
Abstract
Advanced next generation sequencing approaches have started to reveal the cellular and molecular complexity of the microenvironment in many tissues. It is challenging to obtain high quality RNA from mineralised tissues. We developed an optimised method of RNA extraction from feline teeth collected in a clinical setting and at post mortem. Teeth were homogenised in phenol-guanidinium solution at near-freezing temperatures and followed by solid-phase nucleic acid extraction utilising a commercially available kit. This method produced good RNA yields and improved RNA quality based on RNA integrity numbers equivalent (RINe) from an average of 3.6 to 5.6. No correlation was found between RNA purity parameters measured by A260:280 or A230:260 ratios and degree of RNA degradation. This implies that RNA purity indicators cannot be reliably used as parameters of RNA integrity. Two reference genes (GAPDH, RPS19) showed significant changes in expression levels by qPCR at low and moderate RINe values, while RPL17 was stable at all RINe values tested. Furthermore, we investigated the effect of quantity and quality of RNA on the quality of the resultant RNA sequencing (RNA-Seq) data. Thirteen RNA-seq data showed similar duplication and mapping rates (94 to 95%) against the feline genome regardless of RINe values. However one low yield sample with a high RINe value showed a high duplication rate and it was an outlier on the RNA-seq multidimensional scaling plot. We conclude that the overall yield of RNA was more important than quality of RNA for RNA-seq quality control. These results will guide researchers who wish to perform RNA extractions from mineralised tissues, especially if collecting in a clinical setting with the recognised restraints that this imposes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Teeth consist of connective tissues and highly specialised cells that produce a unique extracellular matrix composed of organic matrix proteins and inorganic minerals. Alveolar bone and deciduous teeth resorption during tooth eruption and shedding is a complex process that is tightly regulated at the molecular level (Ten Cate and Nanci 2003; Bei 2009). In permanent teeth, mineralised tissues such as enamel, cementum and dentine do not turn over or have limited regeneration capacities. The hard tissues of the tooth can be resorbed under pathologic conditions such as inflammation, mechanical trauma or idiopathic tooth resorption (TR) (Darcey and Qualtrough 2013). The exact mechanisms behind the pathological resorption observed in idiopathic TR are still unclear. Gene expression profiling of dental samples is of great interest to help map physiological pathways as well as identifying the pathways involved in pathological conditions at the cellular and molecular level. Recent studies of the oral and dental transcriptome have started to unveil the molecular complexity of the dental microenvironment (Simmer et al. 2014; Hu et al. 2015). Whilst in vitro studies are fundamental to the study of these cellular and molecular events and genetically engineered rodents have been important models in dentistry, there is a necessity to utilise human or companion animal derived tissues from clinical archives. Since both ethical and logistical considerations can limit the sourcing of human and companion animal derived tissues from clinics, it is important to maximise utility of collected tissue samples for research purposes.
As high-throughput sequencing techniques are now used more frequently and the cost of such studies is continuing to decrease, it is likely that more clinical veterinary studies will be able to utilise these evolving and cutting-edge approaches. Advanced high-throughput systems especially next generation sequencing, require high quality of RNA. However, in some cases it can be hard to avoid RNA degradation during sample collection. For example, due to the practical limitations, samples may be stored at suboptimal conditions for a period of time. Certain tissue types, in particular mineralised tissues, are difficult to homogenise. In addition, the collection procedure itself may accelerate RNA degradation, for example heat generated by a dental bur during tooth extraction can be detrimental to RNA quality.
Routinely, total RNA yield, purity and integrity are evaluated for RNA quality control. The most rapid and commonly used method for quantification of nucleic acids is to measure ultraviolet (UV) absorption of RNA sample by spectrophotometry (Manchester 1996; Desjardins and Conklin 2010). One commonly applied metric for the assessment of RNA purity is to calculate the ratio of absorbance at 260 and 280 nm (the A260:280 ratio) and at 260 and 230 nm (the A260:230 ratio). The development of microfluidic capillary electrophoresis has allowed the assessment of RNA quality with low volumes of sample (1 μl) through direct trace observation and automated calculation of the 28S:18S ratio (Schroeder et al. 2006). Currently the RNA Integrity Number (RIN) algorithm is the industry standard for RNA quality assessment.
Degradation of RNA might be a minor problem in hybridization-based microarray, in which each expressed gene is measured by a few short and distinct probes. One microarray study reported that only a small portion of probes (0.67%, 275/41,000) were significantly affected by RNA degradation and biological differences far outweighed RNA degradation (Opitz et al. 2010). In recent years, sequencing based platforms like RNA-seq have been extensively applied, in which RNA degradation can be more problematic as degraded RNA can distort yield of sequencing reads which represents abundance of transcripts. Thus RNA degradation could be a major source of variation when measuring gene expression using RNA-seq data (Wang et al. 2016). There is still an ongoing controversy as to what extent gene expression results are affected by RNA degradation in order to decide which partially degraded samples to include in an analysis. In particular, mineralised tissues are notoriously difficult as it can be difficult to obtain high quality RNA with the conventional phenol- chloroform protocols or commercial kits. Several protocols have been suggested to improve RNA quality from mineralised tissue including preserving samples in liquid nitrogen (Carter et al. 2012). The purpose of this study was to develop an improved protocol for processing samples and extracting RNA from hard tissues collected in a clinical setting with the limitations that this entailed. Here we evaluate the quantity and quality of RNA extracted from feline tissues focusing on teeth from TR free and TR affected cats for next generation sequencing. We also assessed the impact of RNA degradation on reference gene expression by quantitative PCR and on the quality of the RNA sequencing data.
