
Abstract
Between January, 2013 and December, 2014, there was a lumpy skin disease (LSD) outbreak that affected cattle in different localities of Zimbabwe. The outbreak resulted in severe economic losses to the livestock industry. A retrospective study was conducted by examining stored veterinary records of the LSD outbreak at the Central Veterinary Laboratory (CVL) in Harare, Zimbabwe. Over the 2-year period, a total of 10,038 cases and 880 deaths (8.77 %) were recorded. LSD was reported from all regions of the country, with the highest incidence occurring in Mashonaland West (30.95 %) and Midlands province (14.59 %). The frequency of reported outbreaks was highest in March and April, with the lowest reported cases occurring in November. A total of 25 representative specimens (skin biopsies) were collected from nodular skin lesions of infected cattle, and after viral DNA isolation, the P32 gene was successfully amplified, by using PCR, in 88 % (22/25) of all assayed specimens. Out of the 22 samples that showed amplification, 16 (73 %) were selected for DNA sequencing, and from these, 13 sequences were submitted to GenBank and assigned accession numbers: KX033494, KX033495, KX033496, KX033497, KXO33498, KX033499, KX033500, KX033501, KX033502, KX033503, KX033504, KX033505 and KX033506. Phylogenetic analyses of the 13 sequences was done by using MEGA 7 and showed that the viruses formed two major clusters implying that at least two strains of LSDV are in circulation in Zimbabwe. This study provides the first report on the incidence and molecular characterisation of LSDV in Zimbabwe.
Similar content being viewed by others

Introduction
Lumpy skin disease (LSD) is an infectious, eruptive, occasionally fatal disease of cattle caused by the lumpy skin disease virus (LSDV), a member of the capripoxvirus genus. LSDV is sometimes referred to as the Neethling virus (Salib and Osman 2011). LSDV, sheep pox virus (SPPV) and goat pox virus (GTPV) fall within the chordopoxvirus (ChPV) subfamily of Poxviridae. The virus mainly affects cattle and zebus but has also been seen in giraffes, African buffalo and impalas.
Lumpy skin disease was first seen as an epidemic in Zambia (then Northern Rhodesia) in 1929 (Morris 1931), and between 1943 and 1945, cases occurred in Botswana (Bechuanaland), Zimbabwe (Southern Rhodesia) and the Republic of South Africa (Davies 1991a). Since then, lumpy skin disease has affected cattle throughout Africa, including countries like Kenya (Davies 1982), Egypt (Salem 1989) and Sudan (Ali and Obied 1977). In sub-Saharan Africa, LSD is now enzootic in all the countries in which it has occurred and has proved impossible to eradicate. Traditionally, lumpy skin disease is found in southern and eastern Africa, but in the 1970s, it extended northwest through the continent into sub-Saharan West Africa. Since 2000, it has spread to several countries of the Middle East including Israel, Iran, Syria and in 2013 was confirmed in Turkey (Al-Salihi and Hassan 2015; Tuppurainen et al. 2015).
Transmission of the LSD virus that is primarily by biting insects, biting flies (Stomoxys calcitrans and Biomyia fasciata) and mosquitoes (Culex mirificens and Aedes natrionus) can be a source for transmission of the disease (Chihota et al. 2001). Other vectors have been suggested; Tuppurainen et al. (2011) found molecular evidence suggesting that LSDV can be transmitted through hard (Ixodid) ticks (Rhipicephalus decoloratus, Rhipicephalus appendiculatus and Amblyomma hebraeum).
Other risk factors associated with the spread of LSD include a warm humid agro-climate, communal grazing/watering and introduction of new animals in a herd (Gari et al. 2010). LSD presents itself as an acute, subacute or inapparent disease with variable severity depending upon capripoxvirus strain and the host breed. LSD can be suspected whenever clinical signs indicate towards persistent fever, wide spread skin nodules (lumps), enlarged peripheral lymph nodes, conjunctivitis, keratitis, corneal opacity and oedema in the brisket and legs (Radostits et al. 2007). Animals recover slowly from the severe disease and may suffer from mastitis, pneumonia and formation of necrotic skin plugs leaving deep holes in the hide (Tuppurainen et al. 2011).
