
Abstract
There is abundant evidence that extracellular ATP and other nucleotides have an important role in pain signaling at both the periphery and in the CNS. At first, it was thought that ATP was simply involved in acute pain, since ATP is released from damaged cells and excites directly primary sensory neurons by activating their receptors. However, neither blocking P2X/Y receptors pharmacologically nor suppressing the expression of P2X/Y receptors molecularly in sensory neurons or in the spinal cord had an effect on acute physiological pain. The focus of attention now is on the possibility that endogenous ATP and its receptor system might be activated in pathological pain states, particularly in neuropathic pain. Neuropathic pain is often a consequence of nerve injury through surgery, bone compression, diabetes or infection. This type of pain can be so severe that even light touching can be intensely painful; unfortunately, this state is generally resistant to currently available treatments. An important advance in our understanding of the mechanisms involved in neuropathic pain has been made by a recent work demonstrating the crucial role of ATP receptors (i.e., P2X3 and P2X4 receptors). In this review, we summarize the role of ATP receptors, particularly the P2X4 receptor, in neuropathic pain. The expression of P2X4 receptors in the spinal cord is enhanced in spinal microglia after peripheral nerve injury, and blocking pharmacologically and suppressing molecularly P2X4 receptors produce a reduction of the neuropathic pain behaviour. Understanding the key roles of ATP receptors including P2X4 receptors may lead to new strategies for the management of neuropathic pain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although the possibility of extracellular ATP being able to evoke pain in the periphery was raised about 30 years ago [1, 2], significant advances in our understanding of the molecular mechanisms by which ATP causes pain have been made quite recently by the discovery of cell-surface receptors for detecting extracellular ATP, and other nucleotides, via P2 receptors on sensory neurons [3–7]. In a subset of primary afferent sensory neurons, ATP or its analogues produce electrophysiological and biological responses via ligand-gated ion-channel receptors, namely P2X receptors (P2XRs) [8–13], and G protein-coupled receptors, namely P2Y receptors (P2YRs) [14–19]. The P2XR and P2YR subclasses have been further divided into seven (P2X1–7Rs) and eight (P2Y1,2,4,6,11–14Rs) subtypes, respectively [6, 20]. mRNA and protein for both P2XRs and P2YRs are expressed in sensory neurons, but accumulating evidence using gene deletion methods, antisense technologies, and selective receptor antagonists now suggests that the molecular targets of ATP on primary sensory neurons could be P2X3Rs, P2X2/3Rs (heteromultimeric P2X2 and P2X3 receptors), P2Y1Rs and P2Y2Rs [11, 17, 19, 21–26]. ATP responses evoked via these receptors in primary sensory neurons could code for the sensation of pain in vivo in laboratory animals [5–7, 11, 12, 23, 24, 26–35]. All cells contain millimolar levels of ATP and release ATP into the extracellular milieu when tissues are damaged [36] or stimulated mechanically [37]. The released ATP activates neighboring sensory nerve endings via P2X and P2YRs [36, 37]. These make ATP an interesting candidate for a tissue-damage signal at the periphery that may activate nociceptive sensory neurons [5–7, 12, 23, 24, 38]. In the dorsal horn of the spinal cord, ATP, presumably released from primary afferent central terminals [39], modulates excitatory (glutamatergic) and inhibitory (GABAergic and glycinergic) neurotransmission in dorsal horn neurons [40–47]. Activation of P2XRs in the spinal cord enhances pain behaviour [29, 48, 49], suggesting that ATP has a role in pain processing in the spinal cord as well. However, blocking P2XRs or P2YRs pharmacologically or suppressing their expression molecularly in sensory neurons or in the spinal cord had little effect on acute physiological pain evoked by heat or mechanical pressure in normal animals [23, 24, 26, 30, 31, 33]. It thus seems likely that endogenous ATP and its receptors system may be activated rather in pathological pain states, particularly in neuropathic pain, than in normal conditions [26, 30, 31, 50].
