
Abstract
Lolium rigidum Gaud. is an annual grass grown for forage but also an economically damaging crop weed. A single genotype somatic embryogenic callus line, VLR1-60, was identified from a herbicide susceptible L. rigidum population, VLR1, and proved to be amenable to Agrobacterium tumefaciens-mediated transformation. Somatic embryogenic calli were continuously induced from the meristematic region of VLR1-60 plants multiplied in vitro and the basic tolerance level of VLR1-60 to hygromycin B was determined. A hygromycin phosphotransferase gene was used as a selectable marker for hygromycin B selection. Somatic embryogenic calli derived from in vitro grown vegetative tillers were co-cultivated with the A. tumefaciens strain EHA105 harbouring binary vector carrying reporter genes and selectable marker in the presence of acetosyringone for 3 days. Inoculated calli were recovered on callus proliferation medium containing Timentin™ but lacking hygromycin and were then subcultured onto media with hygromycin concentrations increased progressively through time for selection of transformed plant cells. Putative transgenic plants were recovered and integration of transgenes was confirmed by Southern hybridization analysis and by detection of DsRed or GUS activity in transgenic plants. The frequency of plant transformation was 1.3 %. The ability to transform L. rigidum will provide opportunities for functional characterization of genes to improve forage quality and increase our understanding of the evolution of herbicide resistance and of the basic genetics underlying traits that make L. rigidum a damaging crop weed.
Similar content being viewed by others


Introduction
Lolium rigidum Gaud., commonly known as annual ryegrass, Wimmera ryegrass or stiff darnel, is a cross-pollinated annual grass native to the Mediterranean region and introduced to Australia in 1880 (Kloot 1983). Being productive and having a high nutritive value, L. rigidum is well adapted to drought and grazing pressure (Anson et al. 1997; San Emeterio et al. 2004). It is sown alone or in mixed pastures with legumes to provide more nutritionally balanced grazing with longer productive periods. In the large temperate, wheat-dominated cropping areas of the Australian wheat belt, L. rigidum was once widely grown as a pasture plant and has now become naturalized, and is often present at high densities. L. rigidum is now Australia’s most economically damaging crop weed (Yu et al. 2004) having evolved resistance to many different chemical herbicides (Heap and Knight 1982; Burnet et al. 1994; Lorraine-Colwill et al. 2001; Broster and Pratley 2006; Powles and Yu 2010). The cost of L. rigidum infestation in Australian agriculture is estimated to be hundreds of millions of dollars annually (Goggin et al. 2012).
Since the first transgenic Lolium perenne (perennial ryegrass) plants were generated by Spangenberg et al. (1995), efforts in Lolium transformation have resulted in successful transformation of L. multiflorum, L. perenne and L. temulentum by biolistic delivery (Ye et al. 1997; Dalton et al. 1999) and Agrobacterium tumefaciens-mediated methods (Bettany et al. 2003; Bajaj et al. 2006; Cao et al. 2006; Altpeter 2006; Sato and Takamizo 2006; Ge et al. 2007; Zhang et al. 2012). This has resulted in progress in the development of traits such as improved digestibility (Tu et al. 2010), salt tolerance (Wu et al. 2005) and cold tolerance (Hisano et al. 2004). Biolistic transformation of L. rigidum (Bhalla et al. 1999) has been previously reported, but the level of transformation efficiency was not provided. A. tumefaciens-mediated transformation is becoming more favoured as it usually results in a greater proportion of low transgene copy-number events, which better enables stable expression of the transgene in subsequent generations (Travella et al. 2005, Wang and Ge 2006). Here, we report an efficient A. tumefaciens-mediated transformation method for a herbicide-susceptible genotype of L. rigidum. This will enable the use of transgenic approaches to elucidate the molecular basis of herbicide resistance in this grass. Effective forward genetics screens for weediness traits such as T-DNA insertional mutagenesis strategies (Hayashi et al. 1992) and reverse genetics approaches that rely on mutating specific genes including RNA interference (Waterhouse and Helliwell 2003) may lead to better weed management strategies and also to improved forage varieties.
Materials and methods
Selection of somatic embryogenic callus genotypes
A naturalised population VLR1 of herbicide-susceptible L. rigidum from Australia (Neve and Powles 2005) was used in this study. Seeds were de-husked and sterilised, and somatic embryogenic callus lines were established according to Bajaj et al. (2006) based on a protocol for perennial ryegrass embryogenesis. Plants derived from somatic embryogenic callus lines that showed good regeneration (named VLR1-1 to VLR1-70) were maintained as separate genotypes and were multiplied on maintenance medium composed of MS basal medium (Murashige and Skoog 1962) supplemented with 3 % (w/v) sucrose and 8.8 μM 6-benzylaminopurine (BAP). Embryogenic calli were reinduced from tillers of each line on callus induction medium (CIM) composed of MS basal medium supplemented with 3 % (w/v) maltose, 22.6 μM 2,4-dichlorophenoxyacetic acid (2,4-D) (pH 5.8) prior to inoculation with Agrobacterium.
