
Abstract
To investigate the molecular epidemiology of Taiwanese Echovirus 30 (E-30) strains, we analyzed the 876 bp sequence of the VP1 gene from 32 Taiwanese strains isolated in 1988–2008, 498 reference sequences, and one Echovirus 21 strain as the out-group. Phylogenetic analysis detected six E-30 genotypes (designated GI-GVI) that had circulated globally during the past five decades. The genotypes varied widely in geographic distribution and circulation half-life. The GI, GII, and GV were ancient genotypes in which the first strains emerged in the 1950s. The GIII was a reemerging genotype, in which strains had first appeared in Colombia in 1995 before reemerging in the New Independent States (NIS) in 2003. The GIV, an emerging genotype that recently appeared in Asia in 2003, was closely related to the ancient genotypes. The GVI was the circulating genotype, which included eight clusters (A–H) that had circulated since 1967. No GVI-A, C, D, or E strains have been identified during the past 10 years. The GVI-B first appeared in China in 1984 and later in Russia and Asia in the 2000s. The GVI-F, G, and H strains, which comprised the prevalent clusters, had been dominant in Asia Pacific area, globally, and Europe, respectively. Taiwanese strains were classified into GVI-D (1988–1989), GVI-F (1993–2004), and GVI-G (1993–2008). The quiescence period of E-30 is longer in Taiwan (5–8 years) than in other countries (3–5 years).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Echovirus 30 (E-30) is a member of the human enterovirus B (HEV-B) species in the Enterovirus genus, Picornaviridae family [1]. This nonenveloped virus remains stable in highly fluctuating environmental conditions, including salinity, pH, and temperature. Because HEV can be secreted from the respiratory tract and from stool for several weeks after infection, it poses a significant threat of transmission, infection, and even epidemic outbreaks [2]. Of all cases of viral aseptic meningitis in children, echovirus accounts for 80–90% [3] while E-30 is one of the most frequently isolated HEVs in aseptic meningitis (AM) cases. Recent increases in E-30 activity associated with recurrent epidemics of AM have been reported worldwide in the past two decades, e.g., Taiwan [4, 5], Japan [6], China [7], US [8, 9], Brazil [10], Spain [11], Germany [12], France [13–15], the European continent [8, 16], and Russia, New Independent States [17].
According to the Centers for Disease Control (CDC)—Taiwan Surveillance System, which was established in 1999, E-30 is among the most common HEV serotypes in Taiwan and has the highest (17.8%) rate of presenting signs of meningitis. Annual isolation rates vary widely from 12.1 and 4.1% observed in 2001 and 2007, respectively, to the very low or nil infection rates observed in intervening years [18]. Although the cyclic pattern of E-30 outbreaks in Taiwan has important public health implications, only the 2001 outbreak has been reported [4, 5]. A local HEV surveillance program was implemented by Kaohsiung Medical University Hospital, Taiwan in 1981. Since then, three E-30 outbreaks (1988, 1993, and 2001) have occurred in southern Taiwan. Until now, the association between outbreaks of E-30 and its antigenic changes due to virus evolution has not been reported.
The VP1 gene comprises the major serotype-specific neutralization epitopes. Since molecular typing based on VP1 gene correlates well with the classical HEV serotype [19], this study analyzed the VP1 gene to elucidate the intra-typical genetic variation among circulating E-30 Taiwanese isolates over a recent 21-year period as well as trends in the circulation of E-30 isolates worldwide.
Materials and methods
Viruses
Thirty-two Taiwanese strains of E-30 isolated in 1988–2008 were obtained from two medical centers and from CDC-Taiwan. All viral strains were confirmed by indirect fluorescence antibody test using the E-30 monoclonal antibody (Chemicon, Cat. No. 3315, Temecula, CA). After amplification in human rhabdomyosarcoma (RD) cell with Eagle minimum essential medium supplemented with 2% fetal bovine serum, the viruses were centrifuged and then stored at −80°C.
