75KD’ and ‘100KD.’
Functional P2X7Rs are expressed in rat PCNs
Rat PCNs express P2X7R mRNA, as well as mRNAs for P2X2, P2X3, P2X5, and P2X6 receptors (Figure 1B). Furthermore, immunoblot analysis indicated that rPCNs express P2X7R protein (Figure 1C). The P2X7R agonists ATP and BzATP (35) caused a dose-dependent increase in [Ca2+]i in rPCNs (Figure 2), suggesting that calcium influx is stimulated by activation of the endogenously expressed P2X7R protein. Calcium responses were also observed by stimulation of rPCNs with 2-methylthio-ADP (data not shown), an agonist of the P2Y1R, indicating that other P2R subtypes besides the P2X7R can regulate increases in [Ca2+]i.

ATP- and BzATP-induced increases in [Ca2+]i in rat PCNs. (A) Single cell calcium assays were performed on rPCNs that were cultured for 7–10 days on poly-d-lysine-coated coverslips. Fura-2 loaded cells were incubated in PSS with the indicated concentration of BzATP or ATP and the maximum increase in [Ca2+]i was determined, as described in the Materials and methods. (B) The percentage of cells responding to BzATP or ATP is shown as the mean ± S.E.M. of results from three experiments.
ATP and BzATP induce nuclear condensation and DNA fragmentation in rPCNs
Treatment of rPCNs with either BzATP (300 µM) or ATP (100 µM) overnight caused nuclear condensation as indicated by increased DAPI staining of nuclei (Figure 3). BzATP-induced nuclear condensation was inhibited by the P2X7R antagonist oATP, consistent with the involvement of P2X7Rs in this process. Moreover, BzATP treatment of rPCNs caused DNA fragmentation, suggestive of apoptotic cell death (Figure 4). BzATP or ATP also caused DNA strand breakage in rPCNs, another indicator of apoptosis, detected as an increase in the number of TUNEL-positive cells (Figure 5). These data suggest that BzATP or ATP can cause apoptosis in rPCNs through a pathway involving P2X7R activation.

ATP/BzATP-induced nuclear condensation mediated by the P2X7R is dependent on caspase-3 activation. (A) Rat PCNs were cultured for 7–10 days, incubated in serum-free HGGMEM for 6 h and stimulated with BzATP (300 µM) or ATP (100 µM) for 16 h or with H2O2 (1 µM) for 2 h. When indicated, 500 mM oATP was added 2 h prior to addition of BzATP. Cells cultured in B27-AO Neurobasal medium (NB) or in serumfree HGGMEM (−) overnight were used as controls. Then, nuclear condensation was determined by DAPI staining and detected by fluorescence microscopy, as described in the Materials and methods. (B) Cells were treated as in (A) except that the data were expressed as a percentage of cells that exhibited DAPI stained nuclei. Data are the means ± S.E.M. of results from at least four experiments, where *P < 0.05, and ***P < 0.001 indicate significant differences from the serum-starved control (−), and where ### P < 0.001 indicates a significant difference from BzATP treatment. (C) rPCNs were treated as in (A) except that 10 µM ZDEVD-FMK, a caspase-3 inhibitor, was added for 1 h prior to BzATP, when indicated.

BzATP stimulates DNA fragmentation in rat PCNs. Rat PCNs were incubated in serum-free HGGMEM for 6 h and then stimulated with BzATP (300 µM) for 16 h or H2O2 (1 mM) for 2 h. Cells cultured in serumfree HGGMEM (−) overnight were used as the control. Then, cells were lysed, total DNA was purified, the DNA concentration was determined, and 3 µg of purified DNA were loaded onto each lane of a 2% (w/v) agarose gel and electrophoresed, as described in the Materials and methods. DNAfragmentation was visualized after electrophoresis of DNA. Apoptotic U937 cells (+++; provided in the Apoptotic DNA-ladder kit) were used as a positive control and ‘M’ indicates the 100 bp DNA ladder marker.

