
Abstract
The non-structural (NS) gene of highly pathogenic avian influenza viruses of the H5N1 subtype (HPAI-H5N1) isolated in Baltic Sea area of Sweden in 2006 was studied. The phylogenetic analysis data demonstrated that two distinct sub-lineages of HPAI-H5N1 were circulating during the outbreak in Northern Europe in Spring 2006. Sub-lineage I viruses fell into the same clade as viruses found in Denmark and Germany and formed a sub-clade which also included viruses isolated in the Russian Federation in late 2005. Sub-lineage II viruses formed a sub-clade closely related to European, Middle Eastern and African isolates reported in 2006. Analysis of the inferred amino acid sequences of the NS1 protein showed a deletion of five amino acids at positions 80–84. No viruses represented in this study contained Glu92 in the NS1 and all isolates contained the avian-like ESKV amino acid sequences at the NS1 C-terminal end. Sub-lineage I isolates contained unique substitutions V194I in NS1 and G63E in Nuclear export protein (NEP).
Similar content being viewed by others

Introduction
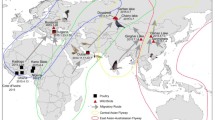
Outbreaks of highly pathogenic avian influenza viruses of the H5N1 subtype (HPAI-H5N1) have been occurring in poultry in southern China since 1996 [1]. During an outbreak of bird flu in Hong Kong in 1997, the virus crossed the species barrier from chicken to humans and killed six of the eighteen people infected [2]. The situation changed in late 2003 when in a very short period HPAI-H5N1 virus caused outbreaks in poultry populations in several countries in East and Southeast Asia, with the first fatal, human case outside Hong Kong. Over 100 million birds died from bird flu or were killed by countries authority in effort to control the outbreak [3]. At that time the world health organization (WHO), largely due to the high mortality rate for infected humans (currently 61%), proposed that the HPAI-H5N1 viruses are strong candidate for causing the next pandemic if the viruses acquire an efficient ability for human-to-human transmission. A seemingly spontaneous outbreak in wild migratory birds at Qinghai Lake in western China and Mongolia in 2005 [4] indicated a dramatic change in a key behaviour of the virus. Earlier outbreaks of H5N1 in wild bird populations were recorded as result of a spillover infection from domestic poultry to wild bird populations [5]. By late 2005 HPAI-H5N1 was detected in poultry in Russia, Turkey and Romania and in wild birds in Croatia. In Early January 2006, the first human case of HPAI-H5N1 outside Southeast Asia was reported from Turkey. The number of affected countries increased dramatically in the first three months of 2006; countries from Africa, Asia and Europe almost simultaneously reported outbreaks of HPAI-H5N1 in wild birds and domestic poultry [6]. In early February 2006 the first wild bird case of HPAI-H5N1 in Northern Europe was observed in Germany, in a Mute Swan (Cygnus olor) on the island of Ruegen, in the southwestern part of the Baltic Sea [7]. Three weeks later HPAI-H5N1 was reported from Denmark [8]. In that same week, the first case of HPAI-H5N1 in Sweden was detected from two Tufted ducks (Aythya fuligula). The ducks were found in ice-free waters in the area around Oskarshamn nuclear power plant by the Baltic Sea, on the east coast of Sweden. As a response to these findings, Swedish authorities, following EU regulations established a protection and monitoring zone in the area. In March 2006, virus was detected from a Mallard (Anas platyrhynchos) on a game bird farm located inside the monitoring zone around the index case. This was the first recorded case of HPAI in Swedish poultry. Between the first case on 24 February 2006 and the last detected case on 21 April 2006, the virus had spread along the Swedish eastern coast, extended northwards up to Stockholm and southwards to the Blekinge archipelago, affecting a coastal area of almost 900 km (Fig. 1).