Viral RNA extraction, RT-PCR, and sequencing
Viral RNA was extracted using QIAmp viral RNA purification kit according to manufacturer’s instructions (Qiagen Chatsworth, CA). Reverse transcription and polymerase chain reaction (RT-PCR) were performed as previously described [20]. The complete VP1 gene (876 bp) of each isolate was amplified by using primers VP3 (008, GCRTGCAATGAYTTCTCWGT) and 2A (011, GCICCIGAYTGITGICCRAA) [8]. Sequencing was performed using the purified PCR products with the ABI Prism Ready Reaction Dideoxy Terminator cycle sequencing kit (Model 3730 version3.4, Applied Biosystems, Foster City CA).
Phylogenetic analysis
Multiple sequence alignments were performed using CLUSTAL W [21]. The Kimura two-parameter model was used as the nucleotide substitution model for estimating pairwise distances [22]. The transition/transversion ratio was also estimated from the data set. Molecular Evolutionary Genetics Analysis, Version 4.0 (MEGA 4) software was used for pairwise comparisons and to construct neighbor-joining (NJ) trees [23]. The reliability of the NJ tree was estimated by bootstrap analysis with 1000 pseudo-replicates. The 876 bp full-length VP1 genes were analyzed in the 32 Taiwanese strains, in the 498 worldwide strains (Table 1), and in the out-group E21 Farina strain.
Sequence diversity analysis in VP1 gene
The substitution rates over amino acid sites were analyzed by DAMBE, ver 5.0.80 [24]. The DNASP program [25] and Nei and Gojobori method [26] were used to estimate the numbers of synonymous and nonsynonymous nucleotide substitutions. The structural guide presented above the alignment is based on that for Echovirus 1 (PDB ID: 1EV1). Three-dimensional (3D) structures were predicted by I-TASSER [27]. The 3D molecular graphs were constructed and aligned using PyMOL (DeLano, WL. The PyMOL Molecular Graphics System, 2002). An Echovirus 1 (Farouk strain, PDB ID: 1EV1) was used as the structural guide [28].
Accession numbers of the nucleotide sequences
Sequence data from the present study were deposited in the DDBJ/EMBL/GenBank nucleotide sequence databases under accession numbers AB505453–AB505457, AB506746, DQ842242–DQ842264, and EF596823–EF596826.
Results
Case profiles and specimens
Notably, almost one-third (28.6%) of the E-30 isolates were detected in cerebrospinal fluid. The remaining were isolated from throat swab (35.7%) or from stool or rectal swab (35.7%). The virus was more common in children aged 5 years and above than in children younger than 5 years (mean age: 5.5 years; range: 6 months–11 years), and the male–female ratio was 1.3:1.
Phylogenetic analysis of VP1 gene
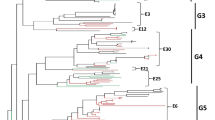
The genetic relationships among 530 worldwide E-30 strains isolated during the past 50 years were inferred by neighbor-joining method based on the 877 nucleotides (2460–3335) of the VP1 gene. The extrapolated genotype demarcation was 15%. Six genotypes (designated GI-GVI) were depicted. All genotypes were supported by high bootstrap values (>80). The dendrogram suggested a monophyletic origin of the worldwide strains. Each genotype also revealed complex internal clades (Fig. 1).

Phylogenetic analysis of 530 worldwide strains of E-30 based on 876 bp VP1 gene sequence. The E21 Farina strain was used as an outgroup. a Rooted tree. Mega 4 software was used to construct the neighbor-joining tree. For major lineages, bootstrap values >80% in 1,000 pseudoreplicates are indicated at the branch nodes. For clarity, the names of most sequences are omitted from the tree. b Unrooted tree. Circle Prototype E-30 Bastianni strain isolated in US in 1958. Triangle viruses isolated in this study
The first strains of GI, GII, and GV emerged in the 1950s (1956, 1957, and 1958, respectively). The GI and GII were geographically distributed in the US and Australia, respectively. The GV consisted of strains isolated in Australia (1969), the Netherlands (1975), and the US (1958–1981), including the prototype Bastinanni strain isolated in the US in 1958. This genotype apparently had a highly diverse and long-term (1960s–1980s) distribution. The GIII first appeared in Colombia (1995) before reemerging in NIS during 2003–2006. The GIV included strains isolated in South Korea and Malaysia during 2003–2004. The GVI included eight clusters (A–H) of strains isolated since 1967. No GVI-A, C, D, or E have been identified in the past decade. The GVI-B strains first appeared in China (1984) before reemerging in Russia (1999–2000, 2004) and Asia, including China, Japan, and Taiwan (2003–2005). The GVI-F was dominant in Asia and Australia. Strains in GVI-G were distributed globally. Strains in GVI-H circulated in Northern and Eastern Europe since the 1990s (Fig. 1; Table 1).