Neuropathic pain, which often develops when nerves are damaged through surgery, bone compression, diabetes or infection, is a type of pathological pain that does not resolve even when the overt tissue damage has healed [51–53]. Neuropathic pain can be so severe that even light contact with clothing can be intensely painful (tactile allodynia — an abnormal hypersensitivity to innocuous stimuli) and is relatively resistant to most current treatments. Accumulating evidence concerning how peripheral nerve injury creates neuropathic pain has suggested that molecular and cellular alterations in primary sensory neurons and in the spinal dorsal horn after nerve injury have important role in the pathogenesis of neuropathic pain [51–53]. While there is an increasing body of evidence suggesting that P2X3Rs in primary sensory neurons have a role in neuropathic pain [26, 30, 31, 54, 55], other P2XR and P2YR subtypes are also beginning to be investigated in terms of their changes in expression using cDNA microarray [56–58]. Recently, we revealed that the P2X4R subtype in the spinal cord is required for the expression of neuropathic pain [59]. Importantly, the expression of P2X4R in the spinal cord is enhanced, and this is highly restricted to microglia that are activated after nerve injury. Until recently, glial cells were generally considered to serve primarily housekeeping roles in the nervous system, but that study directly implicated activated glia, particularly microglia, in the pathogenesis of neuropathic pain [59]. Here we review the progress in the current understanding of how the ATP receptor in spinal microglia participate in the pathophysiology of neuropathic pain.
Microglia and nerve injury-induced pain
Glial cells make up over 70% of the total cell population in the CNS and are classified into astrocytes, oligodendrocytes and microglia. In the adult, microglia are ubiquitously distributed throughout the CNS and represent a morphologically unique type of cell; under normal conditions, microglia have a small soma bearing thin and branched processes [60, 61]. Once activated by neuronal injury, trauma, ischemia, infection, or neurological diseases, microglia show a stereotypic program of changes in morphology, gene expression, function and number [60–62]. Activated microglia change their morphology from a resting, ramified shape into an active, amoeboid shape. The changes in expression of cell-surface molecules (i.e., complement receptor 3, which is recognized by the antibody OX42) and in morphology are widely used as the key diagnostic markers of activated microglia [60, 61].
Clinical evidence that neuropathic pain results from damage to peripheral nerves in humans led to the development of a variety of models for studying neuropathic pain in laboratory animals. In most animal models of neuropathic pain which have been intensively studied so far peripheral nerves are directly damaged [63–67]. Evidence from studies using such models has revealed that peripheral nerve injury leads to a dramatic change in microglia within the spinal dorsal horn [68–72]. Within the first 24 h after peripheral nerve injury, spinal microglia become hypertrophic in their short and thick processes [68]. This is followed by a burst proliferation of microglia with a peak at around 2–3 days after the nerve injury [73]. Activated microglia exhibit upregulated OX42 labeling in the dorsal horn [68, 70–72, 74], which starts to increase as early as 1 day after nerve injury and peaks at around 14 days [70]. The temporal pattern of OX42 upregulation in the dorsal horn correlated with that of the development of tactile allodynia [70], suggesting the role of microglia in neuropathic pain. While there have been many studies showing that activation of microglia in the dorsal horn is correlated with the development of pain hypersensitivity in a wide variety of nerve injury models [68, 70–72, 74, 75], until recently it remained an open question whether spinal microglia play a causal role in neuropathic pain behaviour.
P2X4Rs in spinal microglia are necessary and sufficient for neuropathic pain
A clue to identifying P2X4Rs in the spinal cord as being required for neuropathic pain first came from pharmacological investigations of pain behaviour after nerve injury using the P2XR antagonists TNP-ATP and PPADS [59]. We found that the marked tactile allodynia that develops following injury of a spinal nerve was reversed by acutely administering TNP-ATP intrathecally but was unaffected by administering PPADS. TNP-ATP had no effect on acute pain behaviour in the uninjured state nor on motor behaviour. From the pharmacological profiles of TNP-ATP and PPADS, it was inferred that tactile allodynia depends upon P2X4Rs in the spinal cord. The expression of P2X4R protein, normally low in the naïve spinal cord, progressively increased in the days following nerve injury with a time-course parallel to that of the development of tactile allodynia. Immunohistochemical analysis demonstrated that many small cells in the dorsal horn on the side of the nerve injury were intensely positive for P2X4R protein. These cells were identified as microglia rather than neurons or astrocytes by double immunolablelling using cell-specific markers. The cells expressing P2X4R in the nerve-injured side of the dorsal horn were more numerous than under control conditions and showed high levels of OX42 labeling and morphological hypertrophy, all of which are characteristic markers of activated microglia. Moreover, it was found that reducing the upregulation of P2X4R protein in spinal microglia by means of intrathecally administered antisense oligodeoxynucleotide targeting P2X4R prevented the development of the nerve injury-induced tactile allodynia. Collectively, this evidence implies that P2X4Rs activation is necessary for pain hypersensitivity following nerve injury, and that microglia are required for this hypersensitivity since the expression of these receptors in the dorsal horn is restricted to this type of cell.
