
Abstract
Full-length genome sequencing of pathogenic and attenuated (for chickens) avian coronavirus infectious bronchitis virus (IBV) strains of the same serotype was conducted to identify genetic differences between the pathotypes. Analysis of the consensus full-length genome for three different IBV serotypes (Ark, GA98, and Mass41) showed that passage in embryonated eggs, to attenuate the viruses for chickens, resulted in 34.75–43.66% of all the amino acid changes occurring in nsp 3 within a virus type, whereas changes in the spike glycoprotein, thought to be the most variable protein in IBV, ranged from 5.8 to 13.4% of all changes. The attenuated viruses did not cause any clinical signs of disease and had lower replication rates than the pathogenic viruses of the same serotype in chickens. However, both attenuated and pathogenic viruses of the same serotype replicated similarly in embryonated eggs, suggesting that mutations in nsp 3, which is involved in replication of the virus, might play an important role in the reduced replication observed in chickens leading to the attenuated phenotype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Avian coronavirus infectious bronchitis virus (IBV) causes a highly contagious upper respiratory tract disease in chickens. Live attenuated vaccines are used against the virus but the disease is difficult to control because cross-protection does not usually occur between different serotypes. The respiratory disease caused by this virus can be mild to moderate and can vary depending on the breed of chicken infected as well as the strain of the virus [1]. The virus is worldwide in distribution, and in addition to chickens, IBV has been isolated from peafowl (Galliformes) and other gamma-coronaviruses have been isolated from teal (Anas crecca), geese (Anserinae), pigeons (Columbiformes), and ducks (Anserfiformes) [2].
Coronaviruses are enveloped viruses in the order Nidovirales and are classified based on genome organization and antigenic characteristics as alpha (previously group 1), beta (previously group 2), and gamma (previously group 3)-coronaviruses with the avian coronaviruses belonging to the gamma-coronaviruses. Subgroups within each group have been reported, and recently, comparative full-length genome analysis placed a novel coronavirus from a beluga whale in subgroup 3b and three new coronavirus isolates from passerine birds in subgroup 3c [3]. Infectious bronchitis virus and related isolates as well as turkey coronavirus (TCoV) are assigned to subgroup 3a.
Coronaviruses have a single-stranded positive-sense RNA genome ranging in size from 27 to 30 kb, with a 5′ cap and a 3′ poly-A tail. Transcription occurs through a leader-primed RNA synthesis mechanism that results for IBV in six 3′ co-terminal subgenomic mRNA molecules. Four structural proteins—spike (S), envelope (E), membrane (M), and nucleocapsid (N)—along with the viral RNA make up the enveloped virion. The N protein binds to the viral RNA forming the ribonucleoprotein (RNP) complex. The E and the M protein are membrane bound proteins that play a role in virus assembly [4]. The S glycoprotein on the surface of the virus mediates attachment to the host cell, is responsible for fusion of the host cell membrane and viral envelope, and in IBV, it contains epitopes that define serotype and induce neutralizing antibodies [5]. The S glycoprotein of IBV is post-translationally cleaved into S1 and S2 subunits, and the S1 subunit is reported to have three hypervariable regions [6–8]. Mutations, insertions, deletions, and recombination in S contribute to the genetic diversity of IBV, which is recognized as different genetic or serologic types of the virus [5].
Two polyproteins 1a and 1ab account for approximately two-thirds of the viral genome-coding region and make up the replication transcription complex (RTC). The polyprotein 1ab is translated through a-1 frame-shift translation mechanism that occurs approximately 20–40% of the time [9]. The IBV 1a and 1ab polyproteins are post-translationally cleaved into 15 nonstructural proteins (nsps), nsps 2 through 16 by a papain-like protease (PLP) and the main protease (Mpro), also referred to as the 3C-like protease [10]. IBV does not have an nsp 1 equivalent found in some other coronaviruses. The PLP contained within nsp 3 is divided into PL1 and PL2 papain-like proteases. The PL1 protease, present in other coronaviruses, is truncated and nonfunctional in IBV, thus PL2 cleaves nsps 2, 3, and 4 [11]. The Mpro contained within nsp 5 cleaves nsps 5 through 16. The biological characteristics of many nsps have been previously reported [9, 10, 12–17]. In addition to nsps 3 and 5, which contain proteases PL2 and Mpro, respectively, nsps 2, 4, and 6 contain hydrophobic residues predicted to play a role in anchoring the RTC to the Golgi. Nonstructural proteins 7, 8, 9, and 10 are reported to have RNA-binding activity. Nonstructural protein 11/12 is the RNA-dependent RNA-polymerase, nsp 13 is a RNA helicase, nsp 14 is an exoribonuclease, nsp 15 is an endoribonuclease, and nsp 16 is a methyltransferase.
Adaptation of IBV to different hosts has been associated with changes in the S glycoprotein, suggesting that spike plays a key role in pathogenicity [18, 19]. However, the ectodomain of the S glycoprotein from the Beaudette strain of IBV, an attenuated laboratory strain, was replaced with an S from a pathogenic strain (Mass 41 strain) of the same serotype. This chimeric virus was shown to induce an immune response but remained nonpathogenic in chickens, indicating that the S glycoprotein is not solely responsible for pathogenicity of IBV [2, 20]. In another study, a chimeric IBV was created with the replicase genes 1a and 1ab from the attenuated Beaudette strain, and all of the structural genes from the pathogenic Mass 41 strain including the S gene. This chimeric virus was not pathogenic in chickens, indicating that the replicase proteins also appear to be determinants of IBV pathotype [2, 21]. Genetic differences reported in 1a and S between virulent and avirulent strains of IBV also led others to suggest that the replicase proteins, in addition to S, are involved in the pathotype of the virus [22].
To examine the sequence changes in individual genes associated with attenuation of IBV for chickens, we sequenced and compared the full-length consensus genomes of pathogenic IBV viruses and egg-passaged attenuated (for chickens) viruses from three different serotypes. We also examined the replication of pathogenic and attenuated viruses in embryonated eggs and in chickens to determine whether there are differences in growth rate between the pathotypes.