The GVI-F, G, and H were further divided chronologically into 3, 5, and 3 subclusters, respectively. Strains in GVI-F1 and GVI-F2 consisted of Taiwanese strains isolated in 1993 and Japanese strains isolated in 1997–1998, respectively. The GVI-F3 consisted of strains from Australia (1996–1997), Malaysia (2001, 2004), Japan (2000, 2006), and Taiwan (2001). The GVI-G1 consisted of strains from France (1997), Bahrain (1999, 2000), US (1993, 1997), and Denmark (1997). The GVI-G2 consisted of strains from US (1993–1998), Taiwan (1993), Japan (1993, 1998), Italy (1998), France (1994–2001), Belgium (2000), Netherlands (1996), and Russia and NIS (2000–2003). The GVI-G3, G4, and G5 consisted of strains from China (2002–2004), Jamaica (2004), and Taiwan (2007–2008), respectively. The GVI-H1 consisted of strains isolated from Japan (1991), US (1988–1992), Finland (1989–1990), France (1991–1992), Germany (1993), Netherlands (1993), and Italy (1999). The GVI-H2 consisted of strains isolated from Finland (1992) and Australia (1993). The GVI-H3 consisted of strains isolated from France (1996–2007), Belgium (2000), Germany (1996, 2003), Russia and NIS (1995–2006), north Europe (1994–1997), and Israel (1998–1999) (Fig. 1). These data suggested that the circulating lineages had either been transiently prevalent in a local (e.g., GVI-F1, 2; GVI-G3, 4, 5; and GVI-H2) or had been circulating in a broader area with continuous divergences. All Taiwanese strains were clustered into clades according to year of isolation: GVI-D (1988–1989), GVI-F1 and GVI-G2 (1993), GVI-F3 (2001) and GVI-G5 (2007).
Sequence diversity in VP1 gene
Pairwise comparisons based on analyses of the 876 bp VP1 gene in the 32 Taiwanese strains revealed nucleotide and amino acid identities of 83.1–100.0% and 94.7–100.0%, respectively. Nucleotide and amino acid identities between Taiwanese strains and the Bastianni prototype were 78.2–82.5% and 90.7–94.2%, respectively (data not shown). The amino acid identities among worldwide strains ranged from 89.4 to 100.0%. Pairwise comparisons of nucleotide sequences between strains revealed that worldwide strains had nucleotide identities of 69.4–100.0%; of these, 32.4% had nucleotide identities less than 75% (data not shown). Most pairs with nucleotide sequence identities less than 75% were included in the pairwise comparison between sequences of genotypes I, II, and III versus sequences of genotypes V and VI (Fig. 2).

Pairwise comparison of 876 bp or 292-amino acid sequence in VP1 gene of 530 E-30 strains and pairwise comparison of nucleotide versus amino acid sequence homology among each genotype for all sequences based on VP1 gene; X-axis, nucleotide identity of each genotype; Y axis, amino acid identity of each genotype. The vertical dotted lines mark the 75% demarcation in the pairwise comparison between sequences of genotypes I, II, and III versus sequences of genotypes V and VI
Nucleotide substitutions in VP1 genes occurred predominantly at the third letter of the codon. The dN/dS (nonsynonymous-to-synonymous substitution) ratios among the worldwide and Taiwanese strains were 0.027 and 0.014, respectively. The deduced amino acid sequences of the capsid VP1 gene were aligned and analyzed (Fig. 3). Thirty-eight amino acid substitutions were noted in comparison with the prototype strain Bastianni. Most nonsynonymous changes were concentrated in the following four principal regions of the gene: amino acid positions 1–20, 50–90, 135–165, and 260–292. Comparison of the secondary structure with the functional motif of VP1 revealed that all variable regions were within the N and C terminals or within the loop regions (Fig. 3a–d). The exception was the βB strand (72nd–80th residues). Three-dimensional predictive modeling revealed the largest structural differences in the BC and DE-loop and in the N and C terminus of VP1 (Fig. 3e).