The sufficiency of P2X4R activation in microglia for the development of allodynia was demonstrated by intrathecal administration of activated, cultured microglia in which these receptors had been stimulated in vitro by ATP [59]. In otherwise naïve animals, intrathecal administration of cultured microglia that were preincubated with ATP to activate P2X4Rs on microglia produced tactile allodynia progressively over the 3–5 h following the administration. In contrast, intrathecal administration of unstimulated microglia did not cause allodynia nor did administering vehicle or ATP alone. Microglia also express another subtype of P2XR, P2X7R, but this receptor subtype appears not to be involved because activation of P2X7Rs typically requires a higher concentration of ATP than used [76, 77] and because TNP-ATP, which does not affect P2X7Rs [78], prevents ATP from stimulating microglia to produce allodynia. Moreover, in rats in which tactile allodynia was caused by the ATP-stimulated microglia this allodynia was reversed by administering TNP-ATP [59]. Thus, stimulation of P2X4Rs is required in the tactile allodynia caused by ATP-stimulated microglia and this tactile allodynia therefore resembles that caused by nerve injury. Collectively, these findings indicate that P2X4R stimulation of microglia is not only necessary for tactile allodynia, but is also sufficient to cause the allodynia. Furthermore, this finding makes a strong case that microglia activation is not simply correlated with neuropathic pain behaviour. Rather, microglia within the dorsal horn play an active and ongoing role in the tactile allodynia produced by injury to peripheral nerves.
Possible mechanisms underlying microglial modulation of dorsal horn pain signalling
The variety of biological effects produced by ATP in microglia in in vitro studies using purified microglia in culture may provide hints towards clarifying the mechanisms by which microglia produce altered processing of information in the spinal cord dorsal horn. We have previously shown that ATP stimulates the release of plasminogen in a concentration-dependent manner from 10 to 100 µM with a peak response at 5–10 min after the stimulation [79]. ATP also potently stimulates tumor necrosis factor-α (TNF-α) release following an increase in the TNF-a mRNA expression by activating mitogen-activated protein kinases (MAPKs) [80, 81]. A de novo synthesis and release of interleukin-6 is also induced by ATP through pathways mediated by both Ca2+-dependent protein kinase C and p38 MAPK [82], a member of the MAPK family [83]. Interestingly, we and others have found that nerve injury leads to persistent activation of p38 MAPK, and, importantly, that the activation of p38 MAPK in the spinal cord is entirely restricted to microglia [84, 85]. Furthermore, an inhibitor of this enzyme, SB203580, administered intrathecally, reverses mechanical allodynia following spinal nerve ligation [84, 85]. Also, continuous infusion of SB203580 starting before the nerve injury prevents the development of tactile allodynia [84]. These indicate that p38MAPK could be an important intracellular event in the development of nerve injury-induced pain. Moreover, several cytokines such as IL-1β, IL-6 and TNF-α in the dorsal horn are increased after nerve lesion [86–88] and have been implicated in contributing to nerve-injury pain [75, 86–90].
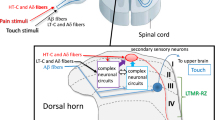
The ability of ATP to release cytokines raises the possibility that microglia might release one, or more, diffusible factor(s) that act either directly or indirectly on neurons (Figure 1). Several cytokines have been reported to alter synaptic transmission in the CNS including the spinal cord [91–93]. For example, the exogenous application of IL-1β enhances NMDA receptor-mediated Ca2+ responses via activating tyrosine protein kinase Src [94], which is known to enhance NMDA receptor activity in dorsal horn neurons [52, 95]. IL-1β also decreases GABAA receptor-mediated currents [96]. More recently, Moriguchi et al. [97] have shown in acute cortical slices that the application of a microglial-conditioned medium potentiates NMDA receptor-mediated postsynaptic responses, but not when the medium is boiled or incubated with proteinase K. Furthermore, they fractionated the medium into six sharp peaks by anion-exchange chromatography and found that the fraction contained a relatively strong protein band with a molecular mass of approximately 70 kDa that showed the most potent enhancing activity on the NMDA receptor-mediated responses. Although they have not identified the molecule(s) yet, they suggested that both heat- and protease-labile molecules released from microglia regulate NMDA receptor-mediated excitatory synaptic transmission in the CNS. Thus, diffusible factors released from activated microglia by activating P2X4Rs may also have modulatory effects on neurons in the pain processing network within the dorsal horn (Figure 1).