Spread of HPAI-H5N1 in east coast of Sweden in 2006. Locations of the studied viruses are indicated. Arrow indicates the location of the index case and letters Indicate provincials’ code
The virulence of influenza viruses is multigenic, but some important factors have been identified. The viral polymerase protein 2 (PB2) with its amino acid at position 627 influences the ability of the virus to replicate in human or mouse cells [9]. The receptor binding efficiency and high cleavability of the haemagglutinin (HA) glycoprotein can influence viral entry and lethal out come of infection [10]. The non-structural (NS1) protein is a multi-functional protein, which also plays a crucial role in viral virulence by countering cellular antiviral activities [11]. It contains two functional domains: the N-terminal RNA-binding domain (residues 1–73) and the C-terminal effector domain (residues 73–237) [12]. It has been suggested that the N-terminal RNA binding domain of the NS1 protein has regulatory activities that are important to prevent interferon-mediated antiviral responses. Binding of the NS1 protein to both single and double stranded RNA might: (a) inhibit activation of interferon-induced protein kinase PKR [13], (b) prevent activation of the 2′-5′oligoadenylate synthetase, which is essential for activation of ribonuclease L (RNase L) system [14], (c) inhibit the activation of interferon regulatory factor 3 (IRF-3) and nuclear factor-kappa B (NF-κB), key regulators of Alpha and Beta interferon (IFN α/β) gene expression, by interfering with the retinoic acid-inducible gene I (RIG-I) [15, 16] and (d) suppress the RNA interference system, by binding to small interfering RNAs [17, 18].
The N-terminal residues 81–113 of the NS1 protein have been shown to bind to eukaryotic translation initiation factor 4GI (eIF4GI), the large subunit of the cap-binding complex eIF4F [19]. By doing so, the NS1 protein recruits eIF4F to the 5′ untranslated region of viral mRNA and activates translation of viral mRNA.
The effector domain of NS1 protein has been associated with regulation of gene expression of the infected cell [20]. It has been shown that the effector domain of the NS1 protein: (a) Inhibits 3′-end processing of cellular pre-mRNA by specific interaction with the 30 kDa subunit of the cleavage and polyadenylation specific factor (CPSF) [21–23] and (b) prevents transport of cellular m-RNA to cytoplasm by interaction with poly(A)-binding protein II (PABII) [24].
The Nuclear export protein (NEP), expressed from a spliced mRNA from the NS gene, consists of 121 amino acids [25] that, in association with the matrix protein 1 (M1), interacts with cellular export factor (CEF1) and mediates the nuclear export of viral ribonucleoprotein (vRNP) complexes [26] by connecting the cellular export machinery with vRNPs [27].
Genetic studies on HPAI-H5N1 have mostly focussed on the Haemagglutinin (HA) gene. Despite the important role of the NS1 gene in virulence of the virus, our knowledge of the gene pool is limited. In this study we analysed the NS gene sequences of 23 HPAI-H5N1 viruses isolated in Baltic Sea area, to gain more detailed knowledge about the genetic variation of HP H5N1 influenza virus.
Materials and methods
Specimens
The Swedish Board of Agriculture coordinated the collection of dead or moribund animals. Post mortem examinations were performed at the National Veterinary Institute in Uppsala. All animals or infected material were handled in government-certified biosafety level 3+ (BSL-3+) facilities. Five hundred and three oropharyngeal swabs were screened for the presence of influenza A viruses by real-time reverse transcription polymerase chain reaction (rRT-PCR) for the matrix protein gene [28]. All positive cases were further examined with an H5 specific reverse transcription polymerase chain reaction (RT-PCR) [29]. Both PCR assays were performed according to the recommendations from the Community Reference Laboratory (CRL; VLA Weybridge). H5 positive samples were further processed by sequence analysis of the haemagglutinin gene at the cleavage site in order to determine the pathogenicity of the isolates.
Twenty-three HPAI-H5N1 positive animals were chosen for this study representing different species, finding date and geographical distribution during the outbreak (Table 1).
RNA extraction and PCR with NS1 gene specific primers
Viral RNA extraction was performed directly from clinical specimens to decrease the risk for introduction of adaptive mutation during virus propagation in embryonated chicken eggs. RNA was extracted in a BSL-3+ laboratory, using Trizol reagent (Invitrogen Corp., Carlsbad, CA) according to the manufacturer’s instructions. The RNA was converted to full-length cDNA using reverse transcriptase (RT). The RT mix comprised 2.5 μl of DMPC water, 5 μl of 5× First Strand buffer (Invitrogen), 0.5 μl of 10 mM dNTP mix (Amersham Biosciences), 2 μl of 50 mM random primers oligonucleotides (pdN6), 2 μl of 40 U/μl RNAguard (Amersham Biosciences), 2 μl of 200 U/μl MMLV reverse transcriptase (Invitrogen) and 5 μl RNA solution with approximately 1 μg/μl RNA concentration in total volume of 25 μl. The reactions were incubated at 42°C for 90 min followed by inactivation of the enzyme at 95°C for 5 min.