Material and methods
Sample collection and tissue processing
Clinical tooth samples were collected from cats presented to the Hospital for Small Animals, The Royal (Dick) School of Veterinary Studies, The University of Edinburgh, UK with full owner’s consent. TR was diagnosed by the combination of oral examination and intraoral dental radiography (Heaton et al. 2004). The TR affected teeth were extracted by a veterinary surgeon using standard dental equipment under full general anaesthesia (Reiter and Soltero-rivera 2014). Further research samples including teeth, maxilla, mandibles and various soft tissues including muscle, liver, intestine and gingiva were collected at post mortem from cats and dogs euthanized for a wide range of medical reasons and donated to the school for research.
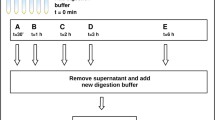
The clinical samples underwent one of the following processes: protocol A) immediately stored in RNAlater® solution (Thermo Fisher Scientific) (Fig. 1, Step 1. A) at room temperature or protocol B) snap frozen in liquid nitrogen before storage at −80 °C (Fig. 1, Step 1. B).

Workflow of tissue processing for RNA extraction
Post mortem samples consisting of mandibles or maxillae underwent the following processes: protocol C) immediately snap frozen in liquid nitrogen and stored at −80 °C until assessment of TR status by radiography (Fig. 1, Step 1. C). To phenotype TR status, frozen jaws were delivered to the radiographic facility in a liquid nitrogen carrier, radiographed, and then immediately put back into the liquid nitrogen. In order to extract the teeth from the frozen jaws, a standard dental extraction technique using dental equipment was used (Reiter and Soltero-rivera 2014). However, RNA extracted using this technique was badly degraded (average RINe = 4.0), which might have been caused by the heat produced by the dental burs during tooth extractions from the frozen samples. An alternative method was therefore developed where teeth were extracted from the alveolar sockets of the jaws using bone cutters and forceps while maintaining cold temperatures by working over dry ice.
Further tissue samples were also collected at post mortem (bones and various soft tissues), and processed according to: protocol D) tissues were trimmed into small pieces, snap frozen in liquid nitrogen and stored at −80 °C until RNA extraction (Fig. 1, Step 1. D).
RNA extraction
Two different RNA isolation protocols were compared: protocol 1 and 2. TRIzol reagent is a commonly used solution for soft tissues and is based on a guanidinium thiocyanate-phenol-chloroform extraction method (Chomczynski and Sacchi 1987). Protocol 1 was a modified version of the manufacturer’s protocol (TRIzol® reagent, Thermo Fisher Scientific, Fig. 1, Step 2). In brief, frozen tissues were placed in a beaded tube (Lysing Matrix D tubes, Fisher) with TRIzol® reagent (1 ml/100 mg of tissue). The sample tube was placed in the FastPrep FP120 (Thermo Savant) to agitate at a speed of 4 m/s for 20 s. Each tube was incubated at room temperature for 5 min before adding 0.2 ml of 1-bromo-3-chloropropane (BCP B9673, Sigma). After vigorously shaking the tube by hand for 15 s, the tube was left to stand at room temperature for 3 min. For phase separation, samples were centrifuged at 12,000 rpm for 15 min at room temperature and the upper aqueous layer was transferred to a new tube. An equal volume of isopropyl alcohol was added and incubated for 10 min on ice. To precipitate the RNA, the tube was centrifuged for 30 min at 12,000 rpm at room temperature. The supernatant was removed and the RNA pellet was washed with 1 ml of 75% ethanol. The sample was briefly vortexed and centrifuged for 30 min at room temperature at 12,000 rpm. The supernatant was removed and the RNA pellet was air-dried for 5 to 10 min before RNA was re-suspended in RNase free water.
Protocol 2 was modified from a previous study (Reno et al. 1997) and optimised for cartilage and tendon in our lab (Clements et al. 2006) (Fig. 1, Step 2). To facilitate disruption and pulverisation of hard tissues, we used a grinding machine consisting of a stainless steel jar and ball (Mixer Mill MM200, Retsch, Germany). The grinding jar and ball were submerged in liquid nitrogen for a few minutes before use to ensure that the sample remained frozen during homogenisation. TRIzol reagent (0.5 ml/100 mg of tissue) was added to the pre-chilled jar and allowed to freeze. Frozen tissue and the pre-chilled ball were placed into the jar and the lid firmly closed. The Mixer Mill MM200 was set at 30 Hz for 2 min to perform radial oscillations in a horizontal position. The pulverized tissue was transferred to a pre-chilled centrifuge tube containing TRIzol® reagent (0.5 ml/100 mg of tissue). After a brief mix, the mixture was left to stand for 30 min before being centrifuged at 12,000 rpm for 10 min at 4 °C. The supernatant was transferred into a clean tube and 200 μl of chloroform was added, mixed and left to stand at room temperature for 10 min. A further centrifugation step of 15 min at 12,000 rpm at 4 °C was performed and the clear supernatant was transferred into a clean tube. An equal volume of 70% ethanol was added to each tube and mixed by pipetting. RNA was washed and DNase digestion was performed according to the manufacturer’s instruction (RNeasy Mini Kit, Qiagen). RNA was eluted with 30 to 50 μl of RNase free water, and stored at −80 °C.