Schematic illustration of possible mechanisms by which P2X4R in activated microglia modulate pain signaling in the dorsal horn. Peripheral nerve injury leads to molecular and cellular alterations in the spinal dorsal horn after nerve injury. Activated microglia after nerve injury increase the expression of P2X4R. The P2X4R are activated by ATP which is presumably released from primary sensory neurons (or astrocytes), and in turn cause a rise in the intracellular calcium and activate p38MAPK. These signals lead to the release of bioactive diffusible factors such as cytokines and chemokines. The diffusible messengers released from microglia may then interact with excitatory and inhibitory synapses of neighboring dorsal horn neurons and enhance the excitability in dorsal horn neurons, which in turn leads to increased transmission in pain signaling neurons. The enhanced transmission in the dorsal horn pain network might be due to the facilitation of glutamatergic synaptic transmission or the suppression of GABA/glycinergic inhibition.
Conclusion
Damage or injury to peripheral nerve leads to changes in the spinal cord that cause the activation of microglia (Figure 1). These activated microglia express P2X4R; endogenous ATP can then activate these receptors and lead to neuropathic pain. Almost all currently known drugs for neuropathic pain were developed to target neurons, and these drugs do not exhibit adequate therapeutic effects in patients with neuropathic pain [98]. We expect that efforts to elucidate how P2X4R signaling in microglia causes neuropathic pain will provide us both with exciting insights into pain mechanisms and with clues to developing new therapeutic agents which may fundamentally change the management of intractable pain. Such strategies could not have been anticipated based on the prevailing neuroncentric view of pain.
Abbreviations
- IL-1β:
-
interleukin-1β
- IL-6:
-
interleukin-6
- MAPK:
-
mitogen-activated protein kinase
- PPADS:
-
pyridoxalphosphate-6-azophenyl-2'4-disulphonic acid
- TNF-α:
-
tumor necrosis factor-α
- TNP-ATP:
-
2'3-0-(2,4,6-trinitrophenyl)adenosine 5-triphosphate
References
Bleehen T, Hobbiger F, Keele CA. Identification of algogenic substances in human erythrocytes. J Physiol 1976; 262: 131-9.
Bleehen T, Keele CA. Observations on the algogenic actions of adenosine compounds on the human blister base preparation. Pain 1977; 3: 367-7.
Chen CC, Akopian AN, Sivilotti L et al. A P2X purinoceptor expressed by a subset of sensory neurons. Nature 1995; 377: 428-1.
Lewis C, Neidhart S, Holy C et al. Coexpression of P2X2 and P2X3 receptor subunits can account for ATP- gated currents in sensory neurons. Nature 1995; 377: 432-.
Burnstock G, Wood JN. Purinergic receptors: Their role in nociception and primary afferent neurotransmission. Curr Opin Neurobiol 1996; 6: 526-2.
Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev 1998; 50: 413-2.
Burnstock G. P2X receptors in sensory neurones. Br J Anaesth 2000; 84: 476-8.
Cook SP, Rodland KD, McCleskey EW. A memory for extracellular Ca2+ by speeding recovery of P2X receptors from desensitization. J Neurosci 1998; 18: 9238-4.
Ueno S, Tsuda M, Iwanaga T et al. Cell type-specific ATP-activated responses in rat dorsal root ganglion neurons. Br J Pharmacol 1999; 126: 429-6.
Li C, Peoples RW, Lanthorn TH et al. Distinct ATP-activated currents in different types of neurons dissociated from rat dorsal root ganglion. Neurosci Lett 1999; 263: 57–60.
Tsuda M, Koizumi S, Kita A et al. Mechanical allodynia caused by intraplantar injection of P2X receptor agonist in rats: Involvement of heteromeric P2X2/3 receptor signaling in capsaicin-insensitive primary afferent neurons. J Neurosci 2000; 20: RC90.
Chizh BA, Illes P. P2X receptors and nociception. Pharmacol Rev 2001; 53: 553-8.
Dunn PM, Zhong Y, Burnstock G. P2X receptors in peripheral neurons. Prog Neurobiol 2001; 65: 107-4.