PCR amplification with NS gene specific primers (Fw primer: 5′ CAA AAA CAT AAT GGA TYC CAA CAC K 3′, Rev primer 5′ ATT AAA TAA GCT GAA AMG AGA A 3′) was performed to amplify the product containing the full-length NS gene. Twenty-five microlitre PCR mix contained 1×Platinum Taq buffer (Invitrogen), 200 μM dNTP, 2.5 mM MgCl2, 240 nM each of Fw primer and Rw primer, 1 U Platinum Taq DNA Polymerase (Invitrogen) and 3 μl cDNA. Reactions were placed in a thermal cycler at 95°C for 2 min, then cycled 35 times between 95°C for 20 sec, at 58°C for 60 sec and at 72°C for 90 sec and were finally kept at 8°C until later use.
The PCR products were treated with shrimp alkaline phosphatase–exonuclease I (ExoSapI) (U.S Biologicals, Swampscott, MA, USA) (5 μl ExoSapI per reaction, 30 min. at 37°C followed by 10 min at 95°C) and utilized for sequencing directly.
Sequence analysis
Sequences of the purified PCR products were determined using gene specific primers and BigDye Terminator version 3.1 chemistry (Applied Biosystems, Foster City, CA), according to the manufacturer’s instructions. Reactions were run on a 3100 DNA analyser (Applied Biosystems). Sequencing was performed at least twice in each direction.
NS1 sequences obtained from GenBank
The NS1 gene sequences of 66 additional HPAI-H5N1 viruses obtained from GenBank were used in phylogenetic studies [30]. Comparison of the amino acid sequence of NS1 was performed by analysing an additional 830 HPAI-H5N1 viruses reported to the GenBank database since 1996.
Phylogenetic and sequence analysis
After sequencing, assembly of sequences, removal of low-quality sequence data, nucleotide sequence translation into protein sequence, additional multiple sequence alignments and processing were performed with the Bioedit software version 7.0.4.1 [31] with an engine based on the Custal W algorithm. Blast homology searches (http://www.ncbi.nlm.nih.gov/blast) were used to retrieve the top fifty homologous sequences for the sequenced gene from the GenBank database. The phylogenetic analysis, based on complete genome nucleotide and amino acid sequences were constructed with Molecular Evolutionary Genetics Analysis (MEGA, version 3.1) software [32] using neighbour-joining tree inference analysis with the Tamura-Nei γ-model, with 2000 bootstrap replications to assign confidence levels to branches. Phylogenetic relationships of sequences were further confirmed by Bayesian analysis in mrbayes-3.1.2 [33]. For Bayesian analysis, six Monte Carlo Markov Chains (MCMC) were simultaneously run for 10000 generations, saving a tree every 100 generations.
The nucleotide sequence data obtained in this study have been submitted to the GenBank database and are available under accession numbers EU121999–EU122021.
Results
Viruses
A total of 75 avian influenza virus infected animals were identified during the period from February 24 to April 21 2006 from the east coast of Sweden (Table 2).
Of the 75 cases, 64 were characterized as H5N1. Sequence analysis of the HA showed that amino acid sequences at the HA-cleavage sites had multiple basic amino acids (PQGERRRKKR) which is the well-recognized typical hallmark of HPAI [34]. The virus was detected in several species of aquatic birds, including Mute Swan (Cygnus olor), Scaup (Aythya marila), Gossander (Mergus merganser), Smew (Mergus albellus), Canada goose (Branta canadensis) and Herring gull (Larus argentatus). The virus was also isolated from predators of aquatic birds, such as Common buzzard (Buteo buteo), European eagle owl (Bubo bubo) and in a wild mink (Mustela vison).