Comparison of 292 amino acid sequences of the VP1 gene in 33 E-30 strains (32 Taiwanese strains and E-30/Bastianni) with E21/Farina as outgroup. The structural guide presented above the alignment is based on that for Echovirus 1 (PDB ID: 1EV1)[28]. Amino acid residues exposed on the outer surface of the virus particle are indicated by a plus sign (+) under the sequences. a Amino acid substitution rate over amino acid residues, worldwide strains. b Pairwise comparison of synonymous and nonsynonymous mutations, Taiwanese strains. Country abbreviations: TW Taiwan, US United States. c Amino acid sequence alignment of the 292 amino acid sequences of the VP1 gene among 33 E-30 strains (32 Taiwanese strains and E-30/Bastianni) with E21/Farina as outgroup. The isolates are identified by accession number, year isolated, country abbreviation, and strain name. Strains obtained from National Cheng Kung University Hospital are indicated by open circles. Sequences obtained from CDC-Taiwan are indicated by solid circles. Only amino acids that differed from those of E-30 prototype Bastianni strain sequence are shown. Identical residues are indicated by small dots. d Plane jelly-roll structure of VP1 protein showing the motif and secondary structure. The numbers correspond to the β-strand residue in a given structural motif. The substitution sites are marked with an  . e Three-dimensional (3D) structure prediction for VP1 alignment of five viral strains, including the prototype AF162711-58-US-Bastianni (GV) strain and four Taiwanese strains. The secondary structures corresponding to the VP1 are indicated
. e Three-dimensional (3D) structure prediction for VP1 alignment of five viral strains, including the prototype AF162711-58-US-Bastianni (GV) strain and four Taiwanese strains. The secondary structures corresponding to the VP1 are indicated
Discussion
Earlier studies of the VP1 gene revealed the temporal dynamic and genetic diversity of E-30. Although research groups in different countries may obtain different phylogenetic classification results because they use different strains or analysis methods, the worldwide E-30 strains isolated during the last half century can be divided into four to eight genotypes [8, 16, 29, 41, 43]. Further refinement of phylogenetic classifications is expected as sequence data for additional worldwide strains accumulate in the Genbank database. Assuming a 15% nucleotide difference is sufficient for genotype demarcation, this study depicted six genotypes. All genotypes were supported by high bootstrap values (>80) (Fig. 1). The genotypes identified in this study were classified as ancient, reemerging, emerging, and circulating. The GI, GII, and GV were ancient genotypes in which the first strains had emerged in the 1950s but have not appeared in the past two decades. Strains of GIII, a reemerged genotype, first appeared in Colombia (1995) before emerging in Georgia, Ukraine, and Uzbekistan after 2000. The GIV was an emerging genotype that recently appeared in Asia in 2003 and had a closer relationship to the ancient genotype GV than to GVI. The GVI was a circulating genotype, which included eight clusters (A–H) of strains that had circulated since 1967. The GVI-A, C, D, and E were classified as dormant clusters because none of them had been identified during the previous 10 years. The GVI-B was a reemerged cluster, which had first appeared in China in 1984 and later in Russia and Asia in the 2000s. The GVI-E, F, and G were the prevalent clusters, which had been dominant in Asia and Australia, globally, and Northern and Eastern Europe, respectively. The GVI-D had been dominant worldwide during 1985–1991, and the GVI-G strains became the dominant strains circulating globally after 1993 (Fig. 1; Table 1). Multiple lineages are known to co-circulate simultaneously [29]. Co-circulations observed in the current study included GI and GV in US during both 1967–1969 and 1974–1976, GVI-A and B in US during 1967–1974, and GVI-C and D in Australia during 1980–1989 (Table 1). Moreover, some lineages appeared intermittently but over extended periods. For example, GVI-B was intermittent in China during 1984–2004; GI and GV was intermittent in US during 1957–1976 and 1958–1981, respectively.