The World Organisation for Animal Health (OIE) categorises LSD as a notifiable disease because of the substantial economic impact of an outbreak (OIE 2010). The disease is more severe in cows at peak lactation and causes a sharp drop in milk yield, often leading to secondary bacterial mastitis. Temporary or permanent infertility may occur in cows and bulls. The emaciation of infected animals and a convalescence period lasting for several months causes a decreased growth rate in beef cattle (Tuppurainen and Oura 2012).
LSD generally has low mortality (less than 10 %) and varying (1–90 %) morbidity (Coetzer 2004; Salib and Osman 2011). However, the morbidity and mortality of the disease may vary considerably, depending on the breed and immunological status of the cattle population and the insect vectors involved in transmission. The abortion rate in pregnant cows may range from 1 to 7 % (Vorster and Mapham 2008).
LSD outbreaks tend to be sporadic, depending upon animal movements, immune status and wind and rainfall patterns affecting vector populations. The incidence of LSD is high during wet seasons when populations of flies are abundant, and the incidence decreases or ceases during the dry season (Gari et al. 2012). LSD has a different geographical distribution from that of sheep and goat pox, suggesting that cattle strains of capripoxvirus do not infect or transmit between sheep and goats (Ahmed and Kawther 2008). A high incidence of LSDV has been reported recently in other countries like Sultanate of Oman (Tageldin et al. 2014) and Ethiopia (Hailu et al. 2014).
The control of LSD can be achieved through vaccination, restriction of animal movement and eradication of infected and exposed animals. However, this requires adequate financial, infrastructural and human resources and information systems. Under the prevailing conditions in Zimbabwe it has not been possible to implement all these strategies, and thus, vaccination has been adopted as the most important practical approach to LSD control for many years. The Kenyan sheep and goat pox vaccine strain KS-1 has been in use because of its advantage of conferring cross-protection to LSD, in accordance with OIE recommendations (Brenner et al. 2009).
PCR methods for the identification of capripoxviruses through the detection of the P32 gene has been previously described (Ireland and Binepal 1998; Tuppurainen et al. 2005; Varshovi et al. 2009). The P32 gene corresponds to an envelope protein and is homologous to the P35 protein encoded by vaccinia virus H3L gene and is located on the membrane surface of a mature intracellular viral particle (Tulman et al. 2001). The P32 gene is highly conserved among capripoxviruses and has been used by researchers as a diagnostic tool for LSDV (El-Kholy et al. 2008) and for SPPV and GTPV (Varshovi et al. 2009). Sequence information generated from sequencing the P32 gene can be used to differentiate SPPV, GTPV and LSDV (Hosamani et al. 2004).
Due to the upsurge in LSDV cases reported to the CVL, Harare, as from 2013, this study was undertaken to investigate the incidence of LSD in Zimbabwe, by using both active disease follow-ups and analysis of retrospective data. We also sought to assay the PCR method that targets the P32 gene for detection and to characterise LSDV isolates by using molecular techniques as we believe this method has not been adopted in Zimbabwe yet.
Materials and methods
Retrospective data
Outbreak data for the entire country covering the period January, 2013 to December, 2014 was retrieved from the Central Veterinary Laboratory in Harare. Active outbreaks were investigated in eight provinces of Zimbabwe namely Manicaland, Mashonaland East, Mashonaland West, Mashonaland Central, Matebeleland North, Matebeleland South, Midlands and Masvingo. The collected data was recorded on Excel (Microsoft Corp., USA) spreadsheets. Stata software version 9 (StataCorp, College Station, Texas, USA) was used for descriptive statistical analysis. Morbidity, mortality and case fatality rates were estimated in accordance with different variable categories. Outbreak frequencies in relation to geography and season were depicted graphically. Maps were generated by using ArcGIS version 9.0 software (ESRI, Redlands, California, USA).
