
Abstract
Stenotrophomonas maltophilia KB2 is known to produce different enzymes of dioxygenase family. The aim of our studies was to determine activity of these enzymes after induction by benzoic acids in cometabolic systems with nitrophenols. We have shown that under cometabolic conditions KB2 strain degraded 0.25–0.4 mM of nitrophenols after 14 days of incubation. Simultaneously degradation of 3 mM of growth substrate during 1–3 days was observed depending on substrate as well as cometabolite used. From cometabolic systems with nitrophenols as cometabolites and 3,4-dihydroxybenzoate as a growth substrate, dioxygenases with the highest activity of protocatechuate 3,4-dioxygenase were isolated. Activity of catechol 1,2- dioxygenase and protocatechuate 4,5-dioxygenase was not observed. Catechol 2,3-dioxygenase was active only in cultures with 4-nitrophenol. Ability of KB2 strain to induce and synthesize various dioxygenases depending on substrate present in medium makes this strain useful in bioremediation of sites contaminated with different aromatic compounds.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Under aerobic conditions aromatic compounds, with the exception of those with deactivated substituent group, undergo degradation through oxygenetic ring fission by catechol 1,2- or 2,3-dioxygenase as well as protocatechuate 3,4- or 4,5-dioxygenase. Presence of deactivated substituents with strong electrophilic properties makes the attack of oxygen on aromatic ring impossible (Pitter 1985). Since the nitro group is a powerful deactivating substituent aerobic degradation of these substrates occurs generally through the reduction as well as regrouping and elimination of the nitrate group following aromatic ring cleavage (Blasco et al. 1999; Schenzle et al. 1997). Transformation of polynitrated aromatic compounds by F420-dependent enzymatic complex and hydride δ-complex formation was also shown (Ebert et al. 1999). Some microorganisms degrade nitrophenols through aromatic ring fission by dioxygenases without prior removal of the nitrate group. However degradation of nitrophenols occurs most often through their hydroxylation to benzenotriol, which is the ring fission substrate for hydroquinone 1,2-dioxygenase (Chauhan et al. 2000; Pakala et al. 2007), Walia et al. (2002) have shown activity of nitrocatechol 2,3- dioxygenase catalyzing transformation of 4-nitrocatechol into 3-nitro-2-hydroxy-6-oxo-hexa-2,4-dienoic acid in Pseudomonas putida strain. In other study Kieboom et al. (2001) have observed induction of 2,3- dioxygenase in Nocardia strain S3 in the presence of 3-nitrocatechol. It has been shown that this enzyme exhibits absolute specificity to catechol C-3 substituents (Kieboom et al. 2001) and clearly suggests dependence between inductor and the activity of 2,3- dioxygenase. Cassidy et al. (1999) observed degradation of nitrophenols by Sphingomonas sp. strain UG30 after induction of hydroquinone dioxygenase by pentachlorophenol. Pentachlorophenol-induced enzyme transformed benzenotriol formed during p-nitrophenol degradation.
Because of high toxicity and resistance to biodegradation, mononitrophenols are insufficient source of carbon and energy for microorganisms and degradation of these compounds requires presence of additional growth substrate, which increases biomass concentration and stimulates degradation of toxic chemicals. As the compounds similar in structure to transformed nitroaromatics accelerate their degradation, for mononitrophenols degradation other phenolic compounds are frequently used as growth substrate (Cho et al. 2000; Leung et al. 1997, 1999; Wan et al. 2007).
In previous works activity of various dioxygenases isolated from strain KB2 together with the ability of this strain to cometabolic transformation of nitroaromatics was observed (Greń et al. 2010; Guzik et al. 2009). Therefore in the present work an attempt to determine activity level of dioxygenases after their induction by different aromatic compounds was made. The main aim of this work was to check the activity of dioxygenases isolated from cultures with nitrophenols and verification how cometabolic conditions influenced synthesis of these enzymes.
Methods
Bacterial strain and growth conditions
Strain KB2 is a gram-negative, aromatic compounds—degrading bacterium that was isolated from activated sludge of a sewage treatment plant in Bytom—Miechowice in Poland as described previously (Guzik et al. 2009). On the basis of morphological and physiochemical characteristics and 16S rRNA gene sequence analysis was identified as Stenotrophomonas maltophilia (NCBI accession number DQ230920).