BzATP or ATP induces DNA strand breakage in rat PCNs. Rat PCNs were cultured for 7–10 days, incubated in serum-free HGGMEM for 6 h, and stimulated with BzATP (300 µM) or ATP (100 µM) for 16 h or with H2O2 for 2 h. Cells cultured in serum-free HGGMEM (−) overnight were used as the control. Then, cells were fixed and DNA strand breakage was determined by TUNEL assay, as described in the Materials and methods. DNA strand breakage was observed as green fluorescence using fluorescence microscopy.
P2X7R-mediated caspase-3 and caspase-9 activation
Caspase-3 is an essential component of apoptotic pathways in many cell types, including neurons [36], and is activated by the sequential release of cytochrome c from mitochondria dria and the activation of caspase-9 [37]. Consistent with a role for caspase-3 in nucleotide-induced neuronal apoptosis, nuclear condensation caused by BzATP was diminished by treatment of rPCNs with the caspase-3 inhibitor Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-FMK (Z-DEVDFMK; Figure 3C). Results also indicated that treatment of rPCNs with either ATP or BzATP significantly increased the cleavage of caspase-3 in a time- and dose-dependent manner (Figure 6A). BzATP-induced caspase-3 cleavage was inhibited by oATP (Figure 6B), the caspase-3 inhibitor Z-DEVD-FMK (Figure 6B), or P2X7 antisense oligonucleotide (Figure 6C). Although recent studies indicate that oATP might act independently from P2 receptor inhibition [38], inhibition of BzATP-induced caspase-3 cleavage by P2X7R antisense oligonucleotide confirms the involvement of the P2X7R. Since no preparation of primary neurons is devoid of glial cells, dual immunofluorescence analysis for neuron-specific NeuN and cleaved caspase-3 was performed to evaluate whether the apoptotic cells were neurons. The majority of cells treated with BzATP were positive for cleaved caspase-3 and NeuN (e.g., pink and white arrows in Figure 6D), indicating that a large percentage of cells undergoing apoptosis were neurons. NeuN immunoreactivity has been reported to decrease under several pathological conditions [39], and therefore NeuN detection may decrease with caspase-3 activation in apoptotic neurons, suggesting that some apoptotic neurons may go undetected.

P2X7Rs mediate caspase-3 activation in rat PCNs. (A) Rat PCNs were incubated in serum-free HGGMEM for 6 h and then the indicated concentration of ATP or BzATP was added for 16 h or the time indicated. Cells cultured in serum-free HGGMEM (−) overnight were used as controls. (B) Cells were treated as in (A) except that 500 µM oATP was added for 2 h or the indicated concentration (1, 10 or 100 µM) of Z-DEVD-FMK was added for 1 h prior to addition of 300 µM BzATP for 16 h. (C) Cells were treated as in (A) except that P2X7 antisense or sense oligonucleotide was added at the indicated concentration in mg/ml for 8 h prior to addition of 300 µM BzATP for 16 h or 1 mM H2O2 for 2 h. Western analysis was performed as described in Materials and methods to determine the relative amounts of full length pan caspase-3, cleaved caspase-3 or P2X7R protein. Data are the means ± S.E.M. of results from at least three experiments, where *P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences from BzATP-treated (B, C) cells. (D) Rat PCNs were serum-starved overnight in the presence (Panels B, D and F ) or absence (Panels A, C, and E) of 300 mM BzATP and then immunofluorescence of the neuron-specific marker NeuN (green; Panels A, B), cleaved caspase-3 (red; Panels C and D), and the nuclear stain TO-PRO (blue; Panels E and F ) were detected as described in the Materials and methods. Arrows (pink or white) indicate apoptotic neurons that are both caspase-3 and NeuN positive, whereas yellow arrows indicate apoptosis in a NeuN-negative cell. Similar results were obtained in three individually isolated groups of cells.
BzATP also caused the cleavage of caspase-9 (Figure 7A), whereas activation of caspase-9 and caspase-3 were significantly inhibited by the caspase-8 inhibitor Ac-IETD-CHO (Figure 7B) or the caspase-9 inhibitor Z-LEHD-FMK (data not shown), indicating that caspase-8 also plays a role in activating downstream caspase family members (i.e., caspase-9/3) of the P2X7R-mediated apoptosis pathway. We also detected an inhibitory effect of the caspase-9 inhibitor, Z-LEHD-FMK, on caspase-8 cleavage (Figure 7C) suggesting that caspase-8 and caspase-9 may regulate caspase-3 activity in a non-linear fashion.

