
Abstract
In order to establish a rapid detection method for Mycoplasma ovipneumoniae, this study used the loop-mediated isothermal amplification (LAMP) technique to carry out nucleic acid amplification and chromatographic visualization via a lateral flow dipstick (LFD) assay. The M. ovipneumoniae elongation factor TU gene (EF-TU) was detected using a set of specific primers designed for the EF-TU gene, and the EF-TU FIP was detected by biotin labeling, which was used in the LAMP amplification reaction. The digoxin-labeled probe specifically hybridized with LAMP products, which were visually detected by LFD. Here, we established the M. ovipneumoniae LAMP-LFD rapid detection method and tested the specificity, sensitivity, and clinical application of this method. Results showed that the optimized LAMP performed at 60 °C for 60 min, and LFD can specifically and visually detect M. ovipneumoniae with a minimum detectable concentration at 1.0 × 102 CFU/mL. The sensitivity of LAMP-LFD was 1000 times that of the conventional PCR detection methods, and the clinical lung tissue detection rate was 86% of 50 suspected sheep infected with M. ovipneumoniae. In conclusion, LAMP-LFD was established in this study to detect M. ovipneumoniae, a method that was highly specific, sensitive, and easy to operate, and provides a new method for the prevention and diagnosis of M. ovipneumoniae infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mycoplasma ovipneumoniae (M. ovipneumoniae) is an important pathogen that causes atypical pneumonia in goats and sheep (Nicholas et al. 2002; Parham et al. 2006; Xin et al. 2012; Besser et al. 2013). In recent years, M. ovipneumoniae infection has been reported in many regions of China, with gradually increasing mortality rates (Handeland et al. 2014; Besser et al. 2014). Sheep infected with M. ovipneumoniae show respiratory disorders, runny noses, weight loss, growth retardation, and primary infection within 1–3 months, depending on the age of the sheep (Besser et al. 2017). The control and prevention of M. ovipneumoniae is difficult and has caused great economic losses to the sheep industry in many countries (Giangaspero et al. 2012; Xue et al. 2017). In addition, upon M. ovipneumoniae infection, sheep become susceptible to other diseases. Therefore, there is an urgent need to develop a rapid and accurate method to detect M. ovipneumoniae. Such a method not only provides a reference for early prevention, diagnosis, and epidemiological investigation but also has a certain core value for farmers.
At present, the main methods for detecting M. ovipneumoniae are pathogen diagnosis, enzyme linked immunosorbent assay (ELISA), and PCR (Jiang et al. 2016) (Jiang et al. 2016; Ziegler et al. 2014; Li et al. 2016; Yang et al. 2014; Kılıc et al. 2013). Furthermore, the research of M. ovipneumoniae has mainly concentrated on the isolation, identification, and detection technology of pathogenic bacteria (Butler et al. 2017). Although pathogen separation is the “gold standard” for M. ovipneumoniae testing, these steps are relatively tedious and time-consuming and can no longer meet the requirements of animal husbandry. Immunological methods mainly rely on the specific reaction between antigen and antibody, and while the sensitivity and specificity of these methods are high, these methods are relatively complex and rely on assorted experimental conditions and other factors; thus, their clinical application is limited. Furthermore, molecular detection methods require expensive laboratory instruments and manipulation, which may not be readily available in remote areas. In 2000, Notomi et al. developed a loop-mediated isothermal amplification (LAMP), which has the advantages of high sensitivity, good specificity, and simple operation (Domesle et al. 2017; Chen et al. 2017). The LAMP amplification principle is based on the use of 4 primers that are designed for 6 specific regions of the target gene using the strand displacement characteristics of Bst DNA polymerase (Velders et al. 2018; Feng et al. 2017; Zheney et al. 2018; Sheu et al. 2018). At certain temperatures, the 4 primers identify 6 specific regions of the target gene and continuously the process of extension replacement. Therefore, use of the appropriate primer is very important for complete LAMP amplification. In addition to focusing on the highly specific identification of target genes at 6 regions, the annealing temperatures of the inner primers F2 (B2) and F1c (B1c) should be higher than those of the outer primers to ensure that the inner primers appropriately bind the target gene pairs. In order to improve the specificity and sensitivity of the LAMP reaction, 1 pair of ring primers can be designed between the primers and the internal and external primers. LAMP can achieve rapid amplification of nucleic acids in a simple constant-temperature device, and the amplification products can be detected using a turbidimeter, electrophoresis, and by adding fluorescent dye into the tube reaction (Park et al. 2017). LAMP detection time is about 60 min, compared with the conventional PCR method; thus it not only reduces reaction time but also increases sensitivity and specificity, with good prospects and development potential. However, LAMP amplification products need special gel-imaging equipment and other instruments through electrophoresis analysis. During the experiment, it is necessary to contact carcinogen EB, and the fluorescent reagent used in fluorescence detection is expensive, which limits the application and popularization of the technology in grass-roots units. The LAMP-LFD method uses a biotin LAMP product hybridized with a digoxin-labeled DNA probe that is complexed with a gold-labeled anti-digoxin antibody. This hybridization product is trapped by a biotin ligand and bound to a lateral flow test strip, forming an immune complex. Non-hybridized digoxin-labeled probes pass through the test line (T) and bind to the sheep anti-mouse immunoglobulin G (IgG) antibody control line (C). LAMP-LFD is a LAMP amplification detection technology, wherein the product of LAMP is detected on a lateral flow test strip by color to determine the experimental results. This method removes dependence on equipment and avoids contact with ethidium bromide. The whole reaction is based on LAMP detection, which can be completed within 5–10 min; therefore, it has good prospects for application.