Liquid cultures of Stenotrophomonas maltophilia strain KB2 were grown in mineral salts medium (MSM) contained 3.78 g Na2HPO4·12H2O, 0.5 g KH2PO4, 5 g NH4Cl, 0.2 g MgSO4·7H2O, 0.01 g yeast extract and 1 ml of trace element solution (TES) per liter. TES contained 3.82 g FeSO4·7H2O, 0.3 g CoSO4·7H2O, 0.08 g MnSO4·H2O, 0.14 g ZnSO4·7H2O, 0.006 g H3BO3, 0.04 NaMoO4·2H2O, 0.08 g NiSO4·7H2O, 0.003 g CuSO4·5H2O, 0.15 g Al2(SO4)3·18H2O, 0.006 Na2WO4·2H2O, 10 ml 32%HCl in 1 l of the solution. The pH was adjusted to 7.1.
In studies on cometabolic transformation of nitrophenols, as well as induction of enzymes, 3 mM benzoate (BA), 4-hydroxybenzoate (4-HB), 3,4-dihydroxybenzoate (3,4-DHB) as growth substrates and 2-nitrophenol (2-NP), 3-nitrophenol (3-NP) or 4-nitrophenol (4-NP) in concentration 1 mM, as cometabolites, were used. Stock solutions of benzoate (20 mM), 4-hydroxybenzoate (20 mM), 3,4-dihydroxybenzoate (20 mM), 2-nitrophenol (10 mM), 3-nitrophenol (10 mM) or 4-nitrophenol (10 mM) were prepared in distilled water.
Cells were proliferated in mineral medium with 3 mM of phenol (at 30°C on a rotary shaker at 130 rpm), harvested by centrifugation (5,000×g at 4°C for 15 min) and washed with fresh sterile medium. Cells prepared in such a way were used as inoculum. Cultures in sterile mineral salt medium supplemented with 3 mM growth substrates and 1 mM of cometabolite were inoculated with previously prepared cells to the final optical density about 0.1 in absorbance scale at λ = 600 nm, and incubated at 30°C with shaking at 130 rpm. Suitable growth substrate (benzoate, 4-hydroxybenzoate, 3,4-dihydroxybenzoate) was introduced into the culture every 72 h. All cultures were grown in triplicates.
Bacterial growth determination
In cultures enriched with nitrophenols culture density was determined indirectly using modified method of Bradford (1976). 0.5 ml of culture was mixed with 0.5 ml KOH and incubated at 100°C for 10 min. After cooling, 0.2 ml of solution were taken, mixed with 1.0 ml of Bradford reagent and incubated for 10 min. Absorbance of sample was measured at 595 nm. Bovine serum albumin was used as a standard.
Substrate determination
Culture supernatant was prepared by centrifugation (10,000×g at 4°C for 20 min) Concentration of aromatic compounds in the culture supernatant was determined by HPLC (Merck HITACHI) equipped with a LiChromospher® RP-18 column (4 × 250 mm) and a DAD detector (Merck HITACHI). The wavelength for detection of substrates, composition of eluent and solvent, as well as the flow rate, were developed separately for each aromatic compound. The mobile phase, in mononitrophenols and benzoic acid determination, was acetonitryl and 1% acetic acid (40:60 v/v) at the flow rate of 1 ml min−1. 4-HB was separated in methanol:1% acetic acid (60:40 v/v) as a mobile phase, at the flow rate of 0.8 ml min−1. For separation of 3,4-DHB methanol:1% acetic acid (30:70 v/v) at the flow rate of 0.8 ml min−1 was used. The detection wavelength was set at 260 nm. Chemical compounds in the supernatant were identified and quantified by comparing HPLC retention times and UV–visible spectra with those of external standards. Concentration of mononitrophenols as well as benzoic acids was expressed in mM and determined by peak area calculations. Data were corrected by substraction of abiotic degradation of aromatic compounds.
Preparation of cell extracts
Cells of Stenotrophomonas maltophilia strain KB2 were harvested after 7 days of culture by centrifugation (4,500g for 15 min at 4°C) and the pellet was washed with 50 mM phosphate buffer, pH 7.0, and resuspended in the same buffer. Cell-free extracts were prepared by sonication of the whole cell suspension (6 times for 15 s) and centrifugation at 9,000g for 30 min at 4°C. Clear supernatant was used as a crude cell extract for enzyme assays.