In Taiwan, this study is the first long-term (21 years) analysis of E-30 molecular epidemiology. The 1988–1989 strains were clustered in GVI-D together with strains from US, Netherlands, France, and Australia, all of which were isolated in the same period (1985–1991). The 1993 strains were clustered in GVI-F1 (Taiwanese strains only) or in GVI-G2 together with those isolated in Japan (1993) and US (1993–1998). The 2000–2001 strains were clustered in GVI-F3 together with strains isolated in Japan (1997–1998 and 2000) and Malaysia (2001). The 2005 strains were clustered in GVI-B together with isolates from China (2003–2004), Japan (2000, 2004 and 2006), and Russia (1999–2000). The 2005–2008 strains were clustered in GVI-G3 together with strains from China (2002–2004) and Jamaica (2004–2005) (Fig. 1; Table 1). The results indicated that prevalent lineages correlations with isolation period were stronger than correlations with geographic origin. The quiescence period of Taiwanese strains was also longer than that in previous reports (5–8 vs. 3–5 years, respectively) [8, 14, 30]. We surmise that, since Taiwan is an island country, the ocean provides a natural barrier that extends the quiescence period.
The common HEV identification scheme follows the 75% and 85% rule, i.e., in a complete or partial VP1 gene, HEVs are considered the same serotype if nucleotide homology and amino acid homology are >75% and 85%, respectively [31]. However, compared to global strains circulating during the past 50 years, the nucleotide and amino acid homologues were <75% (69.4%) and >85% (89.4%), respectively. Despite the widely varying nucleotide sequence, the amino acid sequence was preserved. Degeneration of the genetic code may accelerate as genetic divergence increases. This implies that, unlike the 85% rule for amino acids, the 75% rule for nucleotides may not be universally applicable, especially in a long-term analysis of a viral lineage such as E-30, which has high genetic diversity and rapid turnover [5, 14, 29]. We reported similar results in our previous molecular epidemiology study of CVB3 [32]. A recent report proposed an even more stringent value for VP1 amino acid identity (88%) in the routine typing of HEV-C [33].
All three HEV capsid proteins VP1–VP3 have the wedge-like protein that folds into a β-barrel with the common jelly-roll structural motif (Fig. 3d, e). The jelly-roll structure consists of a canonical core with eight antiparallel β-strands (designated βB–βI) [34]. The β-barrel of VP1, which is the major structure in the ‘pocket’, is lined with hydrophobic residues and forms a viral receptor binding site. Viruses differ mainly in the proteins in the loop insertions between the β-strands and the N and C termini [29]. In the current study, most nonsynonymous changes occurred in the in the N and C terminals or in the BC, DE, and EF loops of the VP1 protein (Fig. 3). The βB (72nd–81st residues) variability observed in this study also agrees with previous reports that the dN/dS ratio is usually low in βB strand [29]. Interestingly, Halim et al. [35] reported that spanning residues 68–82 of VP1 in Coxsackievirus B4 are a potential B cell epitope associated with a serotype-specific, neutralization antigenic site. Taiwanese E-30 strains also have a highly conserved amino acid motif of eleven residues (PALTAVETGHT) close to the N terminus of VP1. Their proximity to the N terminus of VP1 suggests a function involving uncoating or cell entry [36, 37]. In all E-30 strains, the conservation observed in residues 118, 120, 122, 147, 171, 193, 217, and 241 indicates a drug-binding pocket [38].
In conclusion, the genotyping results showed that multiple E-30 genotypes evolved and diverged continuously. Each genotype had a different geographic distribution and circulation half-life. Certain genotypes have spread worldwide (GVI-D, G) whereas others have been implicated only in local outbreaks (GI, II, IV and GVI-A, E). The GI, II, and V were ancient genotypes whereas GIII was an emerging genotype. The GIV and GVI-E appeared transiently while new variants that had evolved from GIII and GVI-B reemerged in recent outbreaks. In the past two decades, GVI-D (1988–1989), GVI-F (1993–2004) and GVI-G (1993–2008) clusters appeared in Taiwan with a longer quiescence period (5–8 years).