Sample collection
Animals were assessed for characteristic clinical signs of LSD, such as visible skin lesions, enlarged lymph nodes and fever by resident veterinarians. Nodule skin biopsies were then collected aseptically from acutely sick cattle, and the tissue samples placed in sterile universal bottles, prior to being transported on ice to the Virology Section at CVL, Harare. All samples were sent to Harare within 8 h of collection; upon arrival at CVL, the samples were stored at −20 °C until processing. Samples were collected from various localities which included Bulawayo, Mutare, Bindura, Rusape and Nyanga. A total of 25 stored skin tissues were used in this study.
Viral DNA extraction
DNA was extracted from tissue samples by using a Quick-gDNA™ Miniprep kit (Zymo Research Corp., USA), according to the manufacturer’s instructions. Prior to DNA extraction, 25–50 mg of skin tissue was frozen by using liquid nitrogen and then ground to a fine powder by using a chilled mortar and pestle. The crushed tissue was then transferred into 1.5-ml microcentrifuge tubes.
PCR amplification of the LSDV P32 gene
PCR amplification was done in a standard 25 μl PCR reaction. The reaction mixture included 12.5 μl of 2× Taq universal mastermix (Thermo Fisher Scientific, USA), 6.5 μl deionised water, 0.5 μl of P32 gene forward primer (5′-TCC-GAG-CTC-TTT-CCT-GAT-TTT-TCT-TAC-TAT-3′) and 0.5 μl of P32 gene reverse primer (5′-TAT-GGT-ACC-TAA-ATT-ATA-TAC-GTA-AAT-AAC-3′) and 5 μl of the DNA template. The primers were developed from the viral attachment protein encoding gene (Ireland and Binepal 1998). Negative controls comprised of a water control. The PCR was run in a thermocycler (Perkin Elmer GeneAmp PCR System 2400, USA) by using the following thermal cycling conditions: initial denaturation at 95 °C for 1 min, followed by 35 cycles of 95 °C for 30 s for denaturation, 55 °C for 30 s for annealing and 72 °C for 1 min for strand extension. Final elongation was done at 72 °C for 5 min followed by a subsequent hold temperature of 4 °C. The amplified products were then run along a 1.5 % ethidium bromide-stained agarose gel with a 100 bp DNA ladder (Invitrogen, USA) in TBE buffer for 1 h at 100 V and then viewed by using a UV transilluminator (Uvitec, UK). A Uvitec gel documentation system (Uvitec, UK) was used to capture the gel image.
Sequencing of amplified PCR products
Sequencing of purified PCR products was performed at Inqaba Biotec, Pretoria, South Africa, by using an automated ABI3500XL Genetic Analyser and BigDye terminator v3.1 cycle sequencing reactions (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. DNA data (chromatographs and sequences) were sent back by email for analysis. The same primers used to carry out the amplification of the P32 gene in the lab were also used in the sequencing reactions. The sequences were supplied in the form of ab1 files, and the sequence analysis was done by using Basic Local Alignment Search Tool (BLAST) in the NCBI databases; phylogenetic analysis was done by using Molecular Evolutionary Genetics Analysis version 7.0 (MEGA 7) (Kumar et al. 2015).
Results
Retrospective data analysis
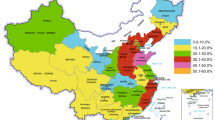
Analysis of retrospective data collected between January, 2013 and December, 2014 indicated that a total of 10,038 cases and 880 deaths (Table 1) were recorded at Central Veterinary Laboratory in Harare, Zimbabwe. The monthly incidence of LSD cases was similar in 2013 and 2014 (Figs. 1 and 2). The frequency of reported outbreaks was higher between January and May (with the highest numbers in March and April) and lower between June and December (with the lowest number in November) (Figs. 1 and 2). The analysis showed that LSD was reported from all regions of the country (Table 1). Over the 2-year period, outbreaks were frequently reported from Mashonaland West, 3107 (30.95 %), and Midlands, 1465 (14.59 %), and Matabeleland North, 499 (4.97 %), had the lowest cases (Table 1). A map showing the nationwide distribution of LSDV was generated by using ArcGIS version 9.0 software (ESRI, Redlands, CA, USA) and is shown in figure A1 in Appendix 1.