Materials and methods
Viruses
Pathogenic and attenuated (for chickens) IBV strains from three different serotypes were used in this study. The pathogenic Arkansas-Delmarva Poultry Industry Ark/Ark-DPI/81 and the Massachusetts strain Mass/Mass41/41 were obtained from Dr. J. Gelb, Jr. (University of Delaware, Newark, DE). The pathogenic Georgia 98 virus, GA98/CWL0470/98 virus, was isolated in our laboratory in 1998 [23]. The pathogenic viruses were propagated in 10-day-old embryonated chicken eggs (Ark/Ark-DPI/81 pass 6, Mass/Mass41/41 pass 8, and GA98/CWL0470/98 pass 8) as previously described [24]. The attenuated viruses of the same strain and serotype were obtained from Intervet and were designated Ark-attenuated (Mildvac-Ark), Mass41-attenuated (Mildvac-H), and GA98-attenuated (Mildvac-Ga-98).
Whole-genome nucleotide and deduced amino acid sequence analysis
Viral RNA extraction, RT-PCR, library construction, and sequencing were conducted as previously described [25]. Briefly, the viruses were filtered through a 0.8-μm filter then through a 0.22-μm filter (Millipore, Billerica, MA) prior to RNA extraction. Viral RNA was purified using the high pure RNA isolation kit according to the manufacturer’s recommendation (Roche Diagnostic Corporation, Foster City, CA) and re-suspended in DEPC-treated water. Reverse transcription (RT) and polymerase chain reaction (PCR) amplification were performed with the Takara RNA LA PCR kit (Takara Bio Inc., Otsu, Shiga, Japan) using a random primer and an amplification primer in a strand displacement amplification reaction following the manufacture’s protocol. The sequence of the random reverse transcription primer was 5′-AGC GGG GGT TGT CGA ATG TTT GAN NNN N-3′, and the amplification primer sequence, which is designed to anneal to the complement of the conserved region on the random primer, was 5′-AGC GGG GGT TGT CGA ATG TTT GA-3′. Both primers were obtained from Integrated DNA Technologies, Inc. (Coralville, IA). For the RT reaction, a master mix was prepared, which included MgCl2 (5 mM), 10× RNA PCR buffer (1×), dNTP mixture (1 mM), RNase inhibitor (1 units/μl), reverse transcriptase (0.25 units/μl), 5′ degenerate primer (2.5 μM), and RNA (5.75 μl/reaction) then 10 μl per sample was aliquoted in a thermocycler tube. The reaction conditions for the RT reaction were 10 min at 30°C for the primer annealing then an hour at 50°C for extension followed by a five-minute incubation at 99°C for inactivation of the enzyme and a five-minute period at 5°C. A PCR master mix—which included at the final concentrations MgCl2 (2.5 mM), 10× LA PCR Buffer (1×), sterilized distilled water (32.25 μl), Takara LA Taq (1.25U/50 μl), and 5′ primer (0.2 μM)—was prepared and 10 μl of the RT reaction was added to 40 μl of the mix. The amplification reaction consisted of a 94°C step for 2 min followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 3 min.
Ten PCR were combined for each virus and purified using the QIAquick PCR purification kit (QIAGEN, Foster City, CA) and then run on a 1% agarose gel to visualize the amplified product. The PCR products were size selected by cutting out amplicons between 500 and 1500 bp from the gel. The amplicons were purified using the QIAquick (QIAGEN) gel purification kit.
The TOPO cloning kit (Invitrogen, Life Technologies, Carlsbad CA) was used to clone the PCR products into the pCR-XL-TOPO vector according to the manufacturer’s recommendations. Then, One Shot TOPO Electrocompetent Escherichia coli cells (Invitrogen) were transformed using 30 μl of competent cells mixed with 2 μl of the ligation reaction and electroporated with settings at 20 kV and 200 Ω using a BioRad (BioRad Gene Pulser, Hercules, CA). The electroporated cells were incubated at 37°C in 480 μl of Super Optimal broth medium for 1 h on a rotary shaker. The cultures were mixed with 70% glycerol and frozen in −80°C until plated on Q-trays (Genetix, Boston, MA) containing liquid broth agar CAT#3002-032 (MP Biomedicals, LLC, Solon, Oh) with 50 μg/ml of kanamycin. The Q-trays were pre-warmed at 37°C before the entire culture (approximately 500 μl) was spread on the plates and incubated overnight at 37°C, then robotically picked with a Q-BOT (Genetix, Boston, MA).
Plasmid DNA from the libraries of cloned cDNA fragments for each virus was isolated using an alkaline lysis method modified for the 96-well format, and incorporating both Hydra and Tomtek robots (http://www.intl-pag.org/11/abstracts/P2c_P116_XI.html). Cycle sequencing reactions were performed using the BigDye™ Terminator® Cycle Sequencing kit Version 3.1 (Applied Biosystems, Foster City, CA) and MJ Research (Watertown, MA) thermocyclers. Finished reactions were filtered through Sephadex filter plates into Perkin-Elmer MicroAmp Optical 96-well plates. A 1/12-strength sequencing reaction on an ABI 3730 was used to sequence each clone from both the 5′ and 3′ ends. Each viral genome was sequenced to approximately 10× coverage. The accuracy of the sequence was ensured by generating data in both the 5′ and the 3′ directions.
Gaps and areas with less than 2× coverage were identified and specific primers were synthesized (IDT) for RT-PCR amplification and sequencing of the ambiguous areas. The RT-PCR was conducted as described above, and the reaction conditions were 42°C for 60 min, 95°C for 5 min, then 10 cycles of 94°C for 30 s, 50°C for 30 s, 68°C for 90 s, followed by 25 cycles of 94°C for 30 s, 50°C for 30 s, 68°C for 90 s + 5 s/cycle added. The final elongation step was 68°C for 7 min, and then, the reaction was cooled to 4°C. The PCR products were sequenced in both directions using the ABI Prism BigDye Terminator v3.0 (Applied Biosystems, Foster City, CA) and the specific primers that were used for amplification at a concentration of 15 ng. The amount of cDNA added to the reaction ranged from 20 to 30 ng, and the sequencing reactions were analyzed on an ABI 3730 (Applied Biosystems).