Role of caspase-8 and caspase-9 in P2X7R-mediated apoptosis in rPCNs. (A) Rat PCNs were incubated in serum-free HGGMEM and BzATP (300 µM) was added for 16 h. Cells cultured in B27-AO Neurobasal medium (NB) or in serum-free HGGMEM (−) overnight were used as controls. (B) Cells were treated as in (A) except that the indicated concentration (µM) of a specific caspase-8 inhibitor, Ac-IETDCHO was added for 1 h prior to addition of 300 µM BzATP. (C) Cells were treated as in (B) except that the indicated concentration of a specific caspase-9 inhibitor, Z-LEHD-FMK, was added. ‘Pan’ ( full-length) and cleaved caspase-8 were detected by Western analysis, as described in the Materials and methods. Data are the means ± S.E.M. of results from at least three experiments, where *P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences from either serum-starved (A) or BzATPtreated (B, C) cells.
Caspase-3 activation by BzATP in rPCNs is dependent upon ERK1/2 or JNK1 phosphorylation
P2X7R-mediated activation of extracellular signal-regulated kinases (ERK1/2) in rat primary astrocytes [40] and SAPK/JNK in human and rodent macrophages occur independently of the activation of caspase-1- or caspase- 3-like proteases [41]. Here, we tested whether ERK1/2 or JNK1 activities were involved in BzATP-induced apoptosis in rPCNs. As indicated in Figure 8, both ERK1/2 and JNK1 were rapidly phosphorylated in response to BzATP or ATP in a dose- and time-dependent manner. Inhibition of ERK1/2 or JNK1 phosphorylation in rPCNs with either the MEK inhibitor UO126 or the JNK inhibitor SP600125, respectively, attenuated caspase-3 activation, indicating the involvement of ERK1/2 and JNK1 in BzATP-induced neuronal cell death (Figure 9).

BzATP or ATP induces ERK1/2 and JNK1 phosphorylation in rPCNs. Rat PCNs were incubated in serum-free HGGMEM for 6 h and treated with the indicated concentration (µM) of BzATP or ATP for 5 min or with 100 µM ATP or 300 µM BzATP for the indicated time period. Cells cultured in serum-free HGGMEM (−) overnight were used as the control. Then, cell lysates were prepared and (A) ERK1/2 or (B) JNK1 phosphorylation was detected by Western analysis, as described in the Materials and methods. Total ERK1/2 and actin were used as protein loading controls for (A) and (B), respectively.

Inhibitors of MEK and JNK decrease BzATP-induced caspase-3 cleavage in rat PCNs. Rat PCNs were incubated in serum-free HGGMEM for 6 h and treated for 30 min with the indicated concentration (µM) of (A) the MEK inhibitor U0126, (B) the JNK inhibitor SP600125 or (A, B) 0.1% (v/v) DMSO as a vehicle control. Then, the cells were incubated with 300 mM BzATP for 16 h, and Western analysis was used to detect cleaved or pan caspase-3, as described in the Materials and methods. Cells cultured in serum-free HGGMEM (−) overnight were used as the control. Data are the means ± S.E.M. of results from at least three experiments, where *P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences from BzATP-treated cells.
Discussion
The results obtained with rat PCNs indicate that activation of the P2X7R by its agonists BzATP or ATP promotes the cleavage of caspases-8/9/3 and the appearance of cellular markers indicative of apoptosis including DNA degradation and nuclear condensation and fragmentation. Since BzATP can activate other P2X receptor subtypes, albeit at higher concentrations than the P2X7R [42], we explored the role of the P2X7R in the apoptotic effects of BzATP in rPCNs using P2X7 antisense oligonucleotide to downregulate P2X7R expression and the P2X7R antagonist oATP. Results indicate that inhibition of P2X7R activity significantly inhibited BzATP-induced nuclear condensation (by P2X7R antagonist oATP; Figure 3) and caspase-3 cleavage (by both oATP and P2X7R antisense oligonucleotide; Figure 6), apoptotic responses that were diminished by incubation of rPCNs with the caspase-3 inhibitor, Z-DEVD-FMK (Figures 3C and 6B). These data strongly suggest that P2X7Rs mediate BzATP-induced apoptosis in rPCNs, although this does not rule out a potential role for other P2 nucleotide receptors.