Incidence of LSD in different provinces per month in the year 2013

Incidence of LSD in different provinces per month in the year 2014
Molecular characterisation of the LSDV
PCR amplification of the LSDV P32 gene
The P32 gene was amplified by using simple PCR, and amplification products consistent with the expected amplicon size (192 bp) were obtained (Fig. 3).

PCR of the LSDV viral attachment gene (P32). Molecular weight marker (100 bp) (Invitrogen, USA) (lane M), negative control (lane N) and positive P32 gene amplicons of expected size (192 bp) (lanes 1–7)
The P32 gene was successfully amplified in 22 (88 %) of all assayed specimens. Out of the 22 samples that showed amplification, 16 (73 %) had strong PCR bands and were selected for DNA sequencing. After editing, 13 sequences were then submitted to GenBank which gave them the following accession numbers: KX033494–KX033506. Table 2 shows information relating to the 13 submitted sequences. Alignment of the 13 sequences by using BLASTn and BLASTx resulted in the identification of LSDV genotypes. All sequences in the alignment of the Zimbabwe outbreak of LSDV isolates were subjected to BLAST search versus the GenBank database. The alignment results confirmed the sequences to be LSDV isolates. The sequences were each compared against an LSDV South African isolate (accession number AF409137).
LSDV genotypes and phylogenetic analysis
Phylogenetic reconstruction of the 13 P32 sequences was done by using MEGA 7 and yielded the phylogenetic tree shown in Fig. 4. The evolutionary history was inferred by using the maximum likelihood method (supporting bootstrap values from 1000 replicates) based on the Tamura-Nei model (Tamura and Nei 1993). The analysis involved 15 nucleotide sequences (13 from the current study and 2 reference sequences). The two reference sequences were obtained from GenBank and included a South African LSDV isolate (AF409137) and an Iranian LSDV isolate (KT253435.1). The derived phylogenetic tree had two main clusters. Cluster I had most of the Zimbabwean isolates, and cluster II had the South African reference strain (AF409137) and one isolate from Mutare (KX033501). One of the LSDV isolates, KX033500, was an outlier and did not cluster with the other Zimbabwean isolates. As expected, the sequence of the Iranian LSDV isolate (KT253435.1) was different from all the other sequences.

Phylogenetic tree of LSDV P32 sequences after multiple sequence alignment by using the maximum likelihood method, supporting bootstrap values from 1000 replicates
Discussion
This study showed that LSD is widely distributed across the whole country (Figure A1). A few cases were recorded in places corresponding to game/wildlife reserves. LSD seems to have already spread extensively in Zimbabwe (Table 1); this means that slaughter policies are inappropriate and extensive vaccination campaigns are recommended (Davies 1991a). Since movement restrictions have already been imposed on livestock in Zimbabwe (in a bid to combat the foot and mouth disease virus), vaccination is probably the best route to follow to try and reduce the morbidity and economic effects of LSD. Follow-up vaccination of calves and revaccination programmes over a period of 2 to 3 years might greatly reduce the incidence of clinical disease (Davies 1991a).
The total number of LSD cases recorded between January, 2013 and December, 2014 (10,038) could be attributed to an increase in naïve cattle due to waning immunity against LSD and an increase in animal movement. Increase in total LSD cases from January to May (Figs. 1 and 2) could have been because of an increase in the vector population during this period as epidemics usually occur in the rainy seasons (Chihota et al. 2001), and this supports the hypothesis that the major route of LSD transmission is by insect vectors (Carn and Kitching 1995). Decrease in total LSD cases between June and December could be attributed to a reduction in the number of insect vectors. Besides transmission by the insect vector, there is also involvement of factors like cyclicity, vaccination status and movement control (Chihota et al. 2001). In this study, vaccination status of the animals could not be ascertained.