Chromatogram files and trace data were read and assembled using SeqMan Pro, and genome annotation was conducted with SeqBuilder (DNASTAR, Inc., v.8.0.2, Madison, WI). Low-quality segments and vector sequence were trimmed from the ends of each sequence and removed from further analysis. Full-length genomes were uploaded to the National Center for Biotechnology Information (NCBI) open reading frame (ORF) finder (http://www.ncbi.nlm.nih.gov/gorf/) to identify ORFs. Nucleotide and deduced amino acid alignments were generated using ClustalW, and phylogenetic trees with 1,000 bootstrap replicates were constructed in the MegAlign program (DNASTAR, Inc.). Hydrophilicity analysis using Hopp-Woods and Kyte-Doolittle were conducted with the Protean program (DNASTAR, Inc.).
Pathogenicity testing in chickens
The viruses were titrated in 10 day of incubation embryonated eggs to obtain a 50% embryo infectious dose (EID50) according to previously published procedures (24). Two-week-old chickens were given 1 × 104 EID50 of virus in 100 μl of PBS equally divided intraocularly and intranasally. Due to isolator availability, different numbers of birds were tested for each virus. Six birds were given Ark/Ark-DPI/81, 20 birds were given Ark attenuated, 10 birds each were given Mass/Mass41/41, Mass attenuated, and GA98 attenuated, and 12 birds were given GA98/CWL0470/98. Each of the negative control groups consisted of 10 birds. Clinical signs and lesions were recorded, and tracheal swabs were collected and placed in 1 ml of ice-cold PBS (pH 7.4) at 5 days post-exposure [26]. The presence of virus in the tracheal swab supernatant was determined by quantitative real-time RT-PCR [27]. Tracheas were collected in 10% neutral buffered formalin, routinely processed into paraffin, and 5-μm sections were cut for hematoxylin and eosin staining. Epithelial hyperplasia, lymphocyte infiltration, and the severity of epithelial deciliation were scored for each trachea with 1 being normal and 4 being severe [28].
Virus replication in embryonated chicken eggs and chicks
As a measure of adaptation, we examined the growth of the Ark/Ark-DPI/81, Ark attenuated, Mass/Mass41/41 and Mass41-attenuated in embryonated eggs and chicks. Because of limited isolator availability, we did not include the GA 98 viruses in this experiment. Virus growth in embryonated eggs was examined by inoculating 1 × 105 EID50 of each virus into 30 eggs at 10 days of incubation via the chorioallantoic route. For each virus, allantoic fluid was harvested from five eggs at 12, 24, 36, 48, 72, and 96 h after inoculation. The amount of virus present in fresh (not previously frozen) allantoic fluid was determined by quantitative real-time RT-PCR [27].
To examine virus growth in chicks, 1 × 105 EID50 of each virus was inoculated into 30 specific pathogen-free chicks at 1 day of age via the ocular/nasal route. Tracheal swabs were collected from each of five birds at 12, 24, 36, 48, 72, and 96 h after inoculation and placed in 1 ml of ice-cold PBS (pH 7.4). Once the birds were swabbed, they were removed from the study. The amount of virus present in the fresh (not previously frozen) tracheal swab supernatant was determined by quantitative real-time RT-PCR [27].
GenBank accession numbers
Sequences generated in this study were submitted to GenBank and assigned the following accession numbers: Ark/Ark-DPI/81 (GQ504720); Ark-attenuated (GQ504721); GA98/CWL0470/98 (GQ504722); GA98-attenuated (GQ504723); Mass/Mass41/41 (GQ504724); and Mass41-attenuated (GQ504725).
Results
Full-length genome sequencing and analysis
The consensus sequence of the full-length genomes of Ark/Ark-DPI/81, Ark-attenuated, GA98/CWL0470/98, GA98-attenuated, Mass/Mass41/41, and Mass41-attenuated were sequenced, and the genome sizes were found to be 27,651 nt, 27,620 nt, 27,638 nt, 27,621 nt, 27,475 nt, and 27,451 nt, respectively. The genome organization consisting of a 5′ untranslated region (UTR), polyproteins 1a and 1ab, spike, 3a, 3b, envelope, membrane, 4b, 5a, 5b, nucleocapsid, and 3′ UTR was the same for all six viruses (Table 1). Gene locations for the nsps in ORF 1a and 1ab are shown in Table 2. The 4b protein, previously recognized in M41 [21], is 94 amino acids long and located downstream from the membrane protein in all the viruses sequenced. A BLAST search was conducted, and we found the protein to have 96% sequence identity with the 4b protein from TCoV (TCoV, GenBank accession number EU022526.1). In addition, a 6b protein downstream of the nucleocapsid protein was similar to the predicted 6b ORF reported for TCoV (GenBank accession number EU022526.1). The 6b ORF was identified in the Ark and GA98 viruses but not in the Mass 41 viruses.
Alignment and phylogenetic analysis of the full-length genomes show that Ark/Ark-DPI/81 has 99.1% sequence identity with Ark-attenuated, GA98/CWL0470/98 has 97.1% sequence identity with GA98-attenuated, and Mass/Mass41/41 has 92.3% sequence similarity with Mass41-attenuated (Fig. 1).