Svichar N, Shmigol A, Verkhratsky A et al. ATP induces Ca2+ release from IP3-sensitive Ca2+ stores exclusively in large DRG neurones. Neuroreport 1997; 8: 1555-.
Koizumi S, Tsuda M, Shigemoto Y et al. Characterization of P2Y receptors in cultured rat dorsal root ganglion neurons. Jpn J Pharmacol 2001; 85: 149.
Sanada M, Yasuda H, Omatsu-Kanbe M et al. Increase in intracellular Ca(2+) and calcitonin gene-related peptide release through metabotropic P2Y receptors in rat dorsal root ganglion neurons. Neuroscience 2002; 111: 413-2.
Tominaga M, Wada M, Masu M. Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA 2001; 98: 6951-.
Molliver DC, Cook SP, Carlsten JA et al. ATP and UTP excite sensory neurons and induce CREB phosphorylation through the metabotropic receptor, P2Y2. Eur J Neurosci 2002; 16: 1850-0.
Moriyama T, Iida T, Kobayashi K et al. Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosci 2003; 23: 6058-2.
Khakh BS, Bao XR, Labarca C et al. Neuronal P2X transmitter-gated cation channels change their ion selectivity in seconds. Nat Neurosci 1999; 2: 322-0.
Collo G, North RA, Kawashima E et al. Cloning OF P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J Neurosci 1996; 16: 2495-07.
Xiang Z, Bo X, Burnstock G. P2X receptor immunoreactivity in the rat cochlea, vestibular ganglion and cochlear nucleus. Hear Res 1999; 128: 190-.
Cockayne DA, Hamilton SG, Zhu QM et al. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature 2000; 407: 1011-.
Souslova V, Cesare P, Ding Y et al. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature 2000; 407: 1015-.
Cockayne DA, Dunn PM, Burnstock G et al. Generation and electrophysiological characterization of P2X2 and P2X2/P2X3 knockout (KO) mice. Soc Neurosci Abstract 2002; 32: 52.12.
Jarvis MF, Burgard EC, McGaraughty S et al. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci USA 2002; 99: 17179-4.
Bland-Ward PA, Humphrey PP. Acute nociception mediated by hindpaw P2X receptor activation in the rat. Br J Pharmacol 1997; 122: 365-1.
Hamilton SG, Wade A, McMahon SB. The effects of inflammation and inflammatory mediators on nociceptive behaviour induced by ATP analogues in the rat. Br J Pharmacol 1999; 126: 326-2.
Tsuda M, Ueno S, Inoue K. In vivo pathway of thermal hyperalgesia by intrathecal administration of alpha,beta-methylene ATP in mouse spinal cord: Involvement of the glutamate-NMDA receptor system. Br J Pharmacol 1999; 127: 449-6.
Dorn G, Abdel’Al S, Natt FJ et al. Specific inhibition of the rat ligand-gated ion channel P2X3 function via methoxyethoxy-modified phosphorothioated antisense oligonucleotides. Antisense Nucleic Acid Drug Dev 2001; 11: 165-4.
Barclay J, Patel S, Dorn G et al. Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J Neurosci 2002; 22: 8139-7.
Honore P, Kage K, Mikusa J et al. Analgesic profile of intrathecal P2X(3) antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain 2002; 99: 11-.
Tsuda M, Shigemoto-Mogami Y, Ueno S et al. Downregulation of P2X3 receptor-dependent sensory functions in A/J inbred mouse strain. Eur J Neurosci 2002; 15: 1444-0.
Inoue K, Tsuda M, Koizumi S. ATP induced three types of pain behaviors, including allodynia. Drug Dev Res 2003; 59: 56–63.
Himaki D, Koizumi S, Mizokoshi A et al. Mechanisms underlying UTP-evoked mechanical allodynia. J Pharmacol Sci 2003; 94: 189.
Cook SP, McCleskey EW. Cell damage excites nociceptors through release of cytosolic ATP. Pain 2002; 95: 41-.
Koizumi S, Fujishita K, Inoue K et al. Ca2+ waves in keratinocytes are transmitted to sensory neurons: The involvement of extracellular ATP and P2Y2 receptor activation. Biochem J 2004; 380: 329-8.
Tsuda M, Koizumi S, Inoue K. Role of endogenous ATP at the incision area in a rat model of postoperative pain. Neuroreport 2001; 12: 1701-.