Retrospective data retrieved at CVL revealed that morbidity was highest in Mashonaland West (30.95 %), followed by Midlands (14.60 %) and lowest in Matebeleland North (4.97 %) (Table 1). The mortality (8.77 %) and morbidity observed during this outbreak were in agreement with other authors who report that the LSD is a disease with high morbidity (1–90 %) and low mortality (<10 %) rates (Davies 1991b; Salib and Osman 2011), and these values can fluctuate according to geography, climate, management conditions, immune status of the animal and breed and strain of virus involved (Tuppurainen and Oura 2012). Differences observed between the mortality rates in different regions (Table 1) could be attributed to variations in the agro-ecological zones and husbandry practices. Similar results were reported by Gari et al. (2010) in a questionnaire-based study of risk factors responsible for the spread of LSD in Ethiopia in which they concluded that the observed differences in prevalence and severity of disease could be linked to diversity of agro-climatic zones and farming practices.
Effective control of LSD requires rapid and accurate laboratory diagnosis supported by clinical findings (Tuppurainen et al. 2005). Accordingly, this work was conducted to try and validate the diagnostic tool that uses PCR and sequencing of the P32 gene to rapidly detect LSDV for possible use in the control of LSD outbreaks in Zimbabwe. Infected animals from which the specimens were collected exhibited typical signs of LSD. The collected specimens (skin biopsies) were used for viral genomic DNA isolation followed by a PCR assay. The PCR assay detected LSDV in 22/25 (88 %) skin biopsies from representative infected cattle. El-Kholy et al. (2008) managed to detect the LSDV in 80 (100 %) of the samples they assayed by using the same assay. The PCR result was fully correlated to field diagnosis based on clinical symptoms. It has been reported that although many other sources for virus detection such as blood, semen and milk were determined, skin biopsies were the best as they contain more viral particles for detection by PCR (Tuppurainen et al. 2005). Therefore, the current approach presents a suitable applicable diagnostic tool for LSDV in Zimbabwe. PCR tends to be the test of choice for rapid detection and identification of a LSD outbreak in strains because serological methods are deemed too time-consuming to be used as primary diagnostic methods even though they are useful for confirming LSD retrospectively (Heine et al. 1999; El-Kholy et al. 2008). Serological assessment of antibodies to a capripoxvirus may sometimes be difficult due to the cross-reactivity encountered with other poxviruses as well as to the low antibody titres elicited in some animals following mild infection or vaccination (Kitching and Hammond 1992).
A total of 16 samples were sequenced by using both the forward and reverse primers used in the PCR reaction, and after editing, 13 sequences were subsequently submitted to GenBank. All the 13 nucleotide sequences were analysed by using BLASTn and BLASTx. The sequencing results confirmed that the obtained isolates were indeed LSDV isolates. The sequences showed significant similarity at nucleotide level to each other and to a South African LSDV isolate (AF409137). Multiple sequence alignments showed high homology percentages (≥99 %) of the nucleotide sequences among local isolates of LSDV. Nevertheless, BLAST searches over the GenBank database and sequence alignments revealed that the isolates of LSDV are highly related (≥95 %) not only with other LSDV strains but also with other capripoxviruses (GTPV and SPPV). These results agree with the theory that all capripoxviruses are genetically related and originated from one ancestral lineage (Tulman et al. 2001). Phylogenetic analysis of the 13 LSDV isolates based on the nucleotide sequence of the P32 gene (Fig. 4) showed that the 13 isolates grouped into two distinct clusters. Cluster I had most of the Zimbabwean isolates (Fig. 4). Cluster I included KX033495, an isolate from Bulawayo (which is a city in the south of Zimbabwe), KX033494 an isolate from Bindura (a town in Mashonaland Central Province) and nine other isolates obtained from Manicaland province. This is significant as it points to the possibility that a similar strain of LSDV could be responsible for most of the LSDV cases circulating in the country. Cluster II had an isolate from Mutare (KX033501) which clustered with the South African reference sequence (AF409137). Another isolate also from Manicaland province (KX033500) was an outlier and was different from all the other Zimbabwean isolates (Fig. 4). This implies that different strains of LSDV could be in circulation in Manicaland. All the Mutare isolates were collected during the same outbreak (Table 2) implying that the observed differences could be genuine. However, there is need for further studies to be done to ascertain this claim. The small size of the envelope protein analysed in this study, and the fact that all strains of capripoxvirus whether of bovine, ovine or caprine origin seem to share a major neutralising site (Coakley and Capstick 1961) which makes cross-protection possible, means that the observed differences may not have any implications in as far as vaccination is concerned (OIE 2010).