Phylogenetic relationship of full-length genomes. Phylogenetic tree showing nucleotide sequence relatedness computed using neighbor-joining and the Nei-Gojobori method. The bootstrap consensus subtree was constructed from 1,000 replicates (percentage of replicate trees in which associated strains clustered together are presented at nodes). The nucleotide sequences were aligned with ClustalW (DNASTAR, Inc.), and the nucleotide substitutions (×100) are shown in the scale at the bottom of the figure
Nucleotide and amino acid sequence differences were identified between each of the pathogenic and attenuated viruses (Table 3). When the genome sequences are compared, there are 249 nucleotide (nt) changes resulting in 62 amino acid changes in the coding regions between the Ark viruses, 629 nt changes resulting in 268 amino acid changes between the GA98 viruses, and 1,805 nt changes resulting in 462 amino acid changes between the Mass 41 viruses (see Table 3 and Supplemental data Tables 5 and 6).
The size of the 5′ UTR is 528 nt for all the viruses (Table 1). The number of nt differences between the Ark viruses for the 5′ UTR was 25 with a 95.6% identity. The GA98 viruses have 6 nt differences with 98.9% identity, and the Mass viruses have 12 nt differences with 98.3% identity in the 5′ UTR (Table 3). The leader junction sequence, nucleotides 57-64 (5′-CTTAACAA), were found to be identical for the Ark and Mass viruses, whereas the GA98/CWL0470/98 pathogenic virus leader junction sequence is 5′-CTCAACAA and the GA98 attenuated virus sequence is 5′-CTTTACAA. The transcriptional regulatory sequences (TRS) were identical in all of the viruses and were 5′-CTGAACAA-3′ for mRNAs 2 and 3, and 5′-CTTAACAA-3′ for mRNAs 4, 5, and 6.
The size of the 3′ UTRs is 273 nt for Ark/Ark-DPI/81 pathogenic and Ark-attenuated, 276 nt for GA98/CWL0470/98, 244 nt for GA98-attenuated, and 322 nt for Mass/Mass41/41, and Mass41-attenuated (Table 1). The number of nt differences within the 3′ UTRs for the Ark viruses is 6 with 98.5% identity. The GA98 viruses have 9 nt differences resulting in 97.1% identity, and the Mass viruses have 2 nt differences with 99.4% identity within the 3′ UTRs (Table 3). The 3′ UTRs contain the S2M motif, which is 41 nt long with a sequence identity of 92.7% or higher between the six viruses.
Protein alignments between pathotypes
Analysis of the locations and number of sequence differences between pathogenic and attenuated viruses of the same serotype for individual nsps in polyproteins 1a and 1a/b (Table 3) shows that nsp 3 has the highest number of amino acid differences among all the nsps. In addition, nsp 3 has the greatest number of differences when coding regions across the entire genome are compared. A schematic representation of nsp 3 and number of amino acid changes in each domain is presented in Fig. 2. The nsp 3 ORF has 43.66% of all amino acid differences observed between Ark/Ark-DPI/81 and Ark-attenuated (including a ten amino acid deletion in the attenuated virus at positions 789–798), 34.75% of all amino acid differences observed between GA98/CWL0470/98 and GA98-attenuated (including an eight amino acid deletion in the pathogenic virus at positions 901–908 and a three amino acid deletion in the pathogenic virus at positions 950–952), and 37.08% of all amino acid differences observed between Mass/Mass41/41 and Mass-attenuated (including a ten amino acid deletion in the attenuated virus at positions 797–806). These changes represent 1.96, 5.18, and 11.06 differences per 100 amino acids within nsp 3 for Ark, GA98 and Mass 41, respectively. We also found a virus subpopulation within the Ark/Ark-DPI/81 strain, which had a ten amino acid deletion in nsp 3 at positions 789–798 similar to the Ark-attenuated virus. The catalytic triad of the PL2 protease, amino acids Cys623, Hys786, Asp802 [29] was conserved among all of the viruses, and a hydrophobicity plot of nsp 3 predicted fours transmembrane regions between amino acids 1,000 and 1,300 (data not shown). The fewest amino acid changes for the nsps between pathogenic and attenuated viruses within a serotype are found in nsps 7–10, which are the RNA-binding proteins.

Schematic representation of nsp 3 with the number of amino acid changes (relative positions indicated by tick marks, vertical black, blue and red lines) and deletions (triangles) between pathogenic and attenuated viruses indicated below. Domain positions are based on the IBV Beaudette nsp 3 sequence (Acc# P27920) alignment from Ziebuhr et al. [11] and include the acidic acid domain, the papain-like protease 1 (PL1) domain, the ADP-ribose binding protein (ADRP), the papain-like protease 2 (PL2), and the Y domain. The black line represents an unknown region between the PL2 and Y domains (Color figure online)
The polyprotein 1ab-1 frame-shift slippery sequence (5′-UUUAAAC) is conserved among all six viruses but the location was found at nt 12,328 for Ark/Ark-DPI/81, nt 12,298 for Ark-attenuated, nt 12,321 for GA98/CWL0470/98, nt 12,360 for GA98-attenuated, nt 12,391 for Mass/Mass41/41 and nt 12,327 for Mass41-attenuated.
The percent amino acid identity for the S glycoprotein is 97.8% for Ark viruses, 96.6% for GA98 viruses, and 97.2% for Mass 41 viruses (Fig. 3). The number of amino acid differences within the S glycoprotein between pathogenic and attenuated viruses are 7, 33, and 27 for Ark, GA98, and Mass 41, respectively (Table 3). The S glycoprotein for the Ark viruses had 9.86% (0.60 differences/100 amino acids) of all amino acid differences, which is the third most variable ORF in the entire genome after nsp 3 and 12. For the GA98 viruses, the S glycoprotein has 13.36% (2.82 differences/100 amino acids) of all amino acid differences, which is the third most variable ORF in the entire genome after nsp 3 and ORF 6b. The S glycoprotein for the Mass 41 viruses has 5.77% of all amino acid differences (2.31 differences/100 amino acids), which was the fourth most variable ORF in the entire genome after nsp 3, 2, and 4. ORF 3b has the fewest number of differences with no differences observed between the Ark viruses, whereas the GA98 and Mass viruses each have one amino acid difference. For ORF 4b, no amino acid differences are observed for the Ark viruses, 16 amino acid differences are observed between the GA98 viruses, and 17 amino acid differences are observed between the Mass 41 viruses. The Ark virus 6b proteins have only one amino acid mutation and are 99.9% similar to each other, whereas the GA98 virus 6b proteins have 43 amino acid mutations, 3 amino acid deletions, and 1 substitution and are only 41.9% similar. Because this protein has not been previously recognized in IBV, a nucleotide BLAST search rather than an amino acid search was conducted and showed that the GA98/CWL0470/98 virus has 98% identity with Mass H120 (FJ888351) and the GA98-attenuated virus has 98% identity with Ark-DPI (EU418976). To determine whether the GA98-attenuated virus 6b sequence was a subpopulation within the GA98/CWL0470/98 virus, two forward primers (GA98A #1 5′-TCACGCTCAAGTTCAAGACCTG-3′, and GA98A #3 5′-CAGCTTTAGGTGAGAATGAACT-3′) and two reverse primers (GA98A #2 5′-TACGATAAAACAAACTAATGAGAA-3′, and GA98A #4 5′-TTGATAGGAAAGCACAGAAATAG-3′) specific for the GA98-attenuated 6b sequence were used in combination in an RT-PCR assay, but no amplicons were observed.