So far, many researchers have reported the use of LAMP-LFD technology to detect pathogenic bacteria (Lalle et al. 2018; Wachiralurpan et al. 2017; Kongkasuriyachai et al. 2017; Huang et al. 2017), but the use of LAMP to detect M. ovipneumoniae is still lacking. Based on the M. ovipneumoniae EF-TU gene, we designed 6 LAMP primers and 1 digoxin-labeled probe to optimize LAMP detection conditions. Then LFD technology was added to establish an accurate and efficient M. ovipneumoniae LAMP-LFD assay. For some small- and medium-sized farms and scattered farmers, especially in remote areas, a fast, practical, sensitive and accurate way to diagnose pneumonia caused by M. ovipneumoniae is established in this study, which may be useful for improving economic conditions.
Materials and methods
Strains and samples
Escherichia coli, Staphylococcus aureus, Salmonella pullorum, Mycoplasma bovis (M. bovis), Mycoplasma hyopneuminiae (M. hyopneuminiae) and Mycoplasma mycoides subsp. Capri (M. mycoides subsp. Capri) were preserved by the laboratory of Microbiology Teaching and Research at Shihezi University, and M. ovipneumoniae strain Y98 was purchased from the China Institute of Veterinary Drugs Control. Twenty M. ovipneumoniae-infected sheep lung tissue were collected from the sheep farms in the surrounding areas of Manasi and Shihezi, and were stored at − 80 °C.
DNA extraction
DNA was extracted from strains and sheep lung tissue according to the instruction of Genomic DNA Extraction Kit (Sangon Biotech Co., Ltd, Shanghai, China). The extracted genomic DNA was evaluated on a Nanodrop and its concentration was calculated, we used ten-fold serial dilutions with sterile water to dilute all Genomic DNA to approximately 101–107 CFU/mL, and stored at − 20 °C until use.
Design of primers and probes
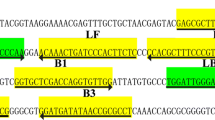
Primers were designed according to the M. ovipneumoniae EF-TU gene published in GenBank in NCBI (NO: JQ990999) combined with the LAMP primer design principle. The Primer Explorer V3 (http://primerexplorer.Jp; Eiken Chemical Co., Ltd., Tokyo, Japan) online Primer design software was used to screen out a set of specific good primers and probes. In addition, Primer 5.0 software was used to design the Primer EF-TUF and EF-TUR (Table 1; Fig. 1), and the expected amplification fragment size was 1209 bp.

LAMP primer design for M. ovipneumoniae EF-TU gene. A The location of M. ovipneumoniae EF-TU primers and probes in the sequence. B The location of M. ovipneumoniae EF-TU primers and probes
LAMP assay
Mycoplasma ovipneumoniae genomic DNA was extracted as a positive template, with 25 µL for the total reaction system comprising of the following: 10× ThermoPol Buffer 2.5 µL, MgSO4 (25 mmol/L) (Sigma, MO, USA) 1.5 µL, dNTPs (10 mmol/L) (Sangon Biotech Co., Ltd, Shanghai, China) 3.5 µL, Bst 2.0 DNA polymerase (8 U/L) (New England Biolabs, Ltd, Beijing, China) 1 µL, 3 µL primers (including inner primers (20 mol/L), outer primers (10 mol/L) and loop primers (15 mol/L) 1 µL), 2.5 µL betaine (10 mol/L) (Sigma, MO, USA), 2 µL positive template, calcein (Sigma, MO, USA) 1 µL, ddH2O. Sterile water was used as the template in the negative control sample.