Enzyme assays
Activity of catechol 1,2-dioxygenase [EC 1.13.11.1] was measured spectrophotometrically by formation of cis,cis-muconic acid at 260 nm (λ260 = 16,800 M−1 cm−1). The reaction mixture contained 20 μl of catechol (50 mM), 67 μl Na2EDTA (20 mM), 893 μl of phosphoric buffer pH 7.4 (50 mM) and 20 μl of crude enzyme extracts in a total volume of 1 ml. When activity of catechol 2,3-dioxygenase was detected, crude enzyme extract was incubated with 5% H2O2 prior to determination of catechol 1,2-dioxygenase. In order to determine catechol 2,3-dioxygenase [EC 1.13.11.2] activity, formation of 2-hydroxymuconic semialdehyde was measured at 375 nm (λ375 = 36,000 M−1 cm−1). The reaction mixture contained 20 μl of catechol (50 mM), 960 μl of phosphoric buffer pH 7.4 (50 mM) and 20 μl of crude extract in a total volume of 1 ml (Hegeman 1966). Activity of protocatechuate 3,4-dioxygenase [EC 1.13.11.3] was measured by protocatechuate depletion at 290 nm in a reaction mixture containing 20 μl of protocatechuate (50 mM), 960 μl of Tris–HCl buffer pH 7.4 (50 mM) and 20 μl of crude extract in a total volume of 1 ml. A molar extinction coefficient of 2,300 M−1 cm−1 was used, which is the difference between λ290 of protocatechuate (3,890 M−1 cm−1) and λ 290 of the product 3-carboxy-cis cis-muconate (1,590 M−1 cm−1). Protocatechuate 4,5-dioxygenase [EC 1.13.11.8] activity was measured spectrophotometrically by formation of 2-hydroxy-4-carboxymuconic semialdehyde at 410 nm (λ410 = 9,700 M−1 cm−1). The reaction mixture contained 20 μl of protocatechuate (50 mM), 960 μl of Tris–HCl buffer pH 7.4 (50 mM) and 20 μl of crude extract in a total volume of 1 ml (Stanier and Ingraham 1954). In order to determine hydroxyquinol 1,2-dioxygenase [EC 1.13.11.37] activity, the formation of 4-hydroxymuconic semialdehyde was measured at 320 nm (λ320 = 11 mM−1cm−1). The reaction mixture contained 200 μl of hydroquinone (50 mM), 700 μl of K–Na phosphate buffer pH 6.6 (100 mM) and 100 μl of crude extract in a total volume of 1 ml (Zaborina et al. 1995). One unit of enzyme activity was defined as the amount of enzyme required to generate 1 μmol of product per minute. Protein concentrations of the crude extract from different inducer-cultured bacteria were determined by the Bradford method using bovine serum albumin (Bradford 1976).
Chemicals
2-nitrophenol, 3-nitrophenol, 4-nitrophenol were obtained from Merck AG, Darmstadt, Germany. 4-Hydroxybenzoic acid and protocatechuic acid were obtained from FLUKA, Fluka AG, Buchs, Switzerland. Benzoic acid and other chemicals were of analytical quality and were obtained from local suppliers.
Results and discussion
Cometabolic degradation of nitrophenols by Stenotrophomonas maltophilia KB2
Within aromatic substrates produced in plants, terpenoids and phenolic compounds, as benzoic acids, are commonly known. These compounds, as secondary metabolites, are produced on the determined growth stage by specific plant tissues or organs (Ornston and Stanier 1966; Singer et al. 2003). They are usually located in leaves by which they enter the soil or are secreted directly into the soil by the root system (Singer et al. 2003). The most important source of these metabolites in soil is decay of plants tissues. Aromatic compounds of plant origin are easily transformed and, as it is known, stimulate bacteria to xenobiotics degradation.
In our studies on mononitrophenols degradation 3 mM benzoate, 4-hydroxybenzoate, 3,4-dihydroxybenzoate as growth substrates were used. Because the ability of strain to metabolise high concentration of xenobiotic is desired quality, in our experiment 1 mM of nitrophenols was used despite the fact that degradation of these substrates at lower concentration (0.05–0.74 mM) was observed (Chauhan et al. 2000; Qui et al. 2007; Walia et al. 2002).