Agrobacterium strains and binary vectors
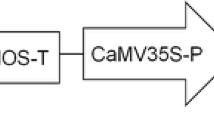
Strains of A. tumefaciens EHA105 carrying the binary vectors pCAMBIA 1305.2 (http://www.cambia.org/daisy/cambia/585) or pDPI000016 (Fig. 1) were used to inoculate somatic embryogenic callus. pDPI000016 was created by Gateway-enabling pPZP200 (Hajdukiewicz et al. 1994) by cloning a Gateway® Conversion System RfA cassette (Life Technologies, Carlsbad, CA, USA) into the PmeI site. A plant selectable marker cassette consisting of the promoter and 5′ untranslated region (UTR) from the actin (act1) gene of Oryza sativa (McElroy et al. 1990) followed by the hygromycin phosphotransferase (hpt) gene was synthesised (Life Technologies). This cassette was terminated with the 3′ UTR, containing transcriptional terminator and polyadenylation sites, from the 35S gene of the cauliflower mosaic virus (CaMV) (Chenault and Melcher 1993). This selection cassette was cloned into the XbaI and KpnI sites of the Gateway-enabled version of pPZP200. A synthetic reporter cassette (Life Technologies) consisting of the promoter, 5′ UTR and first intron from the Zea mays polyubiquitin (Ubi) gene (Toki et al. 1992) driving expression of the DsRed reporter gene (Clontech, Mountain View, CA, USA) was recombined into pDONR221 and sub-cloned into the pPZP200 vector backbone already containing the hph selection cassette to create pDPI000016 (Fig. 1). pCAMBIA1305.2 carries the hygromycin phosphotransferase (hptII) gene driven by a double-enhancer version of the CaMV 35S promoter and the reporter gene GUSPlus™ driven by a CaMV 35S promoter (Fig. 1). The pDPI000016 and pCAMBIA1305.2 binary vectors were introduced into the EHA105 strain of A. tumefaciens by electroporation.

Schematic diagram of the binary plasmid vectors pDPI000016 and pCAMBIA 1305.2. P = promoter and 5′ UTR; T = terminator, polyadenylation signal and 3′ UTR; Os, Oryza sativa; Zm, Zea mays; CaMV, cauliflower mosaic virus; LB, left border; RB, right border
Screening of somatic embryogenic callus lines amenable to Agrobacterium transformation
Meristematic regions of in vitro multiplied vegetative tillers from 70 lines of L. rigidum population VLR1 (VLR1-1 to VLR1-70) were cut into 1 mm pieces, spread onto CIM and cultured in the dark for 4 weeks at 24 °C. Somatic embryogenic calli induced from these tissues were sub-cultured onto CIM after 3 weeks and again 3–4 days before inoculation with A. tumefaciens.
A. tumefaciens cultures containing each binary vector were grown for 48 h at 28 °C in LB (Bertani 1951) solid medium containing 50 mg/L kanamycin and 25 mg/L rifampicin. Suspensions were grown to OD600 0.8–1.0 in liquid MS basal medium supplemented with 2 % (w/v) maltose, 1 % (w/v) glucose, 9 μM 2,4-D, and 400 μM acetosyringone. Somatic embryogenic calli of each line were placed in 50 mL polypropylene centrifuge tubes and incubated in suspension cultures of A. tumefaciens for 20 min with continuous shaking at 100 rpm. Excess liquid was drained and somatic embryogenic calli were blotted on sterile filter paper (Whatman No. 3, Maidstone, England) before placing them on co-cultivation medium (MS basal medium supplemented with 3 % (w/v) sucrose, 9 μM 2, 4-D, 0.44 μM BAP, 400 μM acetosyringone and 0.7 % agar, pH 5.2). Inoculated somatic embryogenic calli were either placed directly onto co-cultivation medium or on top of a layer of sterilized filter paper (Whatman No. 3) placed on the surface of the medium. After 3 days of co-cultivation in the dark at 24 °C, Somatic embryogenic calli were transferred to subculture media (SM), which was CIM medium supplemented with 250 mg/L Timentin™ to supress overgrowth of Agrobacterium and incubated for 2 weeks in the dark at 24 °C. Calli were subculture on the same medium supplemented with 25 mg/L hygromycin B and incubated at the same condition for 2 weeks. Calli were examined at 5 and 28 days after inoculation using a MZ-FLIII fluorescence stereomicroscope (Leica Microsystems, Heidelberg, Germany) to visualise DsRed fluorescence with a filter set (no. 10443423) for DsRed. The somatic embryogenic callus line that yielded the highest frequency of transient (5 dpi) and stable (28 dpi) expression of the reporter genes was selected for use in genetic transformation.