Optimization of reaction temperature and time of the LAMP assay
The PCR tube into the constant temperature water bath was set to 58 °C–62 °C (increasing to 1 °C interval) reaction for 60 min. The temperature was increased to 80 °C for 5 min to inactivate the enzyme, and amplification products (7 µL) were run on 3% agarose gel electrophoresis to determine the optimal temperature. Meanwhile, the reaction time was optimized and set for 30–80 min (increasing in 10 min interval) at constant temperature water bath. After that, the amplified products (7 µL) were analyzed on 3% agarose gel electrophoresis, and the best reaction time was determined.
Optimization of the internal and external primer concentration ratio of the LAMP assay
According to the best reaction temperature and time, the ratio of internal and external primers was set up according to 25 µL system: 1:1, 2:1, 3:1 and 4:1, respectively, and LAMP was amplified. The amplified products of 7 µL were determined by 3% agarose gel electrophoresis, and the best ratio of internal and external primers was determined.
Optimal reaction system for the LAMP assay
The 25 µL system was set up, respectively by optimum reaction temperature, time, and the ratio of inner and outer primers, optimization of Mg2+ volume (1.0 µL, 1.5 µL, 2.0 µL, 2.5 µL) and dNTPs volume (1.0 µL, 1.5 µL, 2.0 µL, 2.5 µL), and 7 µL amplification products were run on 3% agarose gel electrophoresis to determine the best reaction system.
LAMP-LFD assay
In the process of hybridization, LAMP amplification products of biotin labeling, and EF-TU-HP-specific hybridization formed biotin and digoxin probe amplification products. The amplification products were added to the sample pad of the LFD strip and migrated by capillary action, where the biotin LAMP product hybridized with a digoxin-labeled DNA probe and complexed with a gold-labeled anti-digoxin antibody. This hybridization product was trapped by a biotin ligand and bound to a lateral flow test strip, forming an immune complex bound to the test line (T). Non-hybridized digoxin-labeled probes passed through the test line (T) and bound to the sheep anti-mouse IgG antibody control line (C) (Extended Data Fig. S1). The experimental procedure and determination method were as follows: 5 µL LAMP amplification product was added to 80 µL buffer, the mixture was added to the sample mat on the test paper strip, and the result was read after 5–10 min. The lateral flow dipstick (LFD) was provided by the Nucleic Acids Labeling and Detecting Lab, Key Laboratory of Ministry of Education with Provincial Co-construction of Local and Ethnic High Incidence in Xinjiang (Shihezi, Xinjiang, China).
The specificity and repeatability of LAMP-LFD
According on the above LAMP reaction conditions and system optimization, DNA was extracted from M. ovipneumoniae, Escherichia coli, Staphylococcus aureus, Salmonella pullorum, M. Bovis, M. hyopneuminiae, M. mycoides subsp. Capri and amplified. The product was evaluated with 3% agarose gel electrophoresis and LFD assay was performed. The reaction was repeated at least 3 times for LAMP-LFD verification.
Sensitivity of LAMP-LFD
Sensitivity of LAMP-LFD was investigated with different concentrations of M. ovipneumoniae genomic DNA as template and sterile water as negative control, LAMP amplification was carried out using the reaction system and reaction conditions of the above optimization. Meanwhile, the PCR reaction was conducted with EF-TUF and EF-TUF as the upstream and downstream primers, and the two methods were compared. The PCR reaction system was 25 µL, comprising of 2xES Taq MasterMix 12.5 µL, upstream and downstream primer EF-TUF and EF-TUF, respectively 0.4 µL, positive template 2 µL, and ddH2O. The PCR reaction procedure was 95 °C for 5 min; 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 80 s and a final extension of 72 °C for 10 min. The amplification products were tested by electrophoresis and LFD test strips respectively, and the detection sensitivity was the lowest detection concentration of the system.
Clinical application and detection of M. ovipneumoniae
Pathogen separation method detection (gold standard)
Take 0.5 g of lung tissue that suspected M. ovipneumoniae-infected sheep lesions, grind it into minced under sterile conditions, and inoculate the grinded tissue fluid 1:10 in Mo liquid medium and add appropriate amount of ampicillin at 37 °C. Cultured at 5 °C and 5% CO2 for 4–5 days. Passaged when the medium became orange or yellow. Passaging bacteria were inoculated on solid medium after 3 passages, cultured for 7–8 days, and observed papillary colonies under an inverted microscope, and statistical results. If there is a papillary colony, a single colony is picked and inoculated in a liquid culture medium. After the color of the culture medium turns yellow, PCR is performed using Mo-specific primers, and the results are statistically analyzed.
In order to test the feasibility of the established LAMP-LFD assay, we established a reference standard that combined the results of the pathogen separation method and PCR were used to detect the lung tissues of 50 suspected M. ovipneumoniae-infected positive sheep, and the three assays were compared.