Phylogenetic relationships of the spike protein. Phylogenetic tree showing amino acid sequence relatedness computed using neighbor-joining and the Nei-Gojobori method. The bootstrap consensus subtree was constructed from 1,000 replicates (percentage of replicate trees in which associated strains clustered together are presented at nodes). The amino acid sequences were aligned with ClustalW (DNASTAR, Inc.), and the amino acid substitutions (×100) are shown in the scale at the bottom of the figure
Pathogenicity testing in chicks
The data on pathogenicity of the viruses in 2-week-old SPF chicks are presented in Table 4. At 5 days post-inoculation, clinical signs, virus detection and tracheal lesions were observed in each of the groups given the pathogenic viruses. None of the birds given attenuated viruses had clinical signs, tracheal lesions, or virus detected in the trachea with the exception of 1 of 20 birds given the Ark-attenuated virus, which had mild clinical signs and virus detected in the trachea. The negative control group did not have clinical signs, tracheal lesions, or virus detected in the trachea.
Virus replication in embryonated eggs and in chickens
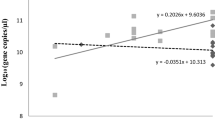
Due to isolator availability, we were only able to test 2 pathogenic and 2 attenuated viruses (Ark and Mass) in chickens. The average genome copy number, a measure of virus replication, was calculated by quantitative real-time RT-PCR, and no statistical difference in the amount of virus in allantoic fluid harvested from inoculated embryonated eggs was observed between the Ark/Ark-DPI/81 and Ark-attenuated viruses at any of the time points tested (Fig. 4a). The Ark-attenuated virus, which is adapted to embryonated eggs, only killed 5 of 35 embryos by 96 h post-inoculation, whereas Ark/Ark-DPI/81 did not kill any of the embryos (0/35), and all of the surviving embryos for both viruses had lesions consistent with IBV replication by 96 h post-inoculation. For Mass/Mass41/41 and Mass41-attenuated viruses, a statistical difference (P ≤ 0.1) in the amount of virus in embryonated eggs was only observed at 24 h post-inoculation but not at any other sample time (Fig. 4a). The Mass/Mass41/41 virus killed 3 of 35 embryos, and the Mass-attenuated virus killed 1 of 35 embryos by 96 h post-inoculation. Lesions were observed at 96 h post-inoculation in surviving embryos inoculated with the Mass viruses. Embryos examined at all the other time points were alive, and no lesions were observed.

Virus replication expressed as the log10 of viral genome copies. a Virus replication in 10-day-old embryonated eggs. Eggs were inoculated with 1 × 105 50% embryo infectious doses of virus. Time 0 represents allantoic fluid collected from eggs immediately after inoculation. Numbers are the average viral genome copies (n = 5 except where indicated) calculated from the standard curve for the real-time RT-PCR test (Callison et al. [27]). In the Ark-attenuated group, all the embryos died prior to the 96 h time point. b Virus replication in 1-day-old chicks. Chicks were inoculated intranasally/intraocularly with 1 × 105 50% embryo infectious doses of virus. Numbers are the average viral genome copies (n = 5) calculated from the standard curve for the real-time RT-PCR test (Callison et al. [27]). *Indicates that the numbers are statistically different P ≤ 0.1 between Mass/Mass41/41 pathogenic and Mass41-attenuated viruses, and dagger indicates that the numbers are statistically different P ≤ 0.1 between Ark/Ark-DPI/81 pathogenic and Ark-attenuated viruses
Chicks inoculated with virus at 1 day of age showed statistical differences (P ≤ 0.1) in the amount of virus detected in the trachea between the Ark/Ark-DPI/81 and Ark-attenuated viruses at 24, 48, 72, and 96 h post-inoculation with the pathogenic Ark/Ark-DPI/81 having the higher amount of virus at each of the sample times (Fig. 4b). Although not statistically different, the chicks given the pathogenic Ark/Ark-DPI/81 virus also had more virus detected in the trachea than the chicks given the Ark-attenuated virus at 12 and 36 h post-inoculation. Statistical differences (P ≤ 0.1) were observed in the amount of virus detected in the trachea of chicks given the Mass viruses at 24, 36, and 48 h post-inoculation with the pathogenic Mass/Mass41/41 virus having the higher amount of virus. Although not statistically different, the chicks given the pathogenic Mass/Mass41/41 virus also had more virus detected in the trachea than the chicks given the Mass-attenuated virus at 12, 72, and 96 h post-inoculation.