Phylogenetic analysis
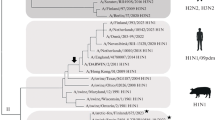
The NS1 gene of the 23 HPAI-H5N1 viruses sequenced in this study and 66 NS1 genes of recently isolated viruses from Asia, Europe and Africa obtained from the GenBank were used for phylogenetic analysis. As Bayesian analysis showed the same phylogenetic relationship as analysis based on neighbour joining using MEGA 3.1, only the results obtained from MEGA are shown here. A phylogenetic tree was constructed on the basis of the complete nucleotide sequence of the NS1 coding region of selected viruses. The phylogenetic analysis data demonstrated that all studied viruses clustered together with Qinghai-like viruses, which have been isolated in Asia, Europe, Africa and the Middle East since 2005. Further analysis showed that two distinct sub-lineages of HPAI-H5N1 viruses circulated during the outbreak in the Baltic Sea area in Spring 2006 (Fig. 2). Our results agree in part with a recent study that reported multiple introductions of HPAI-H5N1 viruses into Europe and Africa [35]. Sub-lineage I viruses were found in animals from the entire infected area on the East coast of Sweden and contain viruses isolated from several species of aquatic birds including a virus found in a Mallard (Anas platyrhynchos) from a game bird farm. Sub-lineage I also include viruses found in predators of aquatic birds like Common buzzard (Buteo buteo), European eagle owl (Bubo bubo) and a wild mink (Mustela vison).

Phylogenetic relationship of NS1 genes of 23 Swedish HPAI-H5N1 viruses isolated during the outbreak in 2006. The protein coding region tree was generated by neighbour-joining analysis with Tamura-Nei γ-model, using MEGA 3.1. Numbers below key nodes indicate the percentage of bootstrap values of 2000 replicates
The phylogenetic analysis data demonstrated that sub-lineage I viruses fell into the same clade as viruses found in Denmark and Germany in 2006 and built a sub-clade with viruses isolated from Mute swans (Cygnus olor) in the Russian Federation and in Croatia in late 2005. Our phylogenetic analysis showed that there is a close relationship between viruses found in Northern Europe and those found in West Africa, mostly from Nigeria (Fig. 3).

Phylogenetic relationship of NS1 genes of 23 Swedish HPAI-H5N1 isolate compared to other H5N1 viruses from Asia, Europe, Middle East and Africa. The protein coding region tree was generated by neighbour-joining analysis with Tamura-Nei γ-model, using MEGA 3.1. Numbers below key nodes indicate the percentage of bootstrap values of 2000 replicates. Swedish isolates are indicated by O and marked in grey
Sub-lineage II circulated over the same time period as sub-lineage I viruses but contains only viruses from one geographical area in Sweden. These viruses were mainly found in the Baltic island of Fårö. Sub-lineage II viruses formed a sub-clade closely related to European, Middle Eastern and African isolates reported in 2006.
Comparisons of the NS gene sequences to sequences in the databases
The similarity of the NS gene of the Swedish isolate was compared with the available sequences in the GenBank database using the Blast program from the National Center for Biotechnology Information, NCBI. Blast homology searches demonstrated that the closest relative to sub-lineage I viruses was a virus found in Germany (A/Swan/Germany/R65/2006) with the highest nucleotide sequence similarity of 99%. The closest relative to viruses in sub-lineage II was a virus found in Italy (A/Cygnus olor/Italy/808/2006) with the highest nucleotide sequence identity of 99%.
Comparison of the entire coding region of the NS1 gene of the A/Tuftedduck/Sweden/V789/06 virus belonging to sub-lineage I with the NS1 gene of the A/Tuftedduck/Sweden/V599/06 virus belonging to sub-lineage II showed 98.9% homologies. Overall the range of nucleotide and amino acid identity between all Swedish isolates were 98.9–100% and 98.6–100%, respectively.
Nucleotide sequence of the NS1 gene of these two viruses differed by seven nucleotides. The inferred amino acid sequence of the NS1 gene for these two viruses differed by two amino acids (A/Tuftedduck/Sweden/V789/06 versus A/Tuftedduck/Sweden/V599/06: A155T and I194V).
Analysis and comparison of inferred amino acid sequences of NS gene
It has been suggested in several studies [12, 24, 36] that the NS protein of influenza virus binds to and sequesters dsRNA produced during viral infection, thereby inhibiting the activation of IFN-mediated antiviral responses. These studies demonstrated that amino acids at the N-terminal RNA-binding domain of NS1 are implicated in this function. The arginine at position 38 and lysine at position 41 contributes to this interaction [37].
Comparison of the amino acid sequences of the Swedish isolates showed 99.5–100% identity in the N-terminal RNA-binding domain and 98–100% identity in C-terminal effector domain of NS1. All the isolates contain arginine at position 38 and lysine at position 41. Analysis of the amino acid sequences of 830 selected viruses from GenBank showed that all contain arginine at position 38 and lysine at position 41.