Results
Optimization of reaction temperature and time of LAMP assay
As detailed in the Methods section, separate experiments were carried out to determine the ideal temperature and reaction time for the LAMP assay. LAMP reaction had the brightest and showed clear trapezoid strip at 60 °C and 60 min (Extended Data Fig. S2A, B). This indicated that LAMP method detection of M. ovipneumoniae only required 60 min, which was faster than PCR detection.
Optimization of the internal and external primer concentration ratio for the LAMP assay
As outlined in the Methods section, the best ratio of internal and external primers (1:1, 2:1, 3:1, and 4:1) was determined once the optimal temperature and reaction time were determined. As shown in Extended Data Fig. S3, the 3:1 ratio of internal and external primers had the best amplification product.
Optimal reaction system of LAMP assay
Upon optimizing the reaction temperature, time, and the ratio of inner and outer primers, we next optimized the MgSO4 volume (1.0 µL, 1.5 µL, 2.0 µL, 2.5 µL) and dNTPs volume (1.0 µL, 1.5 µL, 2.0 µL, 2.5 µL), and 7 µL amplification products were run on 3% agarose gel electrophoresis to determine the best system. The results showed that 2 µL MgSO4 is (Extended Data Fig. 4A) and 2.5 µL dNTPs (Extended Data Fig. 4B) resulted in the highest LAMP amplification.
Reaction parameters were also optimized by single factor optimization experiment to determine the optimal reaction system of 25 µL as follows: 2.5 µL 10× ThermoPol buffer, 2.0 µL MgSO4, 3 µL, 2 µL primers (including internal and external 3:1 in primers and loop primer for 1 µL), 2.5 µL dNTPs, 2.5 µL betaine (10 mol/L), 1 µL Bst 2.0 DNA polymerase, 2 µL positive template, 1 µL calcein, and ddH2O. The PCR tube was placed in a constant temperature water bath at 60 °C for 60 min, 80 °C 5 min, and the enzyme was inactivated.
The establishment of the M. ovipneumoniae LAMP-LFD assay
Mycoplasma ovipneumoniae genomic DNA (concentration of 1.0 × 104 CFU/mL) was extracted and used as the template for the LAMP reaction, and the products were observed by fluorescence dye, 3% agarose gel electrophoresis, and LFD analysis. Results were considered positive when the fluorescent dyes were green, while the negative control was orange (Fig. 2A). The agarose gel electrophoresis showed obvious trapezoidal bands (Fig. 2B), while the negative control had no trapezoidal band. LAMP-LFD results of positive M. ovipneumoniae template appeared in two red strips, respectively located in the test line (T) and control line (C), and negative control was only in one control line (C) a red strip (Fig. 2C). The results showed that the LAMP-LFD assay was effective and feasible.

The establishment of M. ovipneumoniae LAMP-LFD assay. M: DNA Marker; 1: LAMP products; 2: Negative control. A calcein-visual LAMP amplification product. B Electrophoretic analysis of LAMP amplification products. C LAMP-LFD detection result
Specificity and repeatability of LAMP-LFD
Based on the best LAMP reaction system and conditions, biotin-labeled primers in EF-TUFIP LAMP reaction, with four strains test strains and M. ovipneumoniae were used as templates to carry out specificity validation. The results showed that with M. ovipneumoniae genomic DNA as a template, LAMP product after electrophoresis had the same characteristic trapezoidal strips as the LAMP products and probe EF-TU-HP hybrid. Moreover, hybrid products on the LFD strip line position had a clear red strip, indicating that the test result was positive. Meanwhile the other detectable microbial LAMP products were negative, and the EF-TU-HP hybrid product in LFD position was not present in any strip on the line, indicating that the established M. ovipneumoniae LAMP-LFD detection method had good specificity (Table 2; Fig. 3). When the M. ovipneumoniae genomic DNA was repeated as a template, the results were also positive, showing that the test method had good repeatability (Fig. 4).