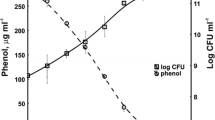
Figure 1 shows that depending on cometabolic system KB2 strain was able to degrade from 0.15 to 0.6 mM of mononitrophenol used after 14 days of cultivation. Moreover, presence of nitrophenols didn’t inhibit degradation of carbon source. Increase of microbial biomass suggests resistance of KB2 strain to these substrates (Fig. 1). During 14 days of experiment only partial cometabolite degradation was observed. Nevertheless possible application of Stenotrophomonas maltophilia KB2 in natural environment could contribute to faster removal of nitroaromatics from degraded area as the bioremediation process takes a long time and the environment undergoes dynamic changes, accompanied by continuous influx of growth substrates.

Cometabolic transformation of nitrophenols in the presence of aromatic compounds of plant origin (arrows indicate introduction of growth substrate into culture; BA-benzoic acid, 4-HB-4-hydroxybenzoic acid, 3,4-DHB-3,4-dihydroxybenzoic acid, 2-NP-2-nitrophenol, 3-NP-3-nitrophenol, 4-NP-4-nitrophenol)
Activity of ring cleavage dioxygenases after induction by various aromatic substrates
KB2 strain is known to synthesize various types of dioxygenases, both catechol and protocatechuate, depending on inductor used. In the previous work on degradation of different phenolic substrates by KB2, synthesis of catechol 1,2- and 2,3- dioxygenase was observed. Activity of catechol 2,3- dioxygenase significantly exceeded catechol 1,2- dioxygenase (Guzik et al. 2009). In the present work activity of catechol 2,3- dioxygenase (4.92 U/mg) was observed only after growing of strain in the presence of phenol, while aromatic hydroxyacids induced synthesis of protocatechuate 3,4- dioxygenase (Table 1). Interesting observation was the lack of protocatechuate dioxygenase activity after induction of KB2 by benzoate (Table 1) that may suggest degradation of this substrate via catechol and induction of additional enzyme—decarboxylase (Peng et al. 2003; Sparnins and Dagleys 1975). Isolation of enzymatic fraction after incubation of KB2 in the presence of mononitrophenols has shown no activity of catechol 2,3- dioxygenase but activity of catechol 1,2- dioxygenase (Table 1). According to the obtained results we assumed that catechol 2,3- dioxygenase was especially sensitive to nitroaromatics and lack of its’ activity might force bacteria to synthesis of hydroquinone 1,2- dioxygenase, enzyme, which is most often induced by nitrophenols (Moonen et al. 2008; Pakala et al. 2007). However activity of hydroquinone 1,2- dioxygenase in the crude enzyme extract of KB2 strain was not observed (Table 1). Activity of protocatechuate 4,5- dioxygenase observed after culturing of KB2 in the presence of 4-NP (Table 1), suggests lower sensitivity of protocatechuate dioxygenases to inhibitory effect of mononitrophenols.
Presence of aromatic ring in the structure of natural phenolic compounds produced by plants (among them benzoic acids) induces synthesis of similar degradation enzymes as xenobiotics do. Such substrates usually are easily degraded by microorganisms according to the variety of degradation systems formed during evolution processes (Mendonca et al. 2004; Münzenberger et al. 2003; Sutherland et al. 1981). Because mononitrophenols were no degraded by KB2 in monosubstrate systems (Greń et al. 2010) we decided to introduce into cometabolic systems benzoic acid, 4-hydroxybenzoic acid and 3,4-dihydroxybenzoic acid. In the presence of 2-NP or 3-NP and 3,4-DHB or 4-HB as a growth substrate activity of protocatechuate 3,4- dioxygenase was observed (Table 2), while in the culture with 4-NP activity of this enzyme was observed only in the presence of 3,4-DHB (Table 2). Even though catechol 1,2- dioxygenase was not active and catechol 2,3- dioxygenase was active only in the presence of 4-NP and 3,4-DHB or benzoate (Table 2), partial degradation of nitrophenols was observed in all cometabolic systems (Fig. 1). As isolation and determination of enzymes activity showed synthesis of protocatechuate dioxygenases by KB2 strain in the presence of nitroaromatics (Table 1) it seems very interesting to determine if such enzymes might take part in nitrophenols degradation, even though degradation of these compounds via protocatechuate pathway was not observed. As it is known aerobic degradation of mononitrophenols proceeds through benzenotriol (Cassidy et al. 1999; Pakala et al. 2007), substrate which has very similar structure to protocatechuate, except the presence of hydroxyl group at C1, instead of carboxyl one (Fig. 2). Additionally it is known that attack of protocatechuate dioxygenases on aromatic ring take place between C3 and C4 or C4 and C5 of catechol ring characteristic for both protocatechuate and benzenotriol (Vaillancourt et al. 2006). According to this knowledge we suggest that in cometabolite systems enzymes of protocatechuic acid degradation might be involved in degradation of benzenotriol formed in the consequence of mononitrophenols degradation. However presented assumptions need to be confirmed by appropriate kinetic parameters determination, especially determination of inhibition constant for aromatic acid/mononitrophenol system as similarity of above-mentioned substrates suggest possibility of their competition for enzyme active site.