Limitations
Potential biases in our study are related to the relatively small sample size of LSDV isolates used in this study. More P32 gene amplicons could have been sequenced.
Recommendations
LSD is now widespread in Zimbabwe and extensive vaccination campaigns should be carried out. We recommend the PCR assay described in this work and potentially the sequencing of the P32 gene to be used for routine identification and characterisation of LSDV isolates in Zimbabwe. The same gene (P32) could become the signature gene in differentiating the capripoxvirus including SPPV and GTPV. Further work needs to be done on the LSDV, SPPV and GTPV to determine their epidemiology in Zimbabwe. The virus investigated in this study needs to be isolated for future manipulations, most importantly the development of a vaccine.
Conclusion
The LSD outbreaks between 2013 and 2014 in Zimbabwe were caused by LSDV. The PCR assay that involves amplification of the P32 gene could prove to be a quick and reliable assay for the detection of LSDV field isolates. Phylogenetic analysis of different LSDV isolates by using the nucleotide sequences of the P32 gene could have practical applications in determining the origins and in the control of LSD.
References
Ahmed, W. and Kawther, S., 2008. Observations on lumpy skin disease in local Egyptian cows with emphasis on its impact on ovarian function, African Journal of Microbiology Research 2, 252–257.
Ali, B. H. and H. M. Obeid., 1977. Investigation of the first out-breaks of lumpy skin disease in Sudan. British Veterinary Journal 133, 184–189.
Al-Salihi, K. A. and I. Q. Hassan., 2015. Lumpy skin disease in Iraq: study of the disease emergence. Transboundary and Emerging Diseases 62, 457–462
Brenner, J., Bellaiche, M., Gross, E., Elad, D., Oved, Z., Haimovitz, M., Wasserman, A., Firedgut, O., Stram, Y., Bumbarov, V. and Yadin, H., 2009, ‘Appearance of skin lesions in cattle populations vaccinated against lumpy skin disease statutory challenge’, Vaccine, 27, 1500–1503.
Carn, V. M. and Kitching, R. P., 1995. An investigation of possible routes of transmission of lumpy skin disease virus (Neethling), Epidemiology and Infection 114, 219–226.
Chihota, C.M., Rennie, L. F., Kitching, R. P. and Mellor, P. S., 2001, ‘Mechanical transmission of lumpy skin disease virus by Ades aegypti (Diptera: Culicidae). Epidemiology and Infection 126, 317–321.
Coakley, W. and Capstick P.B., 1961. Protection of cattle against lumpy skin disease. Factors affecting small scale production of tissue culture propagated virus vaccine, Research in Veterinary Science 2, 369–371.
Coetzer, J., 2004. Lumpy skin disease, in J.A.W Coetzer & R.C Justin (eds), Infectious Diseases of Livestock. 2nd Ed, pp 1268–1276, Oxford University Press, Cape Town, South Africa.
Davies, F.G., 1982. Observations on the epidemiology of lumpy skin disease in Kenya. Journal of Hygiene (London) 88(1):95–102.