Specific analysis results of LAMP-LFD. A LAMP agarose gel electrophoresis. B LAMP-LFD detection. M: DNA Marker; 1: using sterile water as the template for negative control; 2–7: the genomic DNA of Escherichia coli, Staphylococcus aureus, Salmonella pullorum, M. Bovis, M. hyopneumoniae and M. mycoides subsp. Capri were used as template. 8: the genomic DNA of M. ovipneumoniae was used as the template

Repeatability analysis of LAMP-LFD. A LAMP agarose gel electrophoresis; B LAMP-LFD assay. M: DNA Marker; 1–6: the genomic DNA of M. ovipneumoniae (1.0 × 104 CFU/mL) was used as the template; 7: negative control
Sensitivity of LAMP-LFD
Next, we performed sensitivity analysis of LAMP-LFD using different concentrations of M. ovipneumoniae genomic DNA as a template. LAMP-LFD reaction results showed that with the calcein-visual LAMP (Fig. 5A), LAMP agarose gel electrophoresis (Fig. 5B), and LAMP-LFD (Fig. 5C) to detect the sensitivity, the minimum detectable concentration was 1.0 × 102 CFU/mL. The minimum detectable method of PCR was 1.0 × 105 CFU/mL (Fig. 5D), which showed that the sensitivity of LAMP-LFD was 1000 times that of the conventional PCR assay.

Sensitivity analysis of the M. ovipneumoniae LAMP-LFD. A calcium-visual LAMP sensitivity test. B LAMP agrogel electrophoresis method for sensitivity test. C Sensitivity test of LAMP-LFD method; D Sensitivity test of PCR assay. M: DNA Marker; 1–6: different concentrations of positive standard: 1 × 102 CFU/mL; 1 × 103 CFU/mL; 1 × 104 CFU/mL; 1 × 105 CFU/mL; 1 × 106 CFU/mL; 1 × 107 CFU/mL; 7: negative control
Clinical application detection of M. ovipneumoniae
We next determined the clinical application using the established conditions of the LAMP-LFD assay as compared to pathogen separation (gold standard) in 50 suspected M. ovipneumoniae-infected sheep lesions by lung tissue genomic DNA testing. As shown on Table 3, LAMP-LFD assay identified 43 as positive for M. ovipneumoniae, was 100% consistent with the results of the gold standard method. By comparison, conventional PCR assay detected 41 positive samples, which indicated the detection rate was 82%, was 95.35% consistent with the results of the gold standard method, showing that LAMP-LFD could be preliminarily used in the detection of clinical pathogens.
Discussion
In recent years, with the rise in socioeconomic status and quality of life, there has been a higher demand for lamb and wool products, which has increased sheep farming and husbandry (Di et al. 2015). However, the incidence of M. ovipneumoniae targeting sheep has concordantly been on the rise, with epidemic outbreaks reported recently (Bottinelli et al. 2017; Wolff et al. 2018). Therefore, there is a need to establish a rapid, simple, and reliable detection method for M. ovipneumoniae. In this study, a fast and efficient LAMP-LFD assay for detecting M. ovipneumoniae was established by combining LAMP and LFD technology. The method is simple, with a low requirement in experimental conditions, and results are presented visually. The advantages of this method are irreplaceable by other molecular biological methods. LAMP-LFD detection needn’t calcein, compared with the LAMP method; thus it not consider the calcein transport issues, greatly reduces the economic losses caused by transportation process. And water will be polluted by calcein, sewage treatment is more troublesome, causing certain economic losses. The sensitivity of M. ovipneumoniae samples was 5 × 102 CFU/mL, which was 1000 times higher than conventional PCR methods. In this study, M. ovipneumoniae standard strain Y98 was selected for analysis, and the LAMP gene amplification of the EF-TU gene showed positive results, while other pathogenic strains showed negative results. Therefore, this method has good specificity. Various components of the LAMP-LFD reaction were optimized: inside and outside primer concentration ratios and LAMP reaction components, i.e., Mg2+ concentration, dNTPs concentration, temperature, and reaction time. Once the reaction system and the optimal reaction conditions were determined, the results showed that LAMP could effectively complete the amplification within 60 min (Bai et al. 2011), which is faster than other methods. Moreover, the clinical application of the LAMP-LFD assay was tested using 50 suspected M. ovipneumoniae-infected sheep lesions, LAMP-LFD assay identified 43 as positive for M. ovipneumoniae; the parallel PCR identified 41 as positive. LAMP-LFD was 100% consistent with the results of the gold standard method, while PCR was 95.35% consistent, ang it takes about 2.5 h to complete all the steps, pathogen separation (gold standard) takes more time, implying that the LAMP-LFD assay can be applied to the clinical detection of pathogens.
Current detection methods of M. ovipneumoniae include PCR, indirect hemagglutination test, and ELISA (Song et al. 2014; Maksimović et al. 2017; Beser et al. 2012). These methods all have high sensitivity and accuracy but require expensive instruments and have tedious operation steps; thus, it may not be feasible for economically disadvantaged farms and clinics. LAMP-LFD technology has many advantages, such as its simplicity and speed, lack of expensive instruments and equipment, and accuracy of its results and real-time visualization. LAMP-LFD has been applied to food detection, vaccine diagnosis and disease diagnosis by many researchers (Yang et al. 2016; Deng et al. 2015; Rigano et al. 2014; Khunthong et al. 2013; Sun et al. 2014).