References
G. Stanway, F. Brown, P. Christian, T. Hovi, T. Hyypiä, A.M.Q. King, N.J. Knowles, S.M. Lemon, P.D. Minor, M.A. Pallansch, A.C. Palmenberg, T. Skern, in Virus taxonomy eighth report of the international committee on taxonomy of viruses, ed. by C.M. Fauquet, M.A. Mayo, J. Maniloff, U. Desselberger, L.A. Ball (Elsevier/Academic Press, London, 2005), pp. 757–778
B. Rajtar, M. Majek, L. Polanski, M. Polz-Dacewicz, Ann. Agric. Environ. Med. 15, 199–203 (2008)
S. Diedrich, G. Driesel, E. Schreier, J. Med. Virol. 46, 148–152 (1995)
J.R. Wang, H.P. Tsai, S.W. Huang, P.H. Kuo, D. Kiang, C.C. Liu, J. Clin. Microbiol. 40, 4439–4444 (2002)
G.W. Chen, J.H. Huang, Y.L. Lo, K.C. Tsao, S.C. Chang, Arch. Virol. 152, 1807–1817 (2007)
K. Akiyoshi, N. Nakagawa, T. Suga, J. Japanese, Infect. Dis. 60, 66–68 (2007)
Y.N. Zhao, Q.W. Jiang, R.J. Jiang, L. Chen, D.S. Perlin, Emerg. Infect. Dis. 11, 562–567 (2005)
M.S. Oberste, K. Maher, M.L. Kennett, J.J. Campbell, M.S. Carpenter, D. Schnurr, M.A. Pallansch, J. Clin. Microbiol. 37, 3928–3933 (1999)
N. Khetsuriani, L.-F. Ashley, M.S. Oberst, M.A. Pallansch, MMWR Surveill. Summ. 15, 1–20 (2006)
L. Gomes Mde, L.L. Ferreira, R.H. Gomes, L.M. Lamarao, E. da Silveira, S. Rodrigues Lda, C.F. da Silva, E.R. de Almeida, Braz. J. Infect. Dis. 11, 403–406 (2007)
M. Cabrerizo, J. Echevarria, I. González, T. de Miguel, G. Trallero, J. Med. Virol. 80, 1018–1024 (2008)
B. Roth, M. Enders, A. Arents, A. Pfitzner, E. Terletskaia-Ladwig, J. Med. Virol. 79, 956–962 (2007)
J.L. Bailly, A. Beguet, M. Chambon, C. Henquell, H. Peigue-Lafeuille, J. Clin. Microbiol. 38, 2889–2892 (2000)
D. Brunel, N. Leveque, J. Jacques, F. Renois, J. Motte, L. Andreoletti, J. Clin. Virol. 42, 225–228 (2008)
J.L. Bailly, D. Brosson, C. Archimbaud, M. Chambon, C. Henquell, H. Peigue-Lafeuille, J. Med. Virol. 68, 558–567 (2002)
C. Savolainen, T. Hovi, M.N. Mulders, Arch. Virol. 146, 521–537 (2001)
A.N. Lukashev, O.E. Ivanova, T.P. Eremeeva, L.V. Gmyl, J. Clin. Microbiol. 46, 665–670 (2008)
F. Tseng, H. Huang, C. Chi, T. Lin, C. Liu, J. Jian, L. Hsu, H. Wu, J. Yang, Y. Chang, H. Wang, Y. Hsu, I. Su, J. Wang, C.-T.V.R.L.a.S.P. Network, J. Med. Virol. 79, 1850–1860 (2007)
M. Oberste, K. Maher, D. Kilpatrick, M. Pallansch, J. Virol. 73, 1941–1948 (1999)
A. Kitamura, T. Narisawa, A. Hayashi, Y. Ashihara, H. Ishiko, J. Jpn Assoc. Infect. Dis. 71, 715–723 (1997)
M.A. Larkin, G. Blackshields, N.P. Brown, R. Chenna, P.A. McGettigan, H. McWilliam, F. Valentin, I.M. Wallace, A. Wilm, R. Lopez, J.D. Thompson, T.J. Gibson, D.G. Higgins, Bioinformatics 23, 2947–2948 (2007)
M. Kimura, J. Mol. Evol. 16, 111–120 (1980)
K. Tamura, J. Dudley, M. Nei, S. Kumar, Mol. Biol. Evol. 24, 1596–1599 (2007)
X. Xia, Z. Xie, J. Hered. 92, 371–373 (2001)
P. Librado, J. Rozas, Bioinformatics 25, 1451–1452 (2009)
M. Nei, T. Gojobori, Mol. Biol. Evol. 3, 418–426 (1986)