Davies, F. G., 1991a. Lumpy skin disease of cattle: a growing problem in Africa and the Near East, World Animal Review 68, 37–42.
Davies, F. G., 1991b. Lumpy skin disease, an African capripox virus disease of cattle, British Veterinary Journal 147, 489–503.
El-Kholy, A. A., Soliman H. M. T. and Abdelrahman, K. A., 2008. Polymerase chain reaction for rapid diagnosis of a recent lumpy skin disease virus incursion to Egypt, Arab Journal of Biotechnology 11, 293–302.
Gari, G., Waret-Szkuta, A., Grosbois, V., Jacquiet, P. and Roger, F., 2010. Risk factors associated with observed clinical lumpy skin disease in Ethiopia, Epidemiology and Infection 138, 1657–1666.
Gari, G., Grosbois, V., Waret-Szkuta, A., Babiuk, S., Jacquiet, P. and Roger, F., 2012. Lumpy skin disease in Ethiopia: seroprevalence study across different agroclimate zones, Acta tropica 123, 101–106.
Hailu, B., Tolosa, T., Gari, G., Teklue, T.and Beyene, B., 2014,’ Estimated prevalence and risk factors associated with clinical lumpy skin disease in north-eastern Ethiopia, Preventive Veterinary Medicine 115 (1–2), 64–68.
Heine, H. G., Stevens, M. P., Foord, A. J. and Boyle, D. B., 1999. A capripoxvirus detection PCR and antibody ELISA based on the major antigen P32, homologue of the vaccinia virus H3L gene, Journal of Immunological Methods 227, 187–196.
Hosamani, M., Mondal, B., Tembhurne, P.A., Bandyopadhyay, S.K., Singh, R.K. & Rasool, T.J., 2004. Differentiation of sheep pox and goat poxviruses by sequence analysis and PCR-RFLP of P32 gene, Virus Genes 29 (1),73–80.
Ireland, D. C. and Binepal, Y. S., 1998. Improved detection of Capripoxvirus in biopsy samples by PCR, Journal of Virological Methods 74(1), 1–7.
Kitching, R.P. and Hammond, J.M., 1992. Poxvirus, infection and immunity’, in I.M Roitt & P.1 Delves (Eds), Encyclopaedia of immunology 3rd Ed, pp1261- 1264, Academic press, London.
Kumar, S., Stecher, G. and Tamura, K., 2015. MEGA7; Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology & Evolution (Submitted)
Morris, J. P. A., 1931. Pseudourticaria. Department Animal Health Report, northern Rhodesia.
Radostits, O.M., Gay, C.C., Hinchcliff K.W. and Constable, P. D., 2007. Veterinary Medicine: A textbook of diseases of cattle, horses, sheep, pigs and goats, 10th Edn., WB Saunders Co., Philadelphia, USA.
Salem, A S.,1989. Lumpy Skin Disease in Egypt. In O.I.E. Disease Information. Vol 2. No.2.
Salib, F. A. and Osman, A. H., 2011. ‘Incidence of lumpy skin disease among Egyptian cattle in Giza Governorate, Egypt’, Veterinary World 4, 162–167.
Tageldin, M.H., Wallace, D. B., Gerdes, G. H., Putterill, J. F., Greyling, R. R., Phosiwa, M. N., Al-Busaidy, R. M. A. and Al-Ismaaily, S. S., 2014. Lumpy Skin Disease of Cattle: An emerging problem in the Sultanate of Oman, Tropical Animal Health and Production 46, 214–246.
Tamura, K and Nei, M. 1993. Estimation of the number of nucleic acid substitutions in the control region of mitochondrial DNA in humans & Chipanzees. Molecular Biology & Evolution 10, 512–526.
Tulman, E. R., Afonso, C. L., Lu, Z., Zsak, L., Kutish, G. F. and Rock, D. L., 2001. Genome of Lumpy Skin Disease Virus, Journal of Virology 75(15), 7122–7130.