Transformation procedure
A hygromycin B tolerance test of the line selected above was conducted by placing un-inoculated somatic embryogenic calli on CIM supplemented with 0, 10, 25, 50, 75 or 100 mg/L hygromycin B, 50 mg/L being the concentration most commonly used for selection of transgenic shoots from calli for the production of transgenic grasses. The experiment was performed in triplicate with 20 somatic embryogenic calli in each replicate. Cultures were maintained in the dark at 24 °C for 2 weeks. The subsequent selection regime was selected on the basis of survival rates observed in this experiment.
After co-cultivation with A. tumefaciens, somatic embryogenic calli were transferred to SM for 1 week for recovery. Calli were then maintained in the dark at 24 °C and sub-cultured every 2 weeks on SM supplemented with hygromycin B. The first sub-culture was supplemented with 25 mg/L hygromycin B, increasing to 50 and then to 75 mg/L for the two subsequent subcultures. Surviving somatic embryogenic calli were transferred to regeneration medium (MS basal medium supplemented with 3 % (w/v) sucrose and 4.4 μM BAP, 25 mg/L hygromycin B and 250 mg/L Timentin™) and subcultured every 2 weeks until shoots regenerated. The regenerated shoots were transferred onto rooting medium (MS basal medium supplemented with TimentinTH but without hygromycin B) for 10 days. Putative transgenic plants were subsequently transferred to potting mix and grown in a glasshouse (21 °C, 14 h photoperiod/14 °C, 10 h dark period) or a growth cabinet (21 °C, 14 h photoperiod/16 °C, 10 h dark period).
GUS histochemical assay and visualization of DsRed fluorescence
Samples of somatic embryogenic calli inoculated with A. tumefaciens cells harbouring pCAMBIA1305.2 and pDPI00016, and samples of leaves and plantlets from in vitro regenerated putative transgenic plants, were screened for the expression of reporter genes. Samples were incubated in X-Gluc solution (1 mM 5-Bromo-4-chloro-3-indolyl b-D-glucuronide cyclohexylammonium salt (Sigma) in 50 mM sodium phosphate buffer, pH 7, Jefferson 1987) at 37 °C in the dark for 12–16 h to test for uidA enzyme activity and the production of β-glucuronidase. After incubation of samples in 80 % ethanol for 24 h to clear chlorophyll, somatic embryogenic calli were photographed under bright–field illumination using an MZ-FLIII fluorescence stereomicroscope (Leica) equipped with a Leica DFC310 FX image capture system. DsRed fluorescence was visualized using the same stereomicroscope under ultra violet illumination using a DsRed filter set (no. 10443423, excitation: 500–558 nm, emission: 589.5 nm).
Genomic DNA isolation from leaf tissue
30 mg tissue samples of young fully expanded leaves from putative transgenic plants were snap frozen in liquid nitrogen before being freeze dried for 24 h in a Labconco Freezone 4.5® (Labconco, Kansas City, MO). DNA was extracted from lyophilized tissues using the DNeasy® Plant Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions.
PCR analysis of putative transgenic plants
Putative transgenic and isogenic wild type control plants of VLR1-60 were analysed by the polymerase chain reaction (PCR) with primers specific to either the transgene promoter from pDPI000016 (act1 promoter from rice (5′-CTAAGCCTGACGAAGCAGCA-3′ and 5′-ATCTTTGGCCTTGGTAGTTTGG-3′) or to the uidA gene coding sequence contained in pCAMBIA1305.2 (5′-TGACAAGAACCATCCAAG-3′ and 5′-TCCGTTATAGCGATTGAG-3′). Primers targeting an endogenous Histone H3 gene (5′-TGCTTGCCCTTCAGGAGGCT-3′ and 5′-CTGAATGTCCTTGGGCATGAT-3′) were used in a third assay to test the PCR competence of the extracted DNA.