In this study, we used the M. ovipneumoniae EF-TU gene as the detection target and established that the LAMP-LFD assay has strong specificity, high sensitivity, and visual results. Our experimental conditions were optimized at 60 °C under the condition of constant temperature for 60 min, which could complete the detection of M. ovipneumoniae. Furthermore, the requirements for instruments and specific experimental conditions were lower, the sensitivity was 1.0 × 102 CFU/mL, and the method could detect M. ovipneumoniae on-site. Results were revealed by the color strip on the dipstick, which means that the procedure does not require special equipment for detection, increasing the amplification efficiency, convenience, and practicality and avoiding the nonspecific amplification products that often lead to false positives. Therefore, the establishment of this method to detect M. ovipneumoniae provides a practical technology and lays the foundation for the development of a clinical kit, which can be used as a new M. ovipneumoniae detection method in basic-level livestock units.
References
Bai ZD, Shi L, Hu CM, Chen X, Qi JJ, Ba XL, Peng QJ, Chen YY, Chen HC, Guo AZ (2011) Development of a loop-mediated isothermal amplification assay for sensitive and rapid detection of Mycoplasma bovis. Afr J Biotechnol 10:12333–12338. https://doi.org/10.4314/ajb.v10i57
Besser TE, Highland MA, Katherine B, Cassirer EF, Anderson NJ, Ramsey JM, Mansfield K, Bruning DL, Wolff P, Smith JB, Jenks JA (2012) Causes of pneumonia epizootics among bighorn sheep, Western United States, 2008–2010. Emerg Infect Dis 18:406–414. https://doi.org/10.3201/eid1803.111554
Besser TE, Cassirer EF, Highland MA, Wolff P, Justice-Allen A, Mansfield K, Davis MA, Foreyt W (2013) Bighorn sheep pneumonia: sorting out the cause of a polymicrobial disease. Prev Vet Med 108:85–93. https://doi.org/10.1016/j.prevetmed.2012.11.018
Besser TE, Cassirer EF, Potter KA, Lahmers K, Oaks JL, Shanthalingam S, Srikumaran S, Foreyt WJ (2014) Epizootic pneumonia of bighorn sheep following experimental exposure to Mycoplasma ovipneumoniae. PloS ONE 9:e110039. https://doi.org/10.1371/journal.pone.0110039
Besser TE, Cassirer EF, Potter KA, Foreyt WJ (2017) Exposure of bighorn sheep to domestic goats colonized with Mycoplasma ovipneumoniae induces sub-lethal pneumonia. PloS ONE 12:e0178707. https://doi.org/10.1371/journal.pone.0178707
Bottinelli M, Schnee C, Lepri E, Stefanetti V, Filippini G, Gobbi M, Sebastianelli M, Antenucci P, Rampacc E, Coletti M, Passamonti F (2017) Investigation on mycoplasma populations in pneumonic dairy lamb lungs using a DNA microarray assay. Small Ruminant Res 147:96–100. https://doi.org/10.1016/j.smallrumres.2016.12.038
Butler CJ, Edwards WH, Jennings-Gaines JE, Killion HJ, Wood ME, McWhirter DE, Paterson JT, Proffitt KM, Almberg ES, White PJ, Rotella JJ, Garrott RA (2017) Assessing respiratory pathogen communities in bighorn sheep populations: sampling realities, challenges, and improvements. PloS ONE 12:e0180689. https://doi.org/10.1371/journal.pone.0180689
Chen SC, Liu CC, Wang YN, Fu LM, Shih SH (2017) An integrated microfluidic loop-mediated isothermal amplification platform for koi herpesvirus detection. Chem Eng J 334:1828–1834. https://doi.org/10.1016/j.cej.2017.11.119
Deng J, Pei J, Gou H, Ye Z, Liu C, Chen J (2015) Rapid and simple detection of Japanese encephalitis virus by reverse transcription loop-mediated isothermal amplification combined with a lateral flow dipstick. J Virol Methods 213:8–105. https://doi.org/10.1016/j.jviromet.2014.12.006
Di X, Yan M, Min L, Li Y, Luo H, Liu X, Wang YJ (2015) Mycoplasma ovipneumoniae, induces inflammatory response in sheep airway epithelial cells via, a MyD88-dependent TLR signaling pathway. Vet Immunol Immunop 163:57–66. https://doi.org/10.1016/j.vetimm.2014.11.008