Discussion
Many studies have examined sequence changes in the structural proteins of IBV and found that most of the changes associated with adaptation to a particular host or with a particular virus pathotype occur in the spike glycoprotein [18, 19, 30]. But only a few studies have examined changes across the entire genome associated with biological characteristics of the virus [22, 31]. Ammayappan et al. [22] found a total of 17 amino acid changes between the genomes of Ark DPI 11, a pathogenic virus and Ark DPI 101 an attenuated virus, with four amino changes in nsp 3 and six amino acid changes in the S1 glycoprotein. Based on that data, it was suggested that changes in the replicase sequence in addition to structural proteins might play a role in pathogenicity. Fang et al. [31] found 53.06% of all amino acid substitutions across the entire genome were located in the spike glycoprotein following adaptation of an attenuated avian coronavirus to primate cells, suggesting that spike plays a role in host adaptation.
In this study, we analyzed the consensus full-length genome for the pathogenic and attenuated viruses of three different IBV types and showed that within a virus type, 34.75 to 43.66% of all the amino acid changes between the pathotypes occurred in nsp 3, whereas changes in spike ranged from 5.8 to 13.4% of all changes. It should be noted, however, that spike had the highest number of differences between different serotypes of the virus, which is consistent with previous reports [5–8]. A high percentage of differences between pathogenic and attenuated viruses within a serotype in nsp 3 suggests this region plays a key role in pathogenicity. The nsp 3 is a complex protein with multiple domains making it an attractive target for antiviral drug design [9, 32]. It is approximately 1,600 amino acid residues in length and consists of an acidic domain, an ADP-ribose 1 phosphatase, the PL2 protease (a deubiquitinating protease), Y and transmembrane domains. The acidic domain is of unknown function, however; there is some evidence that it possesses nucleic acid binding activity because it is consistently co-purified with single-stranded RNA [33]. Previous studies with other organisms indicate that electrostatic interactions from this type of domain play a key role in ligand binding [34]. Influenza A viruses also contain a polymerase acidic protein (PA) that is required for the transcription and replication activity of the viral polymerase [34]. Differences between pathogenic and attenuated IBV strains within a serotype, including deletions in Ark and Mass41 viruses, were in and around the acidic domain within nsp 3 (Fig. 2). Thus, it is likely that the acidic domain plays a role in attenuation in chickens but the exact function(s) of the amino acids in this domain is unclear. It was interesting that we observed an eight and a three amino acid deletion in the pathogenic virus GA98/CWL0470/98 at positions 901–908 and 950–952, respectively, compared to the GA98-attenuated virus. Since sequence insertions are not likely to occur during the attenuation process, the GA98-attenuated virus possibly represents a minor undetected subpopulation in the pathogenic virus, which was selected by passage in embryonated eggs.
The ADP-ribose-1 phosphatase domain within nsp 3 is relatively conserved between the pathogenic and attenuated strains. This domain has been shown in the Beaudette laboratory attenuated strain of IBV not to function as an ADP-ribose binding protein [35]. However, the triple glycine sequence that forms part of the ADP-ribose binding site (Gly47-Gly48-Gly49), which was not conserved in Beaudette, is conserved in all of the viruses sequenced herein [35]. This suggests that the ADP-ribose-1 protein may be functional in the pathogenic and attenuated IBV viruses and is consistent with the results of the Mass 41-X domain as reported by Xu et al. [14]. The ADP-ribose-1 phosphatase may be important in pathogenicity of IBV because it has been shown to play a role in ADP ribosylation, a post-translational protein modification involved in DNA damage repair and transcription regulation [14]. In addition, it was reported that the ADP-ribose-1 is dispensable for viral replication in tissue culture, suggesting that this domain is involved in regulation of viral replication rather than the actual replication process [36].
The PL2 domain is a papain-like protease that is responsible for the cleavage of the nsp 2/3 and 3/4 sites. Most coronaviruses have two papain-like proteases; however, in IBV the PL1 protease is truncated and is nonfunctional [16]. The structure of the PL2 protease domain was determined to be a “thumb-palm-finger” motif [37]. This domain has also been shown to be a potent IFN antagonist by inhibiting the phosphorylation and nuclear translocation of interferon regulatory factor 3 (IRF-3) causing a disruption in the activation of the type I IFN response through Toll-like receptor 3 (TLR 3) or retinoic acid-inducible gene I (RIG-I) [38]. Although the catalytic triad of the PL2 protease is conserved, amino acid changes between the pathogenic and attenuated viruses are observed in the PL2 protease, which could affect the efficiency of this IFN antagonist leading to altered viral replication in the cell. The disruption of IFN signaling has been shown in many viral infections, including SARS-CoV, dengue virus, and paramyxoviruses [39–41]. The IBV PL2 viral protease was also shown to have characteristics similar to ubiquitin-specific proteases [42]. Deubuquitinating proteases, which remove ubiquitin from proteins that have been marked by cellular mechanisms for ATP-dependent degradation, could be a potential mechanism by which the virus can alter the cellular environment favoring replication.
The Y domain, containing transmembrane domains at its N-terminus, was originally described by Gorbalenya et al. [43] and has been predicted to consist of three domains Y1, Y2, and Y3, which may act together to form an enzymatic function [32]. The transmembrane domain is inserted into the endoplasmic reticulum (ER) membrane co-translationally and plays an important scaffolding role for the replication transcription complex [9]. Recently, it was shown that three transmembrane domains were predicted for the SARS-CoV nsp 3 but only two were found to span the ER membrane orienting the protease domain of nsp 3 on the cytoplasmic side where viral replication occurs [13, 15]. In murine hepatitis virus (MHV), five transmembrane domains were predicted but only two domains were found to span the membrane, also locating the protease domain on the cytoplasm side [13, 15]. Our sequence data for IBV predicts four transmembrane domains within nsp 3. Assuming the protease domain is located on the cytoplasm side of the membrane, we predict that either two or all four transmembrane domains would be used.