Proposed region of attack of protocatechuic dioxygenases of KB2 strain (a the site of intradiol ring cleavage; b the site of extradiol ring cleavage)
Conclusion
Chemical diversity of contaminations introduced daily into natural environment is so huge that it is impossible to rule out the influence of simple as well as composite organic and inorganic compounds on dioxygenases activity. Because of that intensity of bioremediation processes proceed with the use of Stenotrophomonas maltophilia strain KB2 depends on the presence, availability and concentration of different aromatic substrates among them phenolic compounds of plant origin. Such substrates are not only very common in the environment but, as we have shown, induce different protocatechuate dioxygenases, enzymes that may be used in degradation of aromatic acids as well as other compounds with catechol structure. Although further research would be necessary to confirm our theory, wide substrate specificity of examined enzymes allows the use of Stenotrophomonas maltophilia strain KB2 for bioremediation of environments contaminated with different aromatic compounds.
References
Blasco R, Moore E, Wray V, Pieper D, Timmis K, Castillo F (1999) 3-Nitroadipate, a metabolic intermediate for mineralization of 2, 4-dinitrophenol by a new strain of a Rhodococcus species. J Bacteriol 181:149–152
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–258
Cassidy MB, Lee H, Trevors JT, Zablotowicz RB (1999) Chlorophenol and nitrophenol metabolism by Sphingomonas sp UG30. J Industr Microbiol Biotechnol 23:232–241
Chauhan A, Samanta SK, Jai K (2000) Degradation of 4-nitrocatechol by Burkholderia cepacia: a plasmid-encoded novel pathway. J Appl Microbiol 88:764–772
Cho Y-G, Rhee S-K, Lee S-T (2000) Influence of phenol on biodegradation of p-nitrophenol by freely suspended and immobilized Nocardioides sp. NSP41. Biodegradation 11:21–28
Ebert S, Rieger PG, Knackmuss H-J (1999) Function of coenzyme F420 in aerobic catabolism of 2, 4, 6-trinitrophenol and 2, 4-dinitrophenol by Nocardioides simplex FJ2–1A. J Bacteriol 181:2669–2674
Greń I, Wojcieszyńska D, Guzik U, Perkosz M, Hupert-Kocurek K (2010) Enhanced biotransformation of mononitrophenols by Stenotrophomonas maltophilia KB2 in the presence of aromatic compounds of plant origin. World J Microbiol Biotechnol 26:289–295
Guzik U, Greń I, Wojcieszyńska D, Łabużek S (2009) Isolation and characterization of a novel strain of Stenotrophomonas maltophilia possesing various dioxygenases for monocyclic hydrocarbons degradation. Braz J Microbiol 40:285–291
Hegeman GD (1966) Synthesis of enzymes of the mandelate pathways by Pseudomonas putida. Synthesis of enzyme by the wild type. J Bacteriol 91:1140–1154
Kieboom J, van den Brink H, Frankena J, de Bont JAM (2001) Production of 3-nitrocatechol by oxygenase-containing bacteria: optimization of the nitrobenzene biotransformation by Nocardia S3. Appl Microbiol Biotechnol 55:290–295
Leung KT, Tresse O, Errampalli D, Lee H, Trevors JT (1997) Mineralization of p-nitrophenol by pentachlorophenol—degrading Sphingomonas ssp. FEMS Microbiol Lett 155:107–114
Leung KT, Campbel S, Gan Y, White DC (1999) The role of the Sphingomonas species UG30 pentachlorophenol-4-monooxygenaze in p-nitrophenol degradation. FEMS Microbiol Lett 173:247–253
Mendonca E, Martins A, Anselmo A (2004) Biodegradation of natural phenolic compounds as single and mixed substrates by Fussarium flocciferum. J Biotechnol 7:30–37