Nakatsuka T, Gu JG. ATP P2X receptor-mediated enhancement of glutamate release and evoked EPSCs in dorsal horn neurons of the rat spinal cord. J Neurosci 2001; 21: 6522-1.
Li P, Calejesan AA, Zhuo M. ATP P2X receptors and sensory synaptic transmission between primary afferent fibers and spinal dorsal horn neurons in rats. J Neurophysiol 1998; 80: 3356-0.
Gu JG, MacDermott AB. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature 1997; 389: 749-3.
Hugel S, Schlichter R. Presynaptic P2X receptors facilitate inhibitory GABAergic transmission between cultured rat spinal cord dorsal horn neurons. J Neurosci 2000; 20: 2121-0.
Jang IS, Rhee JS, Kubota H et al. Developmental changes in P2X purinoceptors on glycinergic presynaptic nerve terminals projecting to rat substantia gelatinosa neurones. J Physiol 2001; 536: 505-9.
Rhee JS, Wang ZM, Nabekura J et al. ATP facilitates spontaneous glycinergic IPSC frequency at dissociated rat dorsal horn interneuron synapses. J Physiol 2000; 524(Pt 2): 471-3.
Nakatsuka T, Tsuzuki K, Ling JX et al. Distinct roles of P2X receptors in modulating glutamate release at different primary sensory synapses in rat spinal cord. J Neurophysiol 2003; 89: 3243-2.
Nakatsuka T, Furue H, Yoshimura M et al. Activation of central terminal vanilloid receptor-1 receptors and alpha beta-methylene-ATP-sensitive P2X receptors reveals a converged synaptic activity onto the deep dorsal horn neurons of the spinal cord. J Neurosci 2002; 22: 1228-7.
Gu JG. P2X receptor-mediated modulation of sensory transmission to the spinal cord dorsal horn. Neuroscientist 2003; 9: 370-.
Driessen B, Reimann W, Selve N et al. Antinociceptive effect of intrathecally administered P2-purinoceptor antagonists in rats. Brain Res 1994; 666: 182-.
Okada M, Nakagawa T, Minami M et al. Analgesic effects of intrathecal administration of P2Y nucleotide receptor agonists UTP and UDP in normal and neuropathic pain model rats. J Pharmacol Exp Ther 2002; 303: 66–73.
Dai Y, Fukuoka T, Wang H et al. Contribution of sensitized P2X receptors in inflamed tissue to the mechanical hypersensitivity revealed by phosphorylated ERK in DRG neurons. Pain 2004; 108: 258-6.
Woolf CJ, Mannion RJ. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999; 353: 1959-4.
Woolf CJ, Salter MW. Neuronal plasticity: Increasing the gain in pain. Science 2000; 288: 1765-.
Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci 2002; 5 Suppl: 1062-.
Tsuzuki K, Kondo E, Fukuoka T et al. Differential regulation of P2X(3) mRNA expression by peripheral nerve injury in intact and injured neurons in the rat sensory ganglia. Pain 2001; 91: 351-0.
Kennedy C, Assis TS, Currie AJ et al. Crossing the pain barrier: P2 receptors as targets for novel analgesics. J Physiol 2003; 553: 683-4.
Wang H, Sun H, Della Penna K et al. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience 2002; 114: 529-6.
Xiao HS, Huang QH, Zhang FX et al. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci USA 2002; 99: 8360-.
Yang L, Zhang FX, Huang F et al. Peripheral nerve injury induces trans-synaptic modification of channels, receptors and signal pathways in rat dorsal spinal cord. Eur J Neurosci 2004; 19: 871-3.
Tsuda M, Shigemoto-Mogami Y, Koizumi S et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003; 424: 778-3.
Kreutzberg GW. Microglia: A sensor for pathological events in the CNS. Trends Neurosci 1996; 19: 312-.
Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol 1999; 58: 233-7.
Perry VH. Modulation of microglia phenotype. Neuropathol Appl Neurobiol 1994; 20: 177.
Wall PD, Devor M, Inbal R et al. Autotomy following peripheral nerve lesions: Experimental anaesthesia dolorosa. Pain 1979; 7: 103-1.
Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988; 33: 87–107.
Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 1990; 43: 205-8.
Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992; 50: 355-3.
Decosterd I, Woolf CJ. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000; 87: 149-8.