Southern hybridization analysis
DNA was purified from 5 g (fresh weight) of leaf tissue using CTAB method as described by Maguire et al. (1994). After digestion overnight with HindIII for which there is a single recognition site in the T-DNA of both pDPI00016 and pCAMBIA1305.2 (Fig. 1), 10 μg of DNA was separated by electrophoresis on a 0.8 % agarose gel and transferred to Hybond-N+ nylon membranes (Amersham Biosciences UK Limited, Little Chalfont, Bucks HP7 9NA, UK) using a standard Southern blotting protocol. Membranes were probed using the DIG Easy Hyb System (Roche, South San Francisco, CA) following the manufacturer’s instructions. A probe to detect integration of T-DNA transferred from pDPI000016 was amplified from pDPI000016 using primers targeting the DsRed gene (5′-ACATCCTCAGCCCACAGTTC-3′ and 5′-GTAGTAGCCTGGGAGCTGGA-3′) (Fig. 1). A probe to detect integration of T-DNA transferred from pCAMBIA 1305.2 was amplified from pCAMBIA 1305.2 using the following primers targeting the uidA gene (5′-AAGGATCAGCGTATCGTGCT-3′ and 5′-GCTTTGCCTTGAAAGTCCAC-3′) (Fig. 1). Hybridisation of both probes was performed at 52 °C overnight. A CPD-star chemiluminescent detection protocol was used as per manufacturer’s instructions (DIG Luminescent Detection Kit, Roche).
Statistical analysis
All experiments were performed in triplicate with each experiment containing only one variable. Three plates of 60 somatic embryogenic calli from each line were selected at random to analyse transient expression data and three plates of 60 somatic embryogenic calli derived from VLR1-60 were used to assess each concentration of hygromycin B. ANOVA was used for statistical analysis.
Results and discussion
Selection of regenerable somatic embryogenic callus lines of L. rigidum
Like other Lolium species, L. rigidum is an obligate out-crosser and therefore genetic diversity exists within cultivars. A consistent supply of somatic embryogenic calli of the same genotype with high regenerative capacity is required for efficient recovery of transgenic plants and for uniform transformation efficiency. In this study, a suitable somatic embryogenic callus line was selected from the VLR1 herbicide-susceptible population of L. rigidum. To obtain a single genotypic line with good somatic embryogenic callus characteristics, 4,210 seedlings were screened. Of these seedlings, 70 genotypes (VLR1-1 to 70) were selected and their tillers were maintained in vitro (Fig. 2a). Each line produced dry, friable, yellowish, embryogenic calli (Fig. 2b), which regenerated at frequencies ranging from 68 to 100 %. Ten of them with good regeneration were listed in Table 1. Only 1.5 % of seedlings that were screened produced suitable somatic embryogenic callus. This frequency of seedlings with somatic embryogenic callus ability is much lower than the value of 20 % reported for L. perenne v. Tolosa (Bajaj et al. 2006), or 60–91 % reported for Brachypodium distachyon accessions (Christiansen et al. 2005). Similar frequencies (2 and 0.2 % respectively) were observed in Festuca arundinaceae (tall fescue; Gao et al. 2008) and Festuca pratensis (meadow fescue; Gao et al. 2009).

Agrobacterium tumefaciens-mediated transformation of L. rigidum. In vitro donor material of VLR1-60 (a); Somatic embryogenic callus induced from meristamatic region of the in vitro donor VLR1-60 (b); Expression of DsRed in somatic embryogenic callus tissue 5 days (c) and 28 days(d) after inoculation with A. tumefaciens (Arrow indicates DsRed expression); Co-cultivation of somatic embryogeni calli with A. tumefaciens (e); Hygromycin resistant calli obtained 6 weeks after Agrobacterium-mediated transformation and selection.(f); Expression of β-glucuronidase in somatic embryogenic calli tissue 5 days (g) and 28 days (h) after inoculation with A. tumefaciens (Arrow indicates GUS expression); Regeneration of shoots/plantlets from hygromycin resistant calli (i); Hygromycin resistant putative transgenic plants (j); Rooted putative transgenic plants obtained after transferring the regenerated shoots to rooting medium (k); Transgenic and wild type plants grown in soil (l); Expression DsRed in leave of transgenic (T) and wild type (W) plants observed under bright field (m) and UV illumination using DsRed filter set (n) and in stem of transgenic (T) and wild type (W) plants observed under UV illumination using DsRed filter set (o); Expression of β-glucuronidase in transgenic (T) and wild type (W) plants (p)
Ten somatic embryogenic callus lines with regeneration efficiencies higher than 90 % were identified and subsequently used to test for suitability for A. tumefaciens-mediated transformation (Table 1). Plantlets regenerated in vitro were multiplied and maintained as stock for callus induction (Fig. 2a). The meristematic regions of vegetative tillers from each genotype were cut into 1 mm pieces and spread on CIM. This led to vigorous callus production (Fig. 2b).
Establishment of a transformation system for