Domesle KJ, Yang Q, Hammack TS, Ge B (2017) Validation of a Salmonella loop-mediated isothermal amplification assay in animal food. Int J Food Microbiol 264:63. https://doi.org/10.1016/j.ijfoodmicro.2017.10.020
Feng J, Dai Z, Tian X, Jiang X (2017) Detection of Listeria monocytogenes based on combined aptamers magnetic capture and loop-mediated isothermal amplification. Food Control 85:443–452. https://doi.org/10.1016/j.foodcont.2017.10.027
Giangaspero M, Nicholas RA, Hlusek M, Bonfini B, Osawa T, Orusa R, Tatami S, Takagi E, Moriya H, Okura N, Kato K, Kimura A, Harasawa R, Ayling RD (2012) Seroepidemiological survey of sheep flocks from Northern Japan for Mycoplasma ovipneumoniae and Mycoplasma agalactiae. Trop Anim Health Pro 44:395–398. https://doi.org/10.1007/s11250-011-9952-8
Handeland K, Tengs T, Kokotovic B, Vikøren T, Ayling RD, Bergsjø B, Sigurðardóttir Ó, Bretten T (2014) Mycoplasma ovipneumoniae—a primary cause of severe pneumonia epizootics in the Norwegian Muskox (Ovibos moschatus) population. PloS ONE 9:e106116. https://doi.org/10.1371/journal.pone.0106116
Huang HL, Zhu P, Zhou CX, Yan XJ, Zou YX, Lv PW (2017) Detection of Skeletonema costatum based on loop-mediated isothermal amplification combined with lateral flow dipstick. Mol Cell Probe 36:36. https://doi.org/10.1016/j.mcp.2017.08.003
Jiang F, He J, Navarro-Alvarez N, Xu J, Li X, Li P, Wu WX (2016) Elongation factor Tu and heat shock protein 70 are membrane-associated proteins from Mycoplasma ovipneumoniae capable of inducing strong immune response in mice. PloS ONE 11:e0161170. https://doi.org/10.1371/journal.pone.0161170
Khunthong S, Jaroenram W, Arunrut N, Suebsing R, Mungsantisuk I, Kiatpathomchai W (2013) Rapid and sensitive detection of shrimp yellow head virus by loop-mediated isothermal amplification combined with a lateral flow dipstick. J Virol Methods 188:51–56. https://doi.org/10.1016/j.jviromet.2012.11.041
Kılıc A, Eroksuz H, Muz A, Tasdemir B (2013) Identification by culture, PCR, and immunohistochemistry of mycoplasmas and their molecular typing in sheep and lamb lungs with pneumonia in Eastern Turkey. Trop Anim Health Pro 45:1525–1531. https://doi.org/10.1007/s11250-013-0394-3
Kongkasuriyachai D, Yongkiettrakul S, Kiatpathomchai W, Arunrut N (2017) Loop-mediated isothermal amplification and LFD combination for detection of Plasmodium falciparum, and Plasmodium vivax. Methods Mol Biol 1572:431. https://doi.org/10.1007/978-1-4939-6911-1_28
Lalle M, Possenti A, Dubey JP, Pozio E (2018) Loop-mediated isothermal amplification-lateral-flow dipstick (LAMP-LFD) to detect Toxoplasma gondii oocyst in ready-to-eat salad. Food Microbiol 70:137–142. https://doi.org/10.1016/j.fm.2017.10.001
Li Y, Jiang Z, Xue D, Deng G, Li M, Liu X, Wang YJ (2016) Mycoplasma ovipneumoniae induces sheep airway epithelial cell apoptosis through an ERK signalling-mediated mitochondria pathway. Bmc Microbiol 16:222. https://doi.org/10.1186/s12866-016-0842-0
Maksimović Z, De LFC, Amores J, GómezMartín Á, Rifatbegović M (2017) Comparison of phenotypic and genotypic profiles among caprine and ovine Mycoplasma ovipneumoniae strains. Vet Rec 180:180. https://doi.org/10.1136/vr.103699
Nicholas R (2002) Improvements in the diagnosis and control of diseases of small ruminants caused by mycoplasmas. Small Ruminant Res 45:145–149. https://doi.org/10.1016/S0921-4488(02)00095-0
Parham K, Churchward CP, McAuliffe L, Nicholas R, Ayling R (2006) A high level of strain variation within the Mycoplasma ovipneumoniae population of the UK has implications for disease diagnosis and management. Vet Microbiol 118:83–90. https://doi.org/10.1016/j.vetmic.2006.07.005
Park YR, Kim HR, Kim SH, Lee KK, Lyoo YS, Yeo SG (2017) Loop-mediated isothermal amplification assay for the rapid and visual detection of novel porcine circovirus 3. J Virol Methods 253:26. https://doi.org/10.1016/j.jviromet.2017.12.006