A chimera IBV containing the replicase genes 1a and 1a/b from the attenuated Beaudette strain and the structural genes from the pathogenic Mass 41 strain was not pathogenic in chickens, indicating that the replicase proteins appear to be determinants of pathotype in IBV [2, 21]. Our data strongly support these studies and further indicate that changes in nsp 3 play a key role in IBV pathotype. It should also be emphasized that pathogenicity in avian coronaviruses is likely polygenic, since we and others [22] observed amino acid substitutions in other viral proteins including spike. The 6b ORF detected in TCoV (GenBank accession numbers ACB87503 and ACB87504) is identified in Ark and GA98 viruses herein. Only one amino acid difference was observed between the Ark viruses, but 43 differences as well as 3 amino acid deletions and 1 insertion are observed between GA98 viruses. An attempt to identify a subpopulation in the GA98/CWL0470/98 pathogenic virus with the GA98-attenuated gene 6b was unsuccessful. It is not clear why gene 6b is so variable between the GA98 viruses but it appears recombination rather than mutations over time may have played a role. A nucleotide blast analysis indicated that the GA98/CWL040/98 virus was 98% similar to Mass H120 a vaccine virus and the GA98-attenuated virus was 98% similar to Ark-DPI a pathogenic virus, suggesting an origin for those genes. Nonetheless, assuming the 6b ORF is expressed, it apparently does not play a role in defining pathotype.
Interestingly, we find differences between pathogenic and attenuated viruses in the 5′ and 3′ UTRs. The 5′ and 3′ UTRs play key roles in transcription and replication of coronaviruses [44]. However, the differences between the Ark and Mass viruses, which are 25 nt and 12 nt, respectively, for the 5′ UTR, and 6 nt and 2 nt, respectively, for the 3′ UTR did not appear to affect replication as determined in embryonated eggs. The TRS sequences for generation of the subgenomic mRNAs were identical in all of the viruses; however, the leader junction sequences were different for GA98 viruses. Different leader junction sequences could be important for attenuation since efficiency of subgenomic mRNA production would affect growth of the virus [45].
Differences are observed in the amount of virus detected in chickens given viruses with different pathotypes. When the same amount of virus was administered, birds given the attenuated virus compared to birds given the homologous pathogenic virus had less virus detected in the trachea at all sampling times and the difference was statistically significant for most of the time points. Thus, it appears that the amount of IBV replication in the trachea correlates with the ability of the virus to cause disease in chickens. Attachment and entry, and replication of the attenuated virus (for chickens) were not impaired because it grew to the same titer (with the exception of one time point) as the pathogenic virus in 10-day-old embryonated eggs. Inefficient attachment and entry into chicken host cells in vivo could be due to changes in spike. And decreased replication of the attenuated viruses could be due to the inability of the virus to overcome some as yet unidentified innate defense mechanism(s) in chicken cells that is not present in embryonic cells. Domains within nsp 3 associated with the deubiquitinating protease or IFN antagonists are likely candidates for further research.
In summary, we find that most changes associated with attenuation of IBV for chickens are located within nsp 3 and that the attenuated viruses have reduced replication in chickens but not in 10-day-old embryonated eggs. Changes in spike suggest that attachment and entry may have been affected and changes in nsp 3 suggest that the attenuated virus lost the ability to overcome some innate host cell defense mechanism in the mature chicken cell. The exact mechanism(s) surrounding the interaction of virus and host processes affecting virus replication have yet to be determined for IBV, but identifying the sequence changes in the virus responsible for reduced replication and attenuation is an important step in elucidating those mechanisms. Finally, changes observed in nsp 3 and spike as well as in other viral genes support the polygenic nature of pathogenicity in avian coronaviruses.
Abbreviations
- IBV:
-
Infectious bronchitis virus
- RTC:
-
Replication transcription complex
- PLP:
-
Papain-like protease
References
S.P. Mondal, C.J. Cardona, Virology 324, 238–248 (2004)
D. Cavanagh, Vet. Res. 38, 281–297 (2007)
P.C. Woo, S.K. Lau, C.S. Lam, K.K. Lai, Y. Huang, P. Lee, G.S. Luk, K.C. Dyrting, K.H. Chan, K.Y. Yuen, J. Virol. 83, 908–917 (2009)
K.P. Lim, D.X. Liu, J. Biol. Chem. 276, 17515–17523 (2001)
D. Cavanagh, J. Gelb Jr, Infectioius bronchitis, in Diseases of poultry, ed. by Y.M. Saif, A.M. Fadley, J.R. Glisson, L.R. McDougald, L.K. Nolan, D.E. Swayne (Blackwell Publishing, Ames, 2008), pp. 117–135