P2X7R-mediated neuronal apoptosis in vivo requires that ATP be released from neurons or non-neuronal cells to activate cell surface receptors on neighboring neurons. Neuronal P2X7 receptors are targeted to presynaptic terminals in the central and peripheral nervous systems and activation of the P2X7R has been indicated to promote release of vesicular contents from presynaptic terminals [20]. ATP is present at high concentrations in synaptic vesicles and is released as a co-transmitter with noradrenaline, acetylcholine and other neurotransmitters [1]. ATP release also has been demonstrated in a model of spinal cord injury [43]. Thus, ATP release from excited neurons or damaged cells is an assumed consequence of any brain injury or disease (e.g., ischemia, spinal cord injury, or neurodegenerative diseases), suggesting that pro-apoptotic P2X7Rs are likely activated under pathological conditions in vivo. Recent research indicates a lack of P2X7R expression in adult rat hippocampal neurons [44], whereas our studies demonstrate functional P2X7R expression in rPCNs from 18-day old embryos. We have detected P2X7R mRNA in freshly isolated rPCNs (data not shown) and 7–10 day-old cultures (Figure 1), suggesting that the in vitro cell culture procedure alone was not required for P2X7R expression.
Caspase activation is known to play a central role in the execution of apoptosis by causing nuclear condensation, DNA fragmentation and other apoptotic changes. In the mitochondrial-initiated pathway of apoptosis, caspase activation is triggered by the formation of a multimeric Apaf-1/cytochrome c complex that is fully functional in recruiting and activating procaspase-9 [37]. Activated caspase-9 will then cleave and activate downstream caspases such as caspase-3, -6, and -7. In rPCNs, it was not surprising that activation of caspase-3 was significantly inhibited by the caspase-9 inhibitor Z-LEHD-FMK (data not shown), since caspase-9 is known to activate caspase-3. We also found that in rat PCNs, the caspase-8 inhibitor Ac-IETD-CHO partially inhibited caspase-9 and caspase-3 activation (Figure 7B) indicating that caspase-8 cleavage may be an early event in this neuronal apoptosis pathway. Caspase-8 has been reported to play a role in cytochrome c-dependent apoptosis [45] and caspase-8 activation is associated with Fas receptor-induced apoptosis [46] and can result in the cleavage and activation of Bid, a proapoptotic member of the Bcl-2 family [47]. Truncated Bid translocates from the cytoplasm to the mitochondria, where it appears to antagonize the actions of anti-apoptotic Bcl-2, thereby causing an efflux of cytochrome c from the mitochondria and downstream caspase-9/3 activation [47–51]. In human embryonic kidney (HEK) 293 cells expressing P2X7Rs, extracellular ATP induced apoptosis associated with activation of poly (ADP-ribose) polymerase (PARP) and a decrease in Bax protein expression [29]. We examined the effect of ATP/BzATP on the expression of Bcl-2 family members in rPCNs but failed to detect a significant change in Bax and Bcl-2 protein expression (data not shown). Evidence from cell-free and in vitro expression systems has indicated that caspase-3 can induce cleavage and activation of the upstream initiator caspase-8 [52–54]. Our data indicated that inhibition of caspase-9 cleavage decreased caspase-8 cleavage (Figure 7C), consistent with the ability of downstream caspases to regulate the cleavage of initiator caspases.