Rigano LA, Malamud F, Orce IG, Filippone MP, Marano MR, Amaral AM, Castagnaro AP, Vojnov A (2014) Rapid and sensitive detection of Candidatus, Liberibacter asiaticus by loop mediated isothermal amplification combined with a lateral flow dipstick. Bmc Microbiol 14:86. https://doi.org/10.1186/1471-2180-14-86
Sheu SC, Tsou PC, Lien YY, Lee MS (2018) Development of loop-mediated isothermal amplification (LAMP) assays for the rapid detection of allergic peanut in processed food. Food Chem 257:67–74. https://doi.org/10.1016/j.foodchem.2018.02.124
Song W, Song Q, He L, Zhou Y, Zhao J (2014) The establishment and application of a semi-nested PCR assay for the detection of Mycoplasma ovis. Small Ruminant Res 119:176–181. https://doi.org/10.1016/j.smallrumres.2014.03.001
Sun YL, Yen CH, Tu CF (2014) Visual detection of canine parvovirus based on loop-mediated isothermal amplification combined with enzyme-linked immunosorbent assay and with lateral flow dipstick. J Vet Med Sci 76:509–516. https://doi.org/10.1292/jvms.13-0448
Velders AH, Schoen C, Saggiomo V (2018) Loop-mediated isothermal amplification (LAMP) shield for Arduino DNA detection. Bmc Res Notes 11:93. https://doi.org/10.1186/s13104-018-3197-9
Wachiralurpan S, Sriyapai T, Areekit S, Kaewphinit T, Sriyapai P, Santiwatanakul S, Chansiri K (2017) Development of a rapid screening test for listeria monocytogenes, in raw chicken meat using loop-mediated isothermal amplification (LAMP) and lateral flow dipstick (LFD). Food Anal Method 10:3763–3772. https://doi.org/10.1007/s12161-017-0949-4
Wolff PL, Blanchong JA, Nelson DD, Plummer PJ, McAdoo C, Cox M, Besser TE, Muñoz-Gutiérrez J, Anderson CA (2018) Detection of Mycoplasma ovipneumoniae in pneumonic mountain goat (Oreamnos americanus) kids. J Wildlife Dis. https://doi.org/10.7589/2018-02-052
Xin J, Li Y, Nicholas RA, Chen C, Liu Y, Zhang MJ, Dong H (2012) A history of the prevalence and control of contagious bovine pleuropneumonia in China. Vet J 191:166–170. https://doi.org/10.1016/j.tvjl.2011.02.011
Xue D, Li Y, Jiang Z, Deng G, Li M, Liu XM, Wang YJ (2017) A ROS-dependent and Caspase-3-mediated apoptosis in sheep bronchial epithelial cells in response to Mycoplasma ovipneumoniae infections. Vet Immunol Immunop 187:55–63. https://doi.org/10.1016/j.vetimm.2017.04.004
Yang F, Dao X, Rodriguez-Palacios A, Feng X, Tang C, Yang XN, Yue H (2014) A real-time PCR for detection and quantification of Mycoplasma ovipneumoniae. J Vet Med Sci 76:1631–1634. https://doi.org/10.1292/jvms.14-0094
Yang Y, Li Q, Wang S, Chen X, Du A (2016) Rapid and sensitive detection of Babesia bovis and Babesia bigemina by loop-mediated isothermal amplification combined with a lateral flow dipstick. Vet Parasitol 219:71–76. https://doi.org/10.1016/j.vetpar.2016.02.004
Zheney M, Kaziyev Z, Kassenova G, Zhao L, Liu W, Liang L, Li G (2018) Real-time fluorescence loop-mediated isothermal amplification assay for direct detection of egg drop syndrome virus. BMC Vet Res 14:49. https://doi.org/10.1186/s12917-018-1364-9
Ziegler JC, Lahmers KK, Barrington GM, Parish SM, Kilzer K, Baker K, Besser TE (2014) Safety and Immunogenicity of a Mycoplasma ovipneumoniae Bacterin for domestic sheep (Ovis aries). PloS ONE 9:e95698. https://doi.org/10.1371/journal.pone.0095698
Acknowledgements
We greatly appreciate the time reviewers have spent on this manuscript to provide their useful comments. This study was funded by the National Natural Science Foundation of China (No. 31360226).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, J., Cao, J., Zhu, M. et al. Loop-mediated isothermal amplification-lateral-flow dipstick (LAMP-LFD) to detect Mycoplasma ovipneumoniae. World J Microbiol Biotechnol 35, 31 (2019). https://doi.org/10.1007/s11274-019-2601-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-019-2601-5