Amino acid at the N-terminus of effector domain
Analysis of the amino acid sequence of the studied isolates showed a deletion of five amino acids at positions 80–84 which is similar to that found in H5N1 viruses of all genotypes, except for Gs/Gd, X 0 and X 0 –X 3 Chen and co-workers [38] reported the first GenBank record of such a deletion in 2004, which was from a virus isolated in early 2000 from a duck in southern China (A/Duck/Zhejiang/52/2000). In human cases, such a deletion was reported in H5N1 viruses isolated in Hong Kong early 2003 (A/Hong Kong/212/2003).
Amino acid at position 92
No viruses sequenced in this study contained Glutamic acid at position 92 of the NS1 protein. The amino acid Glu92 in the NS1 protein observed in H5N1/97 influenza viruses is implicated in its ability to modulate the cytokine response and has been associated with the high virulence of these viruses in pigs [39]. Of total of 830-recorded sequences at the GenBank database only 26 contain Glu92, mostly isolated in Hong Kong in 1997.
Amino acid at position 149
It has been suggested that the amino acid at the position 149 of NS1 protein of HPAI-H5N1 affects the ability of the virus to antagonize the induction of IFN α/β(in chicken embryo fibroblasts [40]. All isolates sequenced in this study possessed the amino acid Ala149 in their NS1 protein, which is important for the ability of the virus to replicate in chickens.
Amino acid at the CPSF-binding site
The NS1 protein’s interaction with cleavage and polyadenylation specificity factor (CPSF) inhibits 3′-end processing of cellular pre-mRNA [21–23]. This function is mediated by two distinct domains: one around residue 186 [23] and the other around residues 103 and 106 [41]. All isolates sequenced in this study possessed the amino acid Glu186, Phe103 and Met106 in their NS1 protein. It is noteworthy that NS1 proteins of H5N1/1997 viruses isolated from both avian and human sources had the 102LMLI107 sequence motif while the majority of the viruses isolated from the year 2000 onwards had the 102FMLM107 sequence motif.
Amino acid at the PABII-binding site
The NS1 protein prevents transport of cellular mRNA to the cytoplasm by interaction with poly(A)-binding protein II (PABII) [24]. Amino acids 215–237 have been identified as the binding site for PABII [13]. Comparison among the amino acid sequences of the Swedish isolates showed 93–100% identity at the PABII-binding site.
PDZ-motif at the C-terminal of NS1
Based on large-scale sequence analysis of avian influenza viruses, Obenauer and co-workers [42] suggested that the NS1 protein contains a PDZ-binding motif at the C-terminal end of the protein. PDZ domains are protein-interacting domains present once or multiple times within certain proteins and these domains are involved in the cell signalling, assembly of large protein complexes or intracellular trafficking. NS1 protein might interact with host cellular signalling pathways by binding to PDZ domains on the host’s proteins. All the Swedish isolates possessed a PDZ-binding motif, ESKV, at the C-terminal end of the protein that is similar to other HPAI-H5N1 viruses isolated in Europe, Middle East and Africa since 2006.
Amino acid of NEP
Another product encoded by the NS gene through splicing of a portion of the transcripts for protein expression is the 14-kDa nuclear export protein. It has been suggested that tryptophan at position 78 is involved in NEP-M1 interaction that mediates the nuclear export of viral ribonucleoprotein complexes [27]. All Swedish isolates sequenced in this study possessed the amino acid TRP78 in their NEP. The NEP of the studied isolates consists of 121 amino acids. All the NEP from sub-lineage I viruses contain substitution G63E.
Discussion
One of the products of the non-structural gene of influenza type A viruses, the NS1 protein, plays a role as a virulence factor in the infected cell by preventing the induction of the interferon systems [11], thereby effectively inhibiting the activation of the host’s innate defence against the virus. Despite the important role of the NS1 gene in the virulence of the virus, our knowledge of the current gene pool is limited. Therefore, in this study we analysed the NS gene sequences of 23 HPAI-H5N1viruses, isolated during the outbreak in Baltic Sea area to gain detailed knowledge about the genetics of HP H5N1 influenza virus.
The NS1 gene of HPAI-H5N1 viruses isolated in Sweden consists of 225 amino acids. Analysis of the amino acid sequences of the Swedish isolates showed a deletion of five amino acids at positions 80–84 which is similar to that found in H5N1 viruses of all genotypes, except for Gs/Gd, X 0 and X 0 –X 3 . The exact functional consequence, if any, of this deletion is however not known.