Tuppurainen, E.S.M. and Oura, C.A.L., 2012. Lumpy skin disease: an emerging threat to Europe, the Middle East and Asia, Transboundary and emerging Diseases 59, 40–48. doi: 10.1111/j.1865-1682.2011.01242.x
Tuppurainen, E.S., Venter, E. H. and Coetzer, J. A., 2005. The detection of lumpy skin disease virus in samples of experimentally infected cattle using different diagnostic techniques, Onderstepoort Journal of Veterinary Research 72(2), 153–64.
Tuppurainen, E.S., Stoltsz, W.H., Troskie, M., Wallace, D.B., Oura, C.A., Mellor, P.S., Coetzer, J.A., Venter, E.H., 2011. A potential role for ixodid (hard) tick vectors in the transmission of lumpy skin disease virus in cattle, Transboundary and Emerging Diseases 58(2), 93–104. doi: 10.1111/j.1865-1682.2010.01184.x
Tuppurainen, E. S. M., Venter, E. H., Shisler, J. L., Gari, G., Mekonnen, G. A., Juleff N., Lyons, N. A., De Clercq K., Upton, C., Bowden, T. R., Babiuk, S. and Babiuk L. A. 2015. Capripoxvirus Diseases: Current Status and Opportunities for Control. Transboundary and Emerging Diseases. doi:10.1111/tbed.12444
Varshovi, H.R., Keyvanfar, H., Aghaiypour, K. Pourbakhsh, S.A., Shooshtari, A.H. and Aghaebrahimian, M., 2009. Capripoxvirus identification by PCR based on P32 gene, Archives of Razi Institute 64(1), 19–25.
Vorster, H. and Mapham, H., 2008. Pathology of lumpy skin disease. Livestock Health and Production Reviews 1, Jaargang, 10, 16–21.
World Organisation for Animal Health (OIE) 2010. – Lumpy skin disease. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. OIE, Paris: www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.04.14_LSD.pdf (accessed on 5 August 2015).
Acknowledgments
The authors thank the Central Veterinary Laboratory (CVL), Harare, for provision of isolates, especially Doctor Guri, Dr. Spargo and Dr. Kupa for collection of samples. We thank Dr. P. V. Makhaya for sanctioning the research. Special thanks to Iredale Mutengwa and Stephen Marambe for their technical assistance at CVL. The study was supported by the CVL and the National University of Science and Technology (NUST) Research Board.
Authors’ contributions
This work was conducted by J.M. (NUST), P.M. (NUST, CVL) and B.S. (CVL). P.M. was involved in the project design, was the principle researcher and carried out experimental work in the project. J.M. (NUST) and B.S. (CVL) were responsible for the project design and supervised P.M. (NUST, CVL) in all aspects of the project and also did laboratory work. B.S. (CVL) assisted in obtaining the samples used in this project. All three authors contributed to the writing of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no financial or personal relationship(s) which may have inappropriately influenced them in writing this paper.
Additional information
Significance of work
Lumpy skin disease is one of the most important socio-economic diseases of cattle in Zimbabwe. The disease is responsible for considerable losses in livestock production through loss of body condition, loss of milk production, lowered or complete loss of fertility in bulls and cows, abortion, permanent damage to hides (skin) and incurred costs in the purchasing of drugs to treat sick animals. It is hoped that this study will form or create a basis for further studies on the virus in Zimbabwe and also that the report will be the first or one of the few reports on molecular characterisation of LSDV in Zimbabwe which will contribute to the scarce literature on the subject.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure A1
(DOC 1551 kb)
Rights and permissions
About this article

Cite this article
Mafirakureva, P., Saidi, B. & Mbanga, J. Incidence and molecular characterisation of lumpy skin disease virus in Zimbabwe using the P32 gene. Trop Anim Health Prod 49, 47–54 (2017). https://doi.org/10.1007/s11250-016-1156-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11250-016-1156-9