Y. Zhang, BMC Bioinformatics 9, 40 (2008)
D.J. Filman, M.W. Wien, J.A. Cunningham, J.M. Bergelson, J.M. Hogle, Acta Crystallogr. D 54, 1261–1272 (1998)
J.L. Bailly, A. Mirand, C. Henquell, C. Archimbaud, M. Chambon, F. Charbonne, O. Traore, H. Peigue-Lafeuille, Infect. Genet. Evol. 9, 699–708 (2009)
E.C. McWilliam Leitch, J. Bendig, M. Cabrerizo, J. Cardosa, T. Hyypia, O.E. Ivanova, A. Kelly, A.C. Kroes, A. Lukashev, A. MacAdam, P. McMinn, M. Roivainen, G. Trallero, D.J. Evans, P. Simmonds, J. Virol. 83, 2109–2118 (2009)
M.S. Oberste, M.A. Pallansch, Rev. Med. Microbiol. 16, 163–171 (2005)
P.Y. Chu, G.M. Ke, Y.S. Chen, P.L. Lu, H.L. Chen, M.S. Lee, B.C. Chen, T.S. Huang, Y.C. Li, L.C. Chou, S.Y. Wang, K.H. Lin, Infect. Genet. Evol. 10, 777–784 (2010)
B.A. Brown, K. Maher, M.R. Flemister, P. Naraghi-Arani, M. Uddin, M.S. Oberste, M.A. Pallansch, J. Gen. Virol. 90, 1713–1723 (2009)
M.G. Rossmann, E. Arnold, J.W. Erickson, E.A. Frankenberger, J.P. Griffith, H.J. Hecht, J.E. Johnson, G. Kamer, M. Luo, A.G. Mosser et al., Nature 317, 145–153 (1985)
S. Halim, A.I. Ramsingh, Virology 269, 86–94 (2000)
C.E. Fricks, J.M. Hogle, J. Virol. 64, 1934–1945 (1990)
T. Hovi, M. Roivainen, J. Clin. Microbiol. 31, 1083–1087 (1993)
A.W. Dove, V.R. Racaniello, J. Virol. 74, 3929–3931 (2000)
A. Paananen, C. Savolainen-Kopra, S. Kaijalainen, O. Vaarala, T. Hovi, M. Roivainen, J. Med. Virol. 79, 945–955 (2007)
J.L. Bailly, M. Chambon, C. Henquell, J. Icart, H. Peigue-Lafeuille, J. Clin. Microbiol. 38, 552–557 (2000)
A. Mirand, C. Henquell, C. Archimbaud, H. Peigue-Lafeuille, J.L. Bailly, J. Gen. Virol. 88, 166–176 (2007)
A. Faustini, V. Fano, M. Muscillo, S. Zaniratti, G. La Rosa, L. Tribuzi, C.A. Perucci, Int. J. Infect. Dis. 10, 291–297 (2006)
G. Palacios, I. Casas, D. Cisterna, G. Trallero, A. Tenorio, C. Freire, J. Virol. 76, 4940–4949 (2002)
Acknowledgments
We thank Li-Chiu Chou and Yu-Hwa Lin for their valuable technical assistance. Sequence data from the Taiwan Pathogenic Microorganism Genome Database were kindly provided by CDC-Taiwan with the financial support from the National Research Program for Genome Medicine 98-0324-01-F-20. The interpretation and conclusions contained herein do not represent those of CDC-Taiwan. This study was funded by grants from Centers for Disease Control and Prevention of Taiwan (DOH94-DC-1004) and Kaohsiung Medical University (KMU-M099017). Ethical approval: Not required.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guan-Ming Ke and Kuei-Hsiang Lin equally contributed to this work.
Rights and permissions
About this article
Cite this article
Ke, GM., Lin, KH., Lu, PL. et al. Molecular epidemiology of Echovirus 30 in Taiwan, 1988–2008. Virus Genes 42, 178–188 (2011). https://doi.org/10.1007/s11262-010-0565-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-010-0565-5