G. Koch, A. Kant, J.K.A. Cook, D. Cavanagh, Epitopes of neutralizing antibodies are located within three regions of the S1 spike protein of infectious bronchitis virus, in II. International symposium on infectious bronchitis, ed. by E.F. Kaleta, U. Heffels-Redmann (World Veterinary Poultry Association, Rauischholzhausen, 1991), pp. 154–160
A. Kant, G. Koch, D.J. van Roozelaar, J.G. Kusters, F.A. Poelwijk, B.A. van der Zeijst, J. Gen. Virol. 73, 591–596 (1992)
K.M. Moore, M.W. Jackwood, D.A. Hilt, Arch. Virol. 142, 2249–2256 (1997)
I. Imbert, E.J. Snijder, M. Dimitrova, J.C. Guillemot, P. Lecine, B. Canard, Virus Res. 133, 136–148 (2008)
M.J. van Hemert, S.H. van den Worm, K. Knoops, A.M. Mommaas, A.E. Gorbalenya, E.J. Snijder, PLoS Pathol. 4, e1000054 (2008)
J. Ziebuhr, V. Thiel, A.E. Gorbalenya, J. Biol. Chem. 276, 33220–33232 (2001)
S.G. Fang, H. Shen, J. Wang, F.P. Tay, D.X. Liu, Virology 379, 175–180 (2008)
M. Oostra, E.G. te Lintelo, M. Deijs, M.H. Verheije, P.J. Rottier, C.A. de Haan, J. Virol. 81, 12323–12336 (2007)
L. Li, C. Xue, F. Chen, J. Qin, Q. Xie, Y. Bi, Y. Cao, Vet. Microbiol. 143, 145–154 (2010)
M. Oostra, M.C. Hagemeijer, M. van Gent, C.P. Bekker, E.G. te Lintelo, P.J. Rottier, C.A. de Haan, J. Virol. 82, 12392–12405 (2008)
R.L. Graham, J.S. Sparks, L.D. Eckerle, A.C. Sims, M.R. Denison, Virus Res. 133, 88–100 (2008)
M.M. Cao, H. Ren, P. Zhao, W. Pan, Q.L. Chen, Z.T. Qi, J. Virol. Methods 157, 168–174 (2009)
Y.P. Huang, C.H. Wang, Avian Pathol 36, 59–67 (2007)
S. Liu, Z. Han, J. Chen, X. Liu, Y. Shao, X. Kong, G. Tong, J. Rong, Avian Pathol 36, 231–234 (2007)
T. Hodgson, R. Casais, B. Dove, P. Britton, D. Cavanagh, J. Virol. 78, 13804–13811 (2004)
M. Armesto, D. Cavanagh, P. Britton, PLoS One 4, e7384 (2009)
A. Ammayappan, C. Upadhyay, J. Gelb Jr, V.N. Vakharia, Arch. Virol. 154, 495–499 (2009)
C.W. Lee, M.W. Jackwood, Virus Res. 80, 33–39 (2001)
J.J. Gelb, M.W. Jackwood, Infectious bronchitis, in A laboratory manual for the isolation, identification, and characterization of Avian pathogens, ed. by L. Dufour-Zavala, D.E. Swayne, J.R. Glisson, J.E. Pearson, W.M. Reed, M.W. Jackwood, P. Woolcock (American Association of Avian Pathologists, Kennett Square, 2008), pp. 146–149
M.W. Jackwood, T.O. Boynton, D.A. Hilt, E.T. McKinley, J.C. Kissinger, A.H. Paterson, J. Robertson, C. Lemke, A.W. McCall, S.M. Williams, J.W. Jackwood, L.A. Byrd, Virology 398, 98–108 (2010)
USDA, Title 9, Code of Federal regulations, standard requirements for IBV vaccines. Animal and Plant Health Inspection Service, US National Archives and Records Administration, College Park, MD (1999)
S.A. Callison, D.A. Hilt, T.O. Boynton, B.F. Sample, R. Robison, D.E. Swayne, M.W. Jackwood, J. Virol. Methods 138, 60–65 (2006)
M.W. Jackwood, D.A. Hilt, T.P. Brown, Avian Dis. 47, 627–632 (2003)
K. Ratia, K.S. Saikatendu, B.D. Santarsiero, N. Barretto, S.C. Baker, R.C. Stevens, A.D. Mesecar, Proc. Natl. Acad. Sci. USA 103, 5717–5722 (2006)
D. Cavanagh, J.P. Picault, R. Gough, M. Hess, K. Mawditt, P. Britton, Avian Pathol. 34, 20–25 (2005)
S.G. Fang, S. Shen, F.P. Tay, D.X. Liu, Biochem. Biophys. Res. Commun. 336, 417–423 (2005)
B.W. Neuman, J.S. Joseph, K.S. Saikatendu, P. Serrano, A. Chatterjee, M.A. Johnson, L. Liao, J.P. Klaus, J.R. Yates III, K. Wuthrich, R.C. Stevens, M.J. Buchmeier, P. Kuhn, J. Virol. 82, 5279–5294 (2008)
P. Serrano, M.A. Johnson, M.S. Almeida, R. Horst, T. Herrmann, J.S. Joseph, B.W. Neuman, V. Subramanian, K.S. Saikatendu, M.J. Buchmeier, R.C. Stevens, P. Kuhn, K. Wuthrich, J. Virol. 81, 12049–12060 (2007)
T.S. Guu, L. Dong, P. Wittung-Stafshede, Y.J. Tao, Virology 379, 135–142 (2008)
Y. Piotrowski, G. Hansen, A.L. Boomaars-van der Zanden, E.J. Snijder, A.E. Gorbalenya, R. Hilgenfeld, Protein Sci. 18, 6–16 (2009)
A. Putics, J. Slaby, W. Filipowicz, A.E. Gorbalenya, J. Ziebuhr, Adv. Exp. Med. Biol. 581, 93–96 (2006)
M. Bartlam, Y. Xu, Z. Rao, J. Struct. Funct. Genom. 8, 85–97 (2007)
D. Zheng, G. Chen, B. Guo, G. Cheng, H. Tang, Cell Res. 18, 1105–1113 (2008)
G.A. Versteeg, P.J. Bredenbeek, S.H. van den Worm, W.J. Spaan, Virology 361, 18–26 (2007)
J.R. Rodriguez-Madoz, D. Bernal-Rubio, D. Kaminski, K. Boyd, A. Fernandez-Sesma, J. Virol. 84, 4845–4850 (2010)
A. Ramachandran, C.M. Horvath, J. Interferon Cytokine Res. 29, 531–537 (2009)
H.A. Lindner, N. Fotouhi-Ardakani, V. Lytvyn, P. Lachance, T. Sulea, R. Menard, J. Virol. 79, 15199–15208 (2005)
A.E. Gorbalenya, E.V. Koonin, M.M. Lai, FEBS Lett. 288, 201–205 (1991)
P. Huang, M.M. Lai, J. Virol. 75, 5009–5017 (2001)
A.O. Pasternak, W.J. Spaan, E.J. Snijder, J. Gen. Virol. 87, 1403–1421 (2006)
Acknowledgments
This work was supported by USDA, CSREES award number 2007-35600-17786. The authors appreciate the assistance that was provided by Lauren Byrd, Carey Stewart, and Joshua Jackwood in conducting these studies.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Phillips, J.E., Jackwood, M.W., McKinley, E.T. et al. Changes in nonstructural protein 3 are associated with attenuation in avian coronavirus infectious bronchitis virus. Virus Genes 44, 63–74 (2012). https://doi.org/10.1007/s11262-011-0668-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-011-0668-7