The RNA-binding domain of the Swedish isolates had a closer amino acid sequence identity than the C-terminal effector domain of these viruses. None of the viruses presented in this study contained Glutamic acid at position 92 of the NS1 protein. The amino acid Glu92 in the NS1 protein observed in H5N1/97 influenza viruses is implicated in its ability to modulate the cytokine response and has been associated with the high virulence of these viruses in pigs [38]. Historically, the NS1 gene of influenza viruses has been divided into two separate alleles: A and B [43]. Allele A, includes all influenza viruses circulating in mammalian species and many viruses from avian species, Allele B contains only influenza viruses from avian species. The NS1 gene of the majority of HPAI-H5N1 viruses isolated since 1997 belong to allele A. In contrast, the NS1 gene of the A/Goose/Guangdong/1/96 virus, which was the source of the H5N1/1997 haemagglutinin gene, belonged to allele B. The NS1 genes of HPAI-H5N1 presented in this study all belong to allele A. Our analyses indicated that at least two sub-lineages of HPAI-H5N1 viruses have been circulating in Baltic Sea area during the time of the outbreak. The nucleotide sequence of the NS1 gene of the A/Tuftedduck/Sweden/V789/06 virus belonging to sub-lineage I, differed by seven nucleotides compared to the NS1 gene of the sub-lineage II virus A/Tuftedduck/Sweden/V599/06. All nucleotide substitutions were made by transition. Only two of these substitutions resulted in amino acid changes in the NS1 protein. Sub-lineage I isolates possessed V194I that characterize the isolates from Northern Europe, Nigeria and Niger. This substitution was also present in the sequences obtained from Mute swans (Cygnus olor) isolated in southern Russia in 2005. The NS1 gene of Sub-lineage I viruses has a high sequence-identity with that of viruses isolated in Germany and Denmark while the NS1 gene of sub-lineage II viruses has the highest identity with viruses found in Italy and the Middle East. A Recent study by Salzberg and co-worker [35] identified three different phylogenetic clades of HPAI-H5N1 viruses among isolates from Europe, the Middle East and Africa (EMA). Our analysis of Swedish isolates showed that Sub-lineage I isolates fall into EMA clade 2, while sub-lineage II isolates cluster with EMA clade 1. No host-related differences were observed in our sequence data. The virus was detected in several species of aquatic birds, including Mute Swan (Cygnus olor), Scaup (Aythya marila), Gossander (Mergus merganser), Smew (Mergus albellus), Canada goose (Branta Canadensis) and Herring gull (Larus argentatus). The virus was also isolated from predators of aquatic birds, such as Common buzzard (Buteo buteo), European eagle owl (Bubo bubo) and in a wild mink (Mustela vison).
The NEP of the studied isolates consists of 121 amino acids. All the NEP of the sub-lineage I viruses contains substitution G63E that is similar to viruses found in Denmark and Germany and unique for HPAI-H5N1 Qinghai-like viruses found in Northern Europe.
Our phylogenetic analyses indicate that the NS1 gene of viruses reported in this study and those viruses circulating in Europe, Africa and Middle East since late 2005 share a common ancestor. Our results indicate that most likely the NS1 gene of the virus circulating in Europe originated from an outbreak at Qinghai Lake in China. The NS1 genes of Qinghai-like viruses are evidently separate from HPAI-H5N1 viruses circulating in South East Asia in recent years. Legal or illegal movement of poultry from the endemic area and/or the presence of HPAI-H5N1 virus in migratory birds breeding at the Qinghai Lake, could explain the north west expansion of the outbreak which included an introduction into the poultry population in the Russian Federation and into poultry and wild bird populations in Eastern Europe in late 2005. Sudden westward movement of wild waterfowl due to hard winter condition in early 2006 is the most probable route of introduction of HPAI-H5N1 into the Swedish wild bird population from an infected area in Eastern Europe and the area around the Black sea. Analysis of a greater number of viruses isolated from poultry and wild birds from Eastern Europe might provide better epidemiological data.
The examination of more than 1000 wild birds, comprising a total of 70 species, since Summer 2006 has not yielded any HPAI-H5N1 positive results. So far in 2007, a total of 1500 wild birds, 117 of which were found dead, have tested negative for H5N1.
Regional monitoring activities among the wild bird and domestic bird populations as well as enhanced biosecurity measures among poultry holdings are important measures, which should be taken into consideration to be able to respond to the threat of HPAI-H5N1 to human and animal health.