Moonen MJH, Synowsky SA, van den Berg WAM, Westphal AH, Heck AJR, van den Heuvel RHH, Fraaije MW, van Berkel WJH (2008) Hydroquinone dioxygenase from Pseudomonas fluorescens ACB: a novel member of the family of nonheme-iron(II)-dependent dioxygenases. J Bacteriol 190:5199–5209
Münzenberger B, Hammer E, Wray V, Schauer F, Schmidt J, Strack D (2003) Detoxification of ferulic acid by ectomycorrhizal fungi. Micorrhiza 13:117–121
Ornston LN, Stanier RY (1966) The conversion of catechol and protocatechuate and β-ketoadipate by Pseudomonas putida. J Biol Chem 241:3776–3780
Pakala SB, Gorla P, Pinjari AB, Krovidi RK, Baru R, Yanamandra M, Merrick M, Siddavattam D (2007) Biodegradation of methyl parathion and p-nitrophenol: evidence for the presence of a p-nitrophenol 2-hydroxylase in a gram-negative Serratia sp. strain DS001. Appl Microbiol Biotechnol 73:1452–1462
Peng X, Misawa N, Harayama S (2003) Isolation and characterization of thermophilic Bacilli degrading cinnamic, 4-coumaric and ferulic acid. Appl Environm Microbiol 69:1417–1427
Pitter P (1985) Correlation of microbial degradation rates with the chemical structure. Acta Hydrochim Hydrobiol 13:453–460
Qui X, Zhong Q, Li M, Bai W, Li B (2007) Biodegradation of p-nitrophenol by methyl parathion-degrading Ochrobactrum sp. B2. Int Biodet Biodegrad 59:297–301
Schenzle A, Lenke H, Fischer P, Williams PA, Knackmuss H-J (1997) Catabolism of 3-nitrophenol by Ralstonia eutropha JMP134. Appl Environ Microbiol 63:1421–1427
Singer AC, Crowley DE, Thompson IP (2003) Secondary plant metabolites in phytoremediation and biotransformation. Trends Biotechnol 21:123–130
Sparnins VL, Dagleys A (1975) Alternative routes of aromatic catabolism in Pseudomonas acidovorans and Pseudomonas putida: gallic acid as a substrate and inhibitor of dioxygenases. J Bacteriol 124:1374–1385
Stanier RY, Ingraham JL (1954) Protocatechuic acid oxidase. J Biol Chem 210:799–808
Sutherland JB, Crawford DL, Pometto AL (1981) Catabolism of substituted benzoic acids by Streptomyces. Appl Environ Microbiol 41:442–448
Vaillancourt FH, Bolin JT, Eltis LD (2006) The ins and outs of ring-cleaving dioxygenases. Crit Rev Biochem Mol Biol 41:241–267
Walia S, Shi L, Khan AA, Joshi B, Chaudry R (2002) Sequence analysis and molecular charazterization of a nitrocatechol dioxygenase gene from Pseudomonas putida. J Environ Sci Health B37:379–391
Wan N, Gu J-D, Yan Y (2007) Degradation of p-nitrophenol by Achromobacter xylosoxidans Ns isolated from wetland sediment. Int Biodeter Biodegrad 59:90–96
Zaborina O, Latus M, Eberspacher J, Golovleva LA, Lingens F (1995) Purification and characterization of 6-chlorohydroxyquinol 1, 2-dioxygenase from Streptomyces rochei 303: comparison with an analogous enzyme from Azotobacter sp. Strain GP1. J Bacteriol 177:229–234
Acknowledgments
This work was funded by Polish Ministry of Science and Higher Education funded as a research project from grant-in-aid for scientific research in years 2007-2010. Authors would like to thank Agnieszka Materna for excellent technical assistance.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wojcieszyńska, D., Guzik, U., Greń, I. et al. Induction of aromatic ring: cleavage dioxygenases in Stenotrophomonas maltophilia strain KB2 in cometabolic systems. World J Microbiol Biotechnol 27, 805–811 (2011). https://doi.org/10.1007/s11274-010-0520-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-010-0520-6