We have demonstrated that activation of P2X7Rs in rPCNs increases calcium influx (Figure 2) and stimulates phosphorylation of the intracellular kinases ERK1/2 and JNK1 (Figure 8). Furthermore, inhibition of ERK1/2 or JNK1 phosphorylation decreased BzATP-induced caspase-3 cleavage (Figure 9). Our previous studies demonstrating that P2X7R-mediated ERK1/2 activation was dependent on extracellular Ca2+ [55] suggest a similar role for BzATP-stimulated calcium influx in rPCNs (Figure 2). Recent studies indicate that activation of ERK and phosphatidylinositol 3-kinase/Akt signaling pathways caused inhibition of caspase-3 and cell apoptosis [56]. It also has been reported that inhibition of MEK/ERK signaling is required for the induction of apoptosis by Sulindac metabolites [57]. Nonetheless, our data support the hypothesis that ERK1/2 and JNK1 activation can promote P2X7R-mediated apoptosis in rPCNs, although the mechanism involved requires further investigation.
P2X7Rs can promote neuronal cell death by mediating the release of glutamate and inflammatory factors from cells including astrocytes [18, 58, 59]. Moreover, glutamate can induce ATP release from astrocytes [60], suggesting that ATP and glutamate have reciprocal roles that contribute to the propagation of an apoptotic signal. It also has been reported that the P2X7R modulates macrophage production of TNF-ga, IL-1β and nitric oxide (NO) following LPS exposure [58], consistent with a role for the P2X7R in inflammation [61]. In addition to these factors, structural features within the P2X7R indicate that the mechanism whereby it regulates apoptosis is likely to be complex. There are several functional domains located in the P2X7R C-terminal tail including an LPS-binding domain, a SH3-binding domain, and an overlapping Fcelldeath_ domain, which appear to be important for receptor trafficking and various physiological functions [62], and the role of these domains in P2X7R-mediated apoptosis of neurons warrants further investigation. Our studies with the membrane impermeant fluorescent dye YO-PRO-1 (molecular weight: 375 Da) indicate that BzATP causes pore formation (YO-PRO-1 uptake) in rPCNs (data not shown), consistent with the report that apoptosis in the retinal microvasculature is associated with P2X7 receptor-mediated pore formation [63].
A recent study indicated that spinal cord neurons expressed an abundance of P2X7Rs, and exposure of freshly prepared spinal cord slices to ATP or BzATP evoked highfrequency firing and irreversible increases in the cytosolic Ca2+ concentration [31]. Significant cell death occurred 24 h after spinal cord injury in areas of high ATP release surrounding the epicenter of the injury, while local injection of oATP in the peritraumatic zone strongly reduced both apoptotic cell number and the severity of histological injury, suggesting the involvement of a P2X7R. Other studies indicate that the P2X7R is up-regulated in a mouse model of Alzheimer's disease [64]. Thus, the P2X7R may represent a promising therapeutic target to inhibit neurodegeneration due to injury or disease.
In summary, the data presented indicate that BzATP and ATP can stimulate P2X7Rs in embryonic rat primary cortical neurons leading to the activation of the caspases-8/9/3 and the induction of DNA degradation and nuclear condensation and fragmentation, apoptotic responses that appear to be regulated by ERK1/2 and JNK1. Since neuronal apoptosis occurs in Alzheimer's disease, Parkinson's disease and other neurodegenerative disorders, a better understanding of the mechanisms of P2X7R-mediated apoptosis may lead to novel therapies to prevent apoptosis-related brain injuries.
Abbreviations
- ATP:
-
adenosine 5-triphosphate
- BzATP:
-
2-3-O-(4-benzoylbenzoyl)-ATP
- [Ca2+]i :
-
intracellular free calcium concentration
- ERK:
-
extracellular signal-regulated protein kinase
- JNK:
-
c-Jun NH2-terminal kinase
- MAPK:
-
mitogen-activated protein kinase
- oATP:
-
oxidized ATP
- P2Y2R:
-
P2Y2 nucleotide receptor
- P2X7R:
-
P2X7 nucleotide receptor
- PBS:
-
phosphate buffered saline
- PKC:
-
protein kinase C
- rPCNs:
-
rat primary cortical neurons
- RT-PCR:
-
reverse transcriptase-polymerase chain reaction
- SH3:
-
Src-homology-3
- Src:
-
non-receptor tyrosine kinase
- TNF-α:
-
tumor necrosis factor-α
References
Burnstock G, Cocks T, Kasakov L, Wong HK. Direct evidence for ATP release from non-adrenergic, non-cholinergic (“purinergic- nerves in the guinea-pig taenia coli and bladder. Eur J Pharmacol 1978; 49: 145-.