Measurement of RNA yields, purity and integrity
RNA yield was measured based on absorbance at 260 nm, and the A260:280 and the A260:230 ratios were used to assess the purity of RNA using NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific Inc.). The RNA Integrity Number equivalent (RINe) was determined utilising Agilent RNA ScreenTape assay kit following manufacturer’s protocol on the Agilent 2200 TapeStation. This produces an equivalent RIN (RINe) value which is comparable to RIN generated by the industry standard 2100 Bioanalyzer system for the quality assessment of RNA as an industry standard bioanalyzer system on a microcapillary electrophoretic RNA separation platform (Schroeder et al. 2006). The Agilent software produces a RNA integrity number, the correlating electropherogram, and a gel image for each sample.
Assessment of reference gene expression levels according to RINe values
To investigate the suitability of the feline tooth RNA samples collected in our gene expression study, RNA samples were divided into three categories based on RINe values (High: RINe ≥ 6, Moderate: 5 < RINe < 6, Low: RINe ≤ 5) and quantitative real time RT PCR was performed for each sample. In brief, 400 ng of RNA was reversed transcribed using Omniscript® Reverse Transcription (Qiagen) with random primers at 37 °C for 1 h according to the manufacturer’s instructions. Real-time PCR was performed with high affinity, double stranded DNA-binding dye SYBR green (Takyon™ qPCR MasterMixes for SYBR® assays, Eurogentec) using Stratagene MX3000P qPCR system (Agilent Technologies). The sets of reference gene primers were selected from previously validated feline reference genes (Penning et al. 2007) under universal cycling conditions including pre-incubation at 50 °C for 2 min and 95 °C for 3 min, 40 cycles of amplification at 95 °C for 10 s, 60 °C for 20 s and 72 °C for 30 s, and followed by data analysis at 95 °C for 1 min, 60 °C for 15 s and 95 °C for 15 s. All samples were performed in triplicate and cycling threshold (Ct) values of reference genes were generated to compare gene expression level of each RINe category.
Generation of cDNA libraries and RNA sequencing
Tested RNA samples were sent to our academic facility (Edinburgh Genomics, The University of Edinburgh, UK) for cDNA library production and RNA sequencing. Briefly, one microgram of RNA from each of the thirteen samples and 0.8 μg of RNA from one low quantity sample were used to generate fourteen cDNA libraries using the Illumina TruSeq stranded mRNA sample preparation kit according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Paired-end sequencing was performed using the Hiseq 4000 system (Illumina). The low quality reads were filtered by Phred quality score (Q score 30) (Ewing and Green 1998) and 3′ adapter were trimmed with cutadapt (version 1.8.3) (Martin 2011). All the raw reads have been submitted to the European Nucleotide Archive (ENA) under accession PRJEB24183 (ENA, https://www.ebi.ac.uk/ena).
Mapping to reference genome, generation of mapping statistics, and generation of multidimensional scaling (MDS)
Annotation and alignment were performed using STAR (version 2.5) comparison to the Felis catus genome (version 6.2) in the Ensembl database (Hubbard et al. 2002). Alignment files were generated in the bam format for each sample. The read counts from each sample were generated using HTSeq (version 0.6.0.1) (Anders et al. 2015) with mode ‘union’. Reads were mapped to Felis_catus_6.2 using STAR (version 2.5). Duplicate reads were found using picard tools (version 1.141). Generation of MDS plots were generated using plotMDS function from edgeR package (version 3.12.1) to visualise the level of similarity of individual cases of a dataset.
Statistical analysis for RINe values and reference gene expression by qPCR
Data were analysed using Minitab® 17 Statistical Software (Minitab Ltd., UK). p values below 0.05 were considered statistically significant and specific p values were noted. When data followed a normal distribution, parametric tests were performed. If parametric tests could not be applied, non-parametric testing was performed. One-way ANOVA was used to compare differences between more than two treatment groups. Two sample t-tests or Mann Whitney U-tests were used to compare differences between two groups. Correlations between two groups were assessed using Pearson’s correlation coefficient.