In summary, we analysed a number of HPAI-H5N1 NS1 genes and incorporated our findings into the current information available. Our genetic characterization of the NS gene indicates co-circulation of two sub-lineages of highly pathogenic avian influenza virus of H5N1 subtype in Sweden in 2006. Our result provides useful molecular epidemiological data to understand the dynamics of HPAI-H5N1 evolution and support earlier phylogenetic observations. Furthermore, the ecology of AIV in the wild bird’s population is an area in which research efforts will be concentrated in the future.
References
X. Xu, K. Subbatao, N.J. Cox, Y. Guo, Virology 261, 15–19 (1999)
K. Subbarao, A. Klimov, J. Katz, H. Regnery, W. Lim, H. Hall, M. Perdue, D. Swayne, C. Bender, J. Huang, M. Hemphil, T. Rowe, M. Shaw, X. Xu, K. Fukuda, N.J. Cox, Science 279, 393–396 (1998)
L.D. Sims, J. Domenech, C. Benigno, S. Kahn, A. Kamata, J. Lubroth, V. Martin, P. Roeder, Vet. Rec. 157, 159–164 (2005)
H. Chen, G.J.D. Smith, S.Y. Zhang, K. Qin, J. Wang, K.S. Li, R.G. Webster, J.S.M. Peiris, Y. Guan, Nature 436, 191–192 (2005)
T.M. Ellis, R.B. Bousfield, L.A. Bisset, K.C. Dyrting, G.S.M. Luk, S.T. Tsim, K. Sturm-Ramirez, R.G. Webster, Y. Guan, J.S.M. Peiris, Avian Pathol. 33, 492–505 (2004)
World Health Organization (http://www.who.int/csr/disease/avian_influenza/timeline2007_06_21.pdf). Accessed 21 June 2007
S. Weber, T. Harder, E. Starick, M. Beer, O. Werner, B. Hoffmann, T.C. Mettenleiter, E. Mundt, J. Gen. Virol. 88, 554–558 (2007)
K. Bragstad, P.H. Jorgensen, K. Handberg, A.S. Hammer, S. Kabell, A. Fomsgaard, Virol. J. 4:43 doi: 10.1186/1743-422x-4-43 (2007)
M. Hatta, P. Gao, P. Halfmann, Y. Kawaoka, Science 293, 1840–1842 (2001)
T. Kuiken, E.C. Holmes, J. MacCauley, G.F. Rimmelzwaan, C.S. Williams, B.T. Grenfell, Science 312, 394–397 (2006)
A. Garcia-Sastre, Virology 279, 375–384 (2001)
R.M. Krug, W. Yuan, D.L. Noah, A.G. Latham, Virology 309, 181–189 (2003)
S. Li, J.Y. Min, R.M. Krug, G.C. Sen, Virology 349, 13–21 (2006)
J.Y. Min, R.M. Krug, Proc. Natl. Acad. Sci. USA 103, 7100–7105 (2006)
J. Talon, C.M. Horvath, R. Polley, C.F. Basler, T. Muster, P. Palese, A. Garcia-Sastre, J. Virol. 74, 7989–7996 (2000)
M. Mibayashi, L. Martinez-Sobrido, Y.M. Loo, W.B. Cardenas, M. Gale, A. Garcia-Sastre, J. Virol. 81, 514–524 (2007)
E. Bucher, H. Hemmes, P. De Haan, R. Goldbach, M. Prins, J. Gen. Virol. 85, 983–991 (2004)
W.X. Li, H. Li, R. Lu, F. Li, M. Dus, P. Atkinson, E.W.A. Brydon, K.L. Johanson, A. Garcia-Sastre, L.A. Ball, P. Palese, S.W. Ding, Proc. Natl. Acad. Sci. USA 101, 1350–1355 (2004)
T. Aragon, S. De La Luna, L. Carrasco, J. Ortin, A. Nieto, Mol. Cell. Biol. 20, 6259–6268 (2000)
N. Satterly, P.L. Tsai, J. Van Deursen, D.R. Nussenzveig, Y. Wang, P.A. Faria, A. Levay, D.A. Levy, B.M.A. Fontoura, Proc. Natl. Acad. Sci. USA 104, 1853–1858 (2007)
M.E. Nemeroff, S.M.L. Barabino, Y. Li, W. Keller, R.M. Krug, Mol. Cell 1, 991–1000 (1998)
Z. Chen, Y. Li, R.M. Krug, EMBO J. 18, 2273–2283 (1999)