Burnstock G. Purinergic nerves and receptors. Prog Biochem Pharmacol 1980; 16: 141-4.
Inoue K. Microglial activation by purines and pyrimidines. Glia 2002; 40: 156-3.
Abbracchio MP, Boeynaems J-M, Barnard EA et al. Characterization of the UDP-glucose receptor (renamed here the P2Y14 receptor) adds diversity to the P2Y receptor family. TiPS 2003; 24: 52-.
Burnstock G. P2X receptors in sensory neurones. Br J Anaesth 2000; 84: 476-8.
Norenberg W, Illes P. Neuronal P2X receptors: localisation and functional properties. Naunyn-Schmiedeberg's Arch Pharmacol 2000; 362: 324-9.
von Kugelgen I, Wetter A. Molecular pharmacology of P2Y-receptors. Naunyn-Schmiedeberg's Arch Pharmacol 2000; 362: 310-3.
Khakh BS, Burnstock G, Kennedy C et al. International Union of Pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev 2001; 53: 107-8.
Illes P, Ribeiro AJ. Neuronal P2 receptors of the central nervous system. Eur J Pharmacol 2004; 483: 5–17.
Benham CD. ATP-gated channels in vascular smooth muscle cells. Ann N Y Acad Sci 1990; 603: 275-5.
Di Virgilio F, Chiozzi P, Ferrari D et al. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood 2001; 97: 587–600.
Chessell IP, Simon J, Hibell AD et al. Cloning and functional characterisation of the mouse P2X7 receptor. FEBS Lett 1998; 439: 26–30.
North RA, Surprenant A. Pharmacology of cloned P2X receptors. Annu Rev Pharmacol Toxicol 2000; 40: 563-0.
Surprenant A, Rassendren F, Kawashima E et al. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996; 272: 735-.
Rassendren F, Buell GN, Virginio C et al. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem 1997; 272: 5482-.
Collo G, Neidhart S, Kawashima E et al. Tissue distribution of the P2X7 receptor. Neuropharmacology 1997; 36: 1277-3.
Ferrari D, Chiozzi P, Falzoni S et al. Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J Exp Med 1997; 185: 579-2.
Duan S, Anderson CM, Keung EC et al. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci 2003; 23: 1320-.
Grafe P, Mayer C, Takigawa T et al. Confocal calcium imaging reveals an ionotropic P2 nucleotide receptor in the paranodal membrane of rat Schwann cells. J Physiol 1999; 515: 377-3.
Deuchars SA, Atkinson L, Brooke RE et al. Neuronal P2X7 receptors are targeted to presynaptic terminals in the central and peripheral nervous systems. J Neurosci 2001; 21: 7143-2.
Atkinson L, Batten TF, Moores TS et al. Differential co-localisation of the P2X7 receptor subunit with vesicular glutamate transporters VGLUT1 and VGLUT2 in rat CNS. Neuroscience 2004; 123: 761-.
Weisman GA, Dé BK, Friedberg I et al. Cellular responses to external ATP which precede an increase in nucleotide permeability in transformed cells. J Cell Physiol 1984; 119: 211-.
Virginio C, MacKenzie AB, North RA, Surprenant A. Kinetics of cell lysis, dye uptake and permeability changes in cells expressing the rat P2X7 receptor. J Physiol 1999; 519: 335-6.
Lundy PM, Hamilton MG, Mi L et al. Stimulation of Ca2+ influx through ATP receptors on rat brain synaptosomes: identification of functional P2X7 receptor subtypes. Br J Pharmacol 2002; 135: 1616-6.
Sperlágh B, Köfalvi A, Deuchars J et al. Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat hippocampus. J Neurochem 2002; 81: 1196-11.