Result
Good RNA yields were obtained from tooth samples regardless of homogenisation step used
Obtaining a good yield of RNA from dental tissues is a challenge because individual teeth are relatively small and acellular compared to soft tissues. The wet weight of teeth varied widely and ranged from 10 to 500 mg depending on the type of tooth (incisors, molars, canines or premolars) or to which extent the teeth were affected by TR. In this study, the protocols described produced good yields of total RNA regardless of the homogenisation step used. Samples between 100 and 500 mg of wet weight produced 2.0 to 12 μg of total RNA which was higher than the minimum quantity requirement set for next generation sequencing in our facility (Edinburgh Genomics, http://genomics.ed.ac.uk/resources/sample-requirements). However, some samples weighing less than 50 mg produced low yields of RNA with total RNA less than 1.0 μg (50 to 60 ng/μL). These samples failed to reach the minimum yield of total RNA required (1.2 to 2.2 μg of total RNA) for the assessment of RNA quality for standard RNA-seq (Genomics 2017).
Tissue processing at collection significantly impacted RNA integrity
The tissue processing procedures were performed according to four different protocols depending on the type of tissue and source of samples (Fig. 1). Teeth preserved in RNAlater® solution (protocol A) obtained the lowest RINe values with high variations (3.04 ± 1.8) indicating highly degraded RNA (Fig. 2). This procedure resulted in the expected distinct peaks of 28S and 18S combining into one peak (Fig. 2, A top right panel). Tissue processing protocols B, C and D showed improved RINe values (Fig. 2) but still with partially degraded RNA present. Partial degradation of RNA is indicated by the presence of several extra small peaks to the left of the 18S peak (Fig. 2, bottom right panel). The main difference between protocol A and the other three protocols was the method of tissue storage and RNA stabilisation following collection. While protocol A used RNAlater® solution for RNA stabilisation at room temperature, the other protocols were immediately snap frozen in liquid nitrogen.

Average RINe values obtained from the different sample processing protocols. Left graph reports each RINe value as the average, bars show standard deviation (SD), n = number of samples tested. When teeth were immediately frozen in liquid nitrogen (B: clinical samples, C and D: post mortem samples), RINe values were improved compared to samples stored in RNAlater® at room temperature (A) (p < 0.01 by One-way ANOVA). Right panel shows representative gel images and electropherograms of RNA extracted using protocols A and C. Highly degraded RNA (top right, protocol A) show missing 18S peak and indistinct bands on the electropherogram. Protocol C (bottom right) produced only moderately degraded RNA. The 28S and 18S bands are distinguishable on the electropherograms but several minor peaks can be seen to the left of the 18S peak
The homogenisation steps and tissue type had a significant impact on RNA integrity
Tissue homogenisation is a critical step at which significant RNA degradation can occur. Protocol 1 (Fig. 1, Step 2, homogenisation at room temperature using beaded tubes) resulted in incomplete homogenisation of the mineralised samples. This indicated that this procedure was insufficient for these highly mineralised tissues. When cat teeth were pulverized in cold TRIzol® reagent according to protocol 2 (Fig. 1, Step 2), RINe values were improved. In parallel studies, we also extracted RNA from dog teeth and in agreement with our cat samples we also obtained improved results with protocol 2 for these tooth samples (Table 1). There were two reasons why we wanted to also test dog tissues. One was due to the greater availability of dogs’ teeth to test our protocols, and secondly to test if these protocols were also applicable for dog’s teeth. Dog’s teeth are bigger and harder than cat’s teeth, and testing these teeth ensured a wider application of our protocol for other animals including humans.
To compare the different homogenisation steps on different tissue types, teeth and soft tissues from dogs were used. Regardless of protocol used, RNA from soft tissues consistently achieved higher RINe values (Table 2), suggesting that the less vigorous homogenisation step using beads was sufficient to prevent significant RNA degradation when processing soft tissues. RNA extracted from cat soft tissues such as muscle, liver, intestine and gingiva using protocol 2 also showed improved RINe values (average 6.9) when compared to RNA obtained from hard tissues such as bone and teeth (average 4.9, Table 2). In order to compare across species we processed some dog tissues according to the same protocols; dog soft tissues yielded higher RIN e values (average 7.1 and 6.6 with protocol 1 and 2 respectively) than those of tooth samples (Average 2.7 and 4.7 in Table 2) demonstrating how the protocols used are less critical when working with soft tissues rather than mineralised tissues.
There was no correlation between RNA purity parameters and RNA integrity values
To investigate if there is a correlation between RNA purity and integrity, extracted RNA samples from clinical samples collected according to protocol B (snap frozen in liquid nitrogen) and post mortem samples collected according to protocol C (snap frozen within the jaw) were categorised into High, Moderate, and Low RINe groups according to their RINe values (High: RINe ≥ 6, Moderate: 5 < RINe < 6, Low: RINe ≤ 5, n = 5 for each group (Fig. 3, B). All samples in the high RINe group presented on the gel images and electropherograms with characteristics typical of intact RNA: tall 28S ribosome peaks double the height of the 18S peak, and with only a few small peaks of small RNAs and only minor signs of RNA degradation. The moderate RINe group showed partially degraded ribosome peaks with a moderate height of 28S peaks and extra peaks observed below the 18S peak. In the low RINe group, 28S peaks were shorter than the 18S peaks and there were several degraded RNA productions present (Fig. 3, A). We then compared the A260:280 ratios between the three groups using Spearman’s correlation coefficient. There was no statistically significant difference between any two groups which suggests that the A260:280 ratio does not correlate with or represent RNA integrity (P = 0.781, Spearman’s correlation coefficient). Similar patterns were observed when the A260:230 ratio was used (Fig. 3, Table 3), and no correlation was observed between the A260:280 ratio and the RINe values (P = 0.197, Spearman’s correlation coefficient).