Eriksson NP, Persson JK, Svensson M et al. A quantitative analysis of the microglial cell reaction in central primary sensory projection territories following peripheral nerve injury in the adult rat. Exp Brain Res 1993; 96: 19–27.
Colburn RW, DeLeo JA, Rickman AJ et al. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol 1997; 79: 163-5.
Coyle DE. Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia 1998; 23: 75–83.
Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol 1999; 157: 289–304.
Stuesse SL, Cruce WL, Lovell JA et al. Microglial proliferation in the spinal cord of aged rats with a sciatic nerve injury. Neurosci Lett 2000; 287: 121-.
Gehrmann J, Banati RB. Microglial turnover in the injured CNS: Activated microglia undergo delayed DNA fragmentation following peripheral nerve injury. J Neuropathol Exp Neurol 1995; 54: 680-.
Liu L, Tornqvist E, Mattsson P et al. Complement and clusterin in the spinal cord dorsal horn and gracile nucleus following sciatic nerve injury in the adult rat. Neuroscience 1995; 68: 167-9.
Watkins LR, Milligan ED, Maier SF. Glial activation: A driving force for pathological pain. Trends Neurosci 2001; 24: 450-.
Surprenant A, Rassendren F, Kawashima E et al. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996; 272: 735-.
Khakh BS, Burnstock G, Kennedy C et al. International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev 2001; 53: 107-8.
Virginio C, Robertson G, Surprenant A et al. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol Pharmacol 1998; 53: 969-3.
Inoue K, Nakajima K, Morimoto T et al. ATP stimulation of Ca2+-dependent plasminogen release from cultured microglia. Br J Pharmacol 1998; 123: 1304-0.
Hide I, Tanaka M, Inoue A et al. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem 2000; 75: 965-2.
Suzuki T, Hide I, Ido K et al. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci 2004; 24: 1–7.
Shigemoto-Mogami Y, Koizumi S, Tsuda M et al. Mechanisms underlying extracellular ATP-evoked interleukin-6 release in mouse microglial cell line, MG-5. J Neurochem 2001; 78: 1339-9.
Ono K, Han J. The p38 signal transduction pathway: Activation and function. Cell Signal 2000; 12: 1–13.
Jin SX, Zhuang ZY, Woolf CJ et al. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 2003; 23: 4017-2.
Tsuda M, Mizokoshi A, Shigemoto-Mogami Y et al. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 2004; 45: 89–95.
Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience 2001; 103: 529-9.
Winkelstein BA, Rutkowski MD, Sweitzer SM et al. Nerve injury proximal or distal to the DRG induces similar spinal glial activation and selective cytokine expression but differential behavioral responses to pharmacologic treatment. J Comp Neurol 2001; 439: 127-9.
Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 2003; 306: 624-0.
Robertson B, Xu XJ, Hao JX et al. Interferon-gamma receptors in nociceptive pathways: Role in neuropathic pain-related behaviour. Neuroreport 1997; 8: 1311-.
DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 2001; 90: 1–6.
Kerr BJ, Bradbury EJ, Bennett DL et al. Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci 1999; 19: 5138-8.
Thompson SW, Bennett DL, Kerr BJ et al. Brain-derived neurotrophic factor is an endogenous modulator of nociceptive responses in the spinal cord. Proc Natl Acad Sci USA 1999; 96: 7714-.
Vikman KS, Hill RH, Backstrom E et al. Interferon-gamma induces characteristics of central sensitization in spinal dorsal horn neurons in vitro. Pain 2003; 106: 241-1.
Viviani B, Bartesaghi S, Gardoni F et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 2003; 23: 8692-00.
Yu XM, Askalan R, Keil GJ, II et al. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 1997; 275: 674-.
Wang S, Cheng Q, Malik S et al. Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther 2000; 292: 497–504.
Moriguchi S, Mizoguchi Y, Tomimatsu Y et al. Potentiation of NMDA receptor-mediated synaptic responses by microglia. Brain Res Mol Brain Res 2003; 119: 160-.
Watkins LR, Maier SF. GLIA: A novel drug discovery target for clinical pain. Nat Rev Drug Discov 2003; 2: 973-5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Inoue, K., Tsuda, M. & Koizumi, S. ATP receptors in pain sensation: Involvement of spinal microglia and P2X4 receptors. Purinergic Signalling 1, 95–100 (2005). https://doi.org/10.1007/s11302-005-6210-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-005-6210-4