Papp L, Vizi ES, Sperlagh B. Lack of ATP-evoked GABA and glutamate release in the hippocampus of P2X7 receptor-mice. NeuroReport 2004; 15: 2387-1.
Cavaliere F, Amadio S, Sancesario G et al. Synaptic P2X7 and oxygen/glucose deprivation in organotypic hippocampal cultures. J Cereb Blood Flow Metab 2004; 4: 392-.
Le Stunff H, Auger R, Kanellopoulos J, Raymond MN. The Pro-451 to Leu polymorphism within the C-terminal tail of P2X7 receptor impairs cell death but not phospholipase D activation in murine thymocytes. J Biol Chem 2004; 279: 16918-6.
Wen LT, Caldwell CC, Knowles AF. Poly(ADP-ribose) polymerase activation and changes in Bax protein expression associated with extracellular ATP-mediated apoptosis in human embryonic kidney 293-P2X7 cells. Mol Pharmacol 2003; 63: 706-3
Wang Q, Wang L, Feng YH et al. P2X7 receptor-mediated apoptosis of human cervical epithelial cells. Am J Physiol, Cell Physiol 2004; 287: C1349-8.
Wang X, Arcuino G, Takano T et al. P2X7 receptor inhibition improves recovery after spinal cord injury. Nat Med 2004; 10: 821-.
Suen KC, Lin KF, Elyaman W et al. Reduction of calcium release from the endoplasmic reticulum could only provide partial neuroprotection against beta-amyloid peptide toxicity. J Neurochem 2003; 87: 1413-6.
Kapuściński J, Skoczylas B. Fluorescent complexes of DNA with DAPI 4-6-diamidine-2-phenyl indole.2HCl or DCI 4-6-dicarboxyamide-2-phenyl indole. Nucleic Acids Res 1978; 5: 3775-9.
National Center for Biotechnology Information. Basic Local Alignment Search Tool (BLAST). Available at: http://www.ncbi.nlm.nih.gov/BLAST.
Erb L, Lustig KD, Ahmed AH et al. Covalent incorporation of 3-O-(4-benzoyl)benzoyl-ATP into a P2 purinoceptor in transformed mouse fibroblasts. J Biol Chem 1990; 265: 7424-1.
Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell 1997; 91: 443-.
Li P, Nijhawan D, Budihardjo I et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479-9.
Beigi RD, Kertesy SB, Aquilina G, Dubyak GR. Oxidized ATP (oATP) attenuates proinflammatory signaling via P2 receptor-independent mechanisms. Br J Pharmacol 2003; 140: 507-9.
Unal-Cevik I, Kilinc M, Gursoy-Ozdemir Y et al. Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: A cautionary note. Brain Res 2004; 1015: 169-4.
Panenka W, Jijon H, Herx LM et al. P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci 2001; 21: 7135-2.
Humphreys BD, Rice J, Kertesy SB, Dubyak GR. Stress-activated protein kinase/JNK activation and apoptotic induction by the macrophage P2X7 nucleotide receptor. J Biol Chem 2000; 275: 26792-.
Bianchi BR, Lynch KJ, Touma E et al. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur J Pharmacol 1999; 376: 127-8.
Sim JA, Young MT, Sung HY et al. Reanalysis of P2X7 receptor expression in rodent brain. J Neurosci 2004; 24: 6307-4.
Khera M, Somogyi GT, Kiss S et al. Botulinum toxin A inhibits ATP release from bladder urothelium after chronic spinal cord injury. Neurochem Int 2004; 45: 987-3.
Viswanath V, Wu Y, Boonplueang R et al. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J Neurosci 2001; 21: 9519-8.
Zhuang S, Lynch MC, Kochevar IE. Caspase-8 mediates caspase-3 activation and cytochrome c release during singlet oxygen-induced apoptosis of HL-60 cells. Exp Cell Res 1999; 250: 203-2.
Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501
Kuwana T, Smith JJ, Muzio M et al. Apoptosis induction by caspase-8 is amplified through the mitochondrial release of cytochrome c. J Biol Chem 1998; 273: 16589-4.