RNA purity and integrity. Fifteen RNA samples were divided into three groups: Low, Moderate and High according to their average RINe values. On the left three gel images and electropherograms are shown as representative images of each group (A). Arrow heads on the electropherogram indicate small RNA and arrows indicate degraded RNA productions. The average RINe values from each category was statistically different between the groups (*p < 0.05, Mann-Whitney U-test) (B). In terms of both A260:280 ratio and A260:230 ratio, there were no statistically significant differences between any two groups (P > 0.1, Mann-Whitney U-test) (C and D). Low: RINe ≤5, Moderate: 5 < RINe <6, High: RINe ≥6. Bars indicate standard deviation
RNA degradation levels had an impact on reference gene expression by qPCR
To investigate the impact of RNA degradation on gene expression levels, the three groups were tested for expression of three reference genes, glyceraldehyde-3-phosphatedehydrogenase (GAPDH), ribosomal protein L17 (RPL17) and ribosomal protein S19 (RPS19) using qPCR. The qPCR reactions were performed using the same amount of synthesized cDNA from each RNA sample and cycling threshold (Ct) values for each reference gene were generated. There was no significant difference in the expression of GAPDH between high and moderate RINe groups, however the low RINe group revealed significantly higher Ct values indicating low expression of GAPDH (Fig. 4). For RPS19 the Ct values were significantly different between the high and the moderate RINe groups (Fig. 4). As high Ct value indicates lower gene expression, degraded RNA correlated with a lower expression of reference genes. Whereas expression of GAPDH and RPS19 in this study was affected by RNA degradation, expression of RPL17 was less affected by the level of RNA degradation.

RNA quality and reference gene expression. Expression levels of three reference genes were generated as average of cycling threshold (Ct) with standard error of the mean (bars). (GAPDH * p < 0.05 by two sample t-test, RPS19 *p < 0.05 by two sample t-tests, RPL17 (P > 0.1 by Mann-Whitney U-test). Low: RINe ≤5, Moderate: 5 < RINe <6, High: RINe ≥6. Bars indicate standard deviation
Low yield affects quality of RNA-seq data regardless of RNA integrity
Fourteen RNA samples from TR free (7 samples collected at post mortem according to protocol C) and TR affected teeth (4 samples collected at post mortem according to protocol C and 3 clinical samples collected as described in protocol B) were chosen from the feline tooth resorption study. The quality of cDNA libraries and sequencing was measured by the percentage of duplicated reads and mapped reads (Table 4) (Supplementary Table S1). As cDNA library complexity is determined by duplication rate, high duplication rate is often caused by degraded and fragmented RNA. Interestingly one sample that yielded low quantity of RNA but achieved a high RINe value (RINe = 7.3) showed much higher duplication rate than any of the other samples. After annotation to Felis catus genome, sequencing reads of all libraries except for the one library with the low input RNA mapped to 94 to 95% of the feline genome. The low quantity RNA sample with a high RINe value only mapped 87.53% of the reads to the feline genome. RNA-seq data from each sample as genome wide expression were plotted to investigate the similarity of whole genome expression profile within the given samples (Fig. 5). Interestingly the low yielding RNA sample with a high RIN (Supplementary Table S1) was defined as an outlier from the other samples which might have been caused by the low expression of genes and low quality of the cDNA library due to low yield of RNA rather than quality of RNA (Fig. 5).