Y. Li, Z.Y. Chen, W. Wang, C.C. Baker, R.M. Krug, RNA 7, 920–931 (2001)
Y. Qiu, R.M. Krug, J. Virol. 68, 2425–2432 (1994)
R.A. Lamb, C.J. Lai, Cell 21, 475–485 (1980)
G. Neumann, M.T. Hughes, Y. Kawaoka, EMBO J. 19, 6751–6758 (2000)
H. Akarsu, W.P. Burmeister, C. Petosa, I. Petit, C.W. Muller, W.H. Ruigrok, F. Baudin, EMBO J. 22, 4646–4655 (2003)
E. Spackman, D.A. Senne, L.L. Bulaga, T.J. Myers, M.L. Perdue, L.P. Garber, K. Lohman, L.T. Daum, D.L. Suarez, Avian Dis. 47, 1079–1082 (2003)
M.J. Slomka, V.J. Coward, J. Banks, B.Z. Ouml ndt, I.H. Brown, J. Voermans, G. Koch, K.J. Handberg, P.H. Oslash rgensen, M. Cherbonnel-Pansart, V. Jestin, G. Cattoli, I. Capua, A. Ejdersund, N.P. Thor Eacute, G. Czifra, Avian Dis. 51, 227–234 (2007)
Influenza Virus Resource at National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/genomes/FLU/). Accessed 20 June 2007
T.A. Hall, Nucleic Acids Symposium Series. (Oxford University Press, Oxford, 1999), pp. 95–98
S. Kumar, K. Tamura, M. Nei, Bioinformatics 5, 150–163 (2004)
F. Ronquist, J.P. Huelsenbeck, Bioinformatics 19, 1572–1574 (2003)
D.A. Senne, B. Paniagrahy, Y. Kawaoka, J.E. Pearson, J. Suss, M. Lipkind, H. Kida, R.G. Webster, Avian Dis. 40, 425–437 (1996)
S.L. Salzberg, C. Kingsford, G. Cattoli, D.J. Spiro, D.A. Janies, M.M. Aly, I.H. Brown, E. Couacy-Hymann, G.M. De Mia et al., Emerg. Infect. Dis. 13, 713–718 (2007)
E. Hatada, R. Fukuda, J. Gen. Virol. 73, 3325–3329 (1992)
W. Wang, K. Riedel, P. Lynch, C.Y. Chien, G.T. Montelione, R.M. Krug, RNA 5, 195–205 (1999)
H.L. Chen, G.H. Deng, Z.J. Li, G.B. Tian, Y.B. Li, P. Jiao, L. Zhang, Z. Liu, R.G. Webster, K. Yu, Proc. Natl. Acad. Sci. USA 101, 10452–10457 (2004)
S. Seo, E. Hoffmann, R.G. Webster, Nat. Med. 8, 950–954 (2002)
Z. Li, Y. Jiang, P. Jiao, A. Wang, F. Zhao, G. Tian, X. Wang, K. Yu, Z. Bu, H. Chen, J. Virol. 80, 11115–11123 (2006)
G. Kochs, A. Garcia-Sastre, L. Martinez-Sobrido, J. Virol. 81, 7011–7021 (2007)
J.C. Obenauer, J. Denson, P.K. Mehta, X. Su, S. Mukatira, D.B. Finkelstein, X. Xu, J. Wang, J. Ma, Y. Fan, K.M. Rakestraw, R.G. Webster, E. Hoffmann, S. Krauss, J. Zheng, Z. Zhang, C.W. Naeve, Science 311, 1576–1580 (2006)
S. Ludwig, U. Schultz, J. Mandler, W.M. Fitch, C. Scholtissek, Virology 183, 566–577 (1991)
Acknowledgements
We would like to thank Professor Berndt Klingeborn for helpful scientific discussions and constant support. Many thanks are due to Natasha Dahnberg for help with layout of the figures. Our appreciation also goes to Dr. Neil LeBlanc for critical review of the manuscript. The Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (Formas), financially supported this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zohari, S., Gyarmati, P., Thorén, P. et al. Genetic characterization of the NS gene indicates co-circulation of two sub-lineages of highly pathogenic avian influenza virus of H5N1 subtype in Northern Europe in 2006. Virus Genes 36, 117–125 (2008). https://doi.org/10.1007/s11262-007-0188-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-007-0188-7