Luo X, Budihardjo I, Zou H et al. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481-0.
Schendel SL, Azimov R, Pawlowski K et al. Ion channel activity of the BH3 only Bcl-2 family member, BID. J Biol Chem 1999; 274: 21932-.
Wei MC, Zong WX, Cheng EH et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727-0.
Slee EA, Harte MT, Kluck RM et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol 1999; 144: 281-2.
Wolf BB, Green DR. Suicidal tendencies: Apoptotic cell death by caspase family proteinases. J Biol Chem 1999; 274: 20049-2.
Tang D, Lahti JM, Kidd VJ. Caspase-8 activation and bid cleavage contribute to MCF7 cellular execution in a caspase-3-dependent manner during staurosporine-mediated apoptosis. J Biol Chem 2000; 275: 9303-.
Gendron FP, Neary JT, Theiss PM et al. Mechanisms of P2X7 receptor-mediated ERK1/2 phosphorylation in human astrocytoma cells. Am J Physiol, Cell Physiol 2003; 284: C571-1.
Khreiss T, Jozsef L, Hossain S et al. Loss of pentameric symmetry of C-reactive protein is associated with delayed apoptosis of human neutrophils. J Biol Chem 2002; 277: 40775-1.
Rice PL, Beard KS, Driggers LJ, Ahnen DJ. Inhibition of extracellular-signal regulated kinases 1/2 is required for apoptosis of human colon cancer cells in vitro by Sulindac metabolites. Cancer Res 2004; 64: 8148-1.
Hu Y, Fisette PL, Denlinger LC et al. Purinergic receptor modulation of lipopolysaccharide signaling and inducible nitric-oxide synthase expression in RAW 264.7 macrophages. J Biol Chem 1998; 273: 27170-.
Guerra AN, Fisette PL, Pfeiffer ZA et al. Purinergic receptor regulation of LPS-induced signaling and pathophysiology. Endotoxin Res 2003; 9: 256-3.
Queiroz G, Meyer DK, Meyer A et al. A study of the mechanism of the release of ATP from rat cortical astroglial cells evoked by activation of glutamate receptors. Neuroscience 1999; 91: 1171-1.
Di Virgilio F, Vishwanath V, Ferrari D. On the role of the P2X7 receptor in the immune system. In Abbracchio MP and Williams M (eds): Handbook of Experimental Pharmacology: Purinergic and Pyrimidinergic Signalling Vol.151/II, Berlin Heidelberg New York: Springer 2001, 355-7.
Denlinger LC, Fisette PL, Sommer JA et al. Cutting edge: the nucleotide receptor P2X7 contains multiple protein- and lipid-interaction motifs including a potential binding site for bacterial lipopolysaccharide. J Immunol 2001; 167: 1871-.
Sugiyama T, Kobayashi M, Kawamura H et al. Enhancement of P2X7-induced pore formation and apoptosis: an early effect of diabetes on the retinal microvasculature. Invest Ophthalmol Vis Sci 2004; 45: 1026-2.
Parvathenani LK, Tertyshnikova S, Greco CR et al. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer's disease. J Biol Chem 2003; 278: 13309-7.
Seye CI, Kong Q, Erb L et al. Functional Functional P2Y2 nucleotide receptors mediate uridine s'-triphosphate-induced intimal hyperplasia in collared rabbit carotid arteries. Circulation 2002; 106(21): 2720-.
Acknowledgements
We sincerely thank Xiaolin Zhang, Jennifer Aufder Heide, and the University of Missouri-Columbia Molecular Cytology Core Facility for excellent technical assistance. This study was supported by National Institutes of Health Grants 1 P01-AG18357 and 1 P20-RR15565, the University of Missouri-Columbia Food for the 21st Century Program, and the University of Missouri-Columbia Neuroscience Program (fellowship to Q. Kong).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kong, Q., Wang, M., Liao, Z. et al. P2X7 nucleotide receptors mediate caspase-8/9/3-dependent apoptosis in rat primary cortical neurons. Purinergic Signalling 1, 337–347 (2005). https://doi.org/10.1007/s11302-005-7145-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-005-7145-5