The effect of low yield RNA sample on RNA-seq two-dimensional MDS plot of genome-wide expression profiles. (A) MDS plot showed that the one low quantity RNA sample (highlighted with a square) was displayed as an outlier while other samples were clustered together. (B) When the low yielding RNA sample was excluded from the data set, data from all samples were displayed evenly and clustered. There was no obvious outliers
Discussion
Selection of high quality and quantity RNA is important for downstream gene expression studies using next generation sequencing. The first challenge of extracting RNA from certain types of clinical samples e.g. extracted teeth, is to obtain a good yield of RNA from potentially small quantities of tissue. The wet weights of intact adult teeth range from 100 to 200 mg for mandibular premolars, 400 to 550 mg for canines, and 10 to 40 mg for incisors. Even the larger carnassials (the third molar in the maxillary arcades and the first molar in mandibular arcades) were often less than 500 mg. Like other mineralised tissues, dental tissues are relatively acellular compared to soft tissues as they contain a large amount of hydroxyapatite and collagen (Ten Cate and Nanci 2003). In the cases of these clinical samples, only small quantities of tissues were available due to the loss of dental tissues during the TR disease progression. In some cases, treatment of TR such as partial crown amputation in case of type 2 TR can further reduce availability of tissues (Reiter and Soltero-rivera 2014). TR affected tissues undergo apoptotic processes and stimulate inflammation which can lead to extensive loss of tissues (Dupont and Debowes 2002; Booij-vrieling et al. 2010). Here we compared tissue processing and RNA isolation protocols to maximise the yield of RNA and improve the quality of the RNA obtained from feline teeth. In our study, the average yield of RNA from feline tooth samples with a wet weight of less than 50 mg was below 1.0 μg of total RNA. This quantity of RNA limits the application for expression studies. For example, in many gene expression studies (Delaurier et al. 2002; Kessler et al. 2009; Gunter et al. 2013), an input of 0.5 to 2 μg of RNA for reverse transcription was used. Microarray or RNA sequencing studies require a relatively low amount of RNA due to their high sensitivity. For instance, for cDNA library construction using the TruSeq stranded mRNA kit, 100 ng of RNA can be used as a minimum input according to the manufacturer’s protocol. However, larger quantities of RNA are required to assess the quality and quantity of RNA (e.g. the minimum required by our facility, Edinburgh Genomics is 1.2 μg: 0.2 μg for quality control and 1 μg for library preparation). Therefore the total recommended amount of RNA is 2.2 μg to allow for two attempts at library preparation. In this study, we used 0.8–1 μg of total RNA. As tooth samples with wet weights of 100 to 500 mg yielded more than 1.0 μg of total RNA, we would recommend that gene expression or sequencing studies aim for at least 100 mg of wet weight as a minimum tooth sample size.
Different tissue processing methods were compared based on the RNA quality obtained. As endogenous ribonucleases (RNAse) are widely present in tissues, RNA can be rapidly degraded. To minimize RNA degradation tissue samples are routinely snap frozen by immersion in liquid nitrogen immediately upon collection and then homogenized in the presence of a powerful protein denaturant (chaotropic agent usually guanidinium isothiocyanate). However as liquid nitrogen is often not readily available when collecting clinical samples from most veterinary practices, RNAlater® is a commonly used RNA stabilization solution because of the convenience of tissue storage at room temperature. However, when tooth samples were extracted and stored in RNAlater®, the extracted RNA was highly degraded. This was likely to be due to poor permeability of RNAlater® into the highly mineralised teeth. In order to achieve effective preservation of tissue using this reagent, tissues need to be dissected into small pieces to allow adequate diffusion of the chemical into the tissues (Lader 2012). Other studies have also reported the limited application of RNAlater® in hard tissues (Clements et al. 2006; Carter et al. 2012). Our data showed that snap freezing using liquid nitrogen improved RNA quality. However, the necessity for liquid nitrogen is likely to limit the application of this processing protocol outside of the laboratory environment. In order to collect large number of samples in a clinical setting, the ability to collect and store samples at room temperature would be a great advantage. For example it allows for recruitment of several practices to contribute samples to clinical studies even if they have no access to liquid nitrogen. Our study does however suggest that for soft tissue samples that can be trimmed into small pieces, collection and storage in RNAlater® is likely to be adequate.
In our study, tissue samples that were stored at suboptimal temperatures during phenotyping (assessment of TR status, i.e. became defrosted during the dental radiographic procedure) or were subjected to potentially high temperature during processing (i.e. while extracting the teeth using dental burs at post mortem) also showed marked RNA degradation. By optimising our protocols, we managed to achieve average RINe values of between 5 and 6. The average RIN e value reported in this study is lower than what is reported in other studies using soft tissues. However other studies extracting RNA from mineralised tissues reported similar limitations where RNA from hard tissues was more degraded than RNA extracted from soft tissues (Sun et al. 2012; Carter et al. 2012).
We also investigated if there were any correlation between RNA purity as measured by the A260:280 and A230:260 ratios and RNA integrity as measured by RINe values. The A260:280 ratios were similar in all RNA samples regardless of RINe value. Whilst the A230:260 ratios, were more variable between samples than the the A260:280 ratios, there was no correlation to the RINe values of the samples. This implies that these two indicators cannot be reliably used as parameters of RNA integrity or quality.
To investigate the impact of RNA integrity and quality on gene expression, expression of three reference genes was compared. When equal amount of RNA is reverse-transcribed and analysed by qPCR, the values of Ct of the reference gene is expected to be similar across samples (Conde et al. 2012; Belluoccio et al. 2013; Chapman and Waldenstrom 2015). Also, it should show minimal variability in its expression. Two reference genes, GAPDH and RPS19 showed different expression levels depending on the degradation of RNA with the high RINe value group showing the highest expression with the lowest Ct. Only RPL17 was similar between the three groups. We recognise that reference genes may intrinsically vary between samples or tissues, but these reference genes have been extensively tested and used in our lab on a range of feline tissues and cell lines and have overall proved the most consistent. The difference in reference gene expression might be affected by the extent of degradation of RNA, but it may also imply that certain genes have a more stable gene expression in a specific type of tissue (Penning et al. 2007).
For further investigation of the impact of RNA quality and quantity on RNA sequencing, we evaluated RNA-seq data related to RNA quantity and quality. We collected 13 RNA samples with RNA yields of more than 1.0 μg of total RNA and one clinical sample with a very low yield of total RNA. Thirteen TruSeq stranded mRNA libraries were generated using 1.0 μg of total RNA, while for the low yielding sample only 0.8 μg of total RNA was used. Our data revealed that the RNA-seq data from the low input RNA sample was an outlier which skewed the data set of the whole genome expression profiles. It was documented that degraded RNA samples using poly A selection for preparation of cDNA library are likely to cause 3′ mapping bias that might have an effect on false positives in differential expression and the quantification of duplicate reads (Sigurgeirsson et al. 2014). In our data, duplication rates and percentage of mapped reads were similar across a range of RINe values. The cDNA library complexity and duplication profiles were affected by the low input of RNA rather than by RNA integrity. Therefore the low yield RNA sample was not suitable for RNA-seq analysis and was excluded in the final data analysis. We concluded that the overall yield of the RNA for input into RNA-seq was more important than the quality of RNA for RNA-seq quality control. Recently, there have been technical advances made to allow for low input of RNA or degraded RNA from clinical samples, formalin-fixed paraffin-embedded samples or single cell samples to be used (Deluca et al. 2012; Adiconis et al. 2013; Gallego Romero et al. 2014; Wu et al. 2014). Therefore for gene expression studies, and in particular for high throughput sequencing, RNA extraction protocols, generation of cDNA libraries, RNA-seq and bioinformatics need to be considered and customised. It is likely there will be further opportunities to overcome these limitations in the future.
The goal of this study was to identify improved protocols for RNA extraction from calcified tissues to maximise its application for gene expression studies using precious clinical samples. Here we optimised a protocol to maximise RNA yield with improved quality extracted from feline teeth in a clinical setting and at post mortem. We suggest that minimum amounts of sample are required for a conventional gene expression study. We also identified that it was critical to keep the samples frozen in liquid nitrogen and /or dry ice during the entire tissue processing procedure including the tissue homogenisation process. We further investigated parameters commonly utilised to assess RNA purity and integrity and its correlation to gene expression and RNA-seq. Overall the degradation of RNA was inevitable to some extent when extracting RNA from mineralised tissues such as teeth, but with our optimised protocol we managed to extract RNA with only moderate degradation, and the RNA-seq data from these samples still yielded usable data for further analysis. Furthermore this detailed protocol should have beneficial applications for RNA processing and analysis in mineralised tissue from many animal species either obtained via the clinic or in a research environment.
Change history
27 November 2018
The authors regret that the original version of the above article contained errors in the Figs. 3, 4 and Tables 3 legends. The errors has been corrected.
References
Adiconis X, Borges-rivera D, Satija R, Deluca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, Gnirke A, Pochet N, Regev A, Levin JZ (2013) Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods 10:623–629
Anders S, Pyl PT, Huber W (2015) HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169
Bei M (2009) Molecular genetics of tooth development. Curr Opin Genet Dev 19:504–510
Belluoccio D, Rowley L, Little CB, Bateman JF (2013) Maintaining mRNA integrity during decalcification of mineralized tissues. PLoS One 8:e58154
Booij-vrieling HE, Ferbus D, Tryfonidou MA, Riemers FM, Penning LC, Berdal A, Everts V, Hazewinkel HA (2010) Increased vitamin D-driven signalling and expression of the vitamin D receptor, MSX2, and RANKL in tooth resorption in cats. Eur J Oral Sci 118:39–46
Carter LE, Kilroy G, Gimble JM, Floyd ZE (2012) An improved method for isolation of RNA from bone. BMC Biotechnol 12:5
Chapman JR, Waldenstrom J (2015) With reference to reference genes: a systematic review of endogenous controls in gene expression studies. PLoS One 10:e0141853
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Clements DN, Vaughan-thomas A, Peansukmanee S, Carter SD, Innes JF, Ollier WE, Clegg PD (2006) Assessment of the use of RNA quality metrics for the screening of articular cartilage specimens from clinically normal dogs and dogs with osteoarthritis. Am J Vet Res 67:1438–1444
Conde MC, Nedel F, Campos VF, Smith AJ, Nor JE, Demarco FF, Tarquinio SB (2012) Odontoblast RNA stability in different temperature-based protocols for tooth storage. Int Endod J 45:266–272
Darcey J, Qualtrough A (2013) Resorption: part 1. Pathology, classification and aetiology. Br Dent J 214:439–451
Delaurier A, Allen S, Deflandre C, Horton MA, Price JS (2002) Cytokine expression in feline osteoclastic resorptive lesions. J Comp Pathol 127:169–177
Deluca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G (2012) RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28:1530–1532
Desjardins P, Conklin D (2010) NanoDrop Microvolume Quantitation of Nucleic Acids e2565
Dupont GA, Debowes LJ (2002) Comparison of periodontitis and root replacement in cat teeth with resorptive lesions. J Vet Dent 19:71–75
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II Error probabilities. Genome Res 8:186–194
Gallego Romero I, Pai AA, Tung J, Gilad Y (2014) RNA-seq: impact of RNA degradation on transcript quantification. BMC Biol 12:42
Genomics E (2017) Sample requirements [Online]. Available: https://genomics.ed.ac.uk/resources/sample-requirements [Accessed 08 Aug 2017]
Gunter HM, Fan S, Xiong F, Franchini P, Fruciano C, Meyer A (2013) Shaping development through mechanical strain: the transcriptional basis of diet-induced phenotypic plasticity in a cichlid fish. Mol Ecol 22:4516–4531
Heaton M, Wilkinson J, Gorrel C, Butterwick R (2004) A rapid screening technique for feline odontoclastic resorptive lesions. J Small Anim Pract 45:598–601
Hu S, Parker J, Wright JT (2015) Towards unraveling the human tooth transcriptome: the dentome. PLoS One 10:e0124801
Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Huminiecki L, Kasprzyk A, Lehvaslaiho H, Lijnzaad P, Melsopp C, Mongin E, Pettett R, Pocock M, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-vidal A, Vastrik I, Clamp M (2002) The Ensembl genome database project. Nucleic Acids Res 30:38–41
Kessler Y, Helfer-hungerbuehler AK, Cattori V, Meli ML, Zellweger B, Ossent P, Riond B, Reusch CE, Lutz H, Hofmann-LEHMANN R (2009) Quantitative TaqMan real-time PCR assays for gene expression normalisation in feline tissues. BMC Mol Biol 10:106
Lader ES (2012) Methods and reagents for preserving RNA in cell and tissue samples. Google Patents
Manchester KL (1996) Use of UV methods for measurement of protein and nucleic acid concentrations. Biotechniques 20:968–970
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads 2011, 17
Opitz L, Salinas-riester G, Grade M, Jung K, Jo P, Emons G, Ghadimi BM, Beissbarth T, Gaedcke J (2010) Impact of RNA degradation on gene expression profiling. BMC Med Genet 3:36
Penning LC, Vrieling HE, Brinkhof B, Riemers FM, Rothuizen J, Rutteman GR, Hazewinkel HAW (2007) A validation of 10 feline reference genes for gene expression measurements in snap-frozen tissues. Vet Immunol Immunopathol 120:212–222
Reiter AM, Soltero-rivera MM (2014) Applied feline oral anatomy and tooth extraction techniques: an illustrated guide. J Feline Med Surg 16:900–913
Reno C, Marchuk L, Sciore P, Frank CB, Hart DA (1997) Rapid isolation of total RNA from small samples of hypocellular, dense connective tissues. Biotechniques 22:1082–1086
Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3
Sigurgeirsson B, Emanuelsson O, Lundeberg J (2014) Sequencing degraded RNA addressed by 3' tag counting. PLoS One 9:e91851
Simmer JP, Richardson AS, Wang SK, Reid BM, Bai Y, Hu Y, Hu JC (2014) Ameloblast transcriptome changes from secretory to maturation stages. Connect Tissue Res 55(Suppl 1):29–32
Sun JX, Horst OV, Bumgarner R, Lakely B, Somerman MJ, Zhang H (2012) Laser capture microdissection enables cellular and molecular studies of tooth root development. Int J Oral Sci 4:7–13
Ten Cate AR, Nanci A (2003) Structure of the oral tissues. In: Nanci A, Ten Cate AR (eds) Ten Cate's oral histology : development, structure, and function. Sixth edition. Mosby, St. Louis, Mo
Wang L, Nie J, Sicotte H, Li Y, Eckel-passow JE, Dasari S, Vedell PT, Barman P, Wang L, Weinshiboum R, Jen J, Huang H, Kohli M, Kocher JP (2016) Measure transcript integrity using RNA-seq data. BMC Bioinform 17:58
Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, Quake SR (2014) Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods 11:41–46
Acknowledgements
This work was supported by the BBSRC and Fiona and Ian Russell Seed Corn Grant Fund. In addition, the PhD studentship of S. Lee was funded by The University of Edinburgh under Edinburgh Global Research Scholarship and The Sym Charitable Trust.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure of potential conflicts interest
The authors declare that they have no conflict of interest.
Additional information
The original version of this article was revised: In the legend of Figures 3 and 4, the p value “P < 0.05” should instead have read “p < 0.05” and in the Table 3 the letter “P” of the “P value” should have been small letter “p” and are italicized. Figure 3 legend read as “RINe values” and “RINe ≤ 5, Moderate: 5 < RINe < 6, High: RINe ≥ 6.” contains the subscript letter "e" instead of a superscript letter "e". This should have been read as “RINe values” and ”RINe ≤5, Moderate: 5 < RINe < 6, High: RINe ≥ 6”.
Electronic supplementary material
ESM 1
(DOCX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lee, S., Trivedi, U., Johnson, C. et al. Optimised isolation method for RNA extraction suitable for RNA sequencing from feline teeth collected in a clinical setting and at post mortem. Vet Res Commun 43, 17–27 (2019). https://doi.org/10.1007/s11259-018-9739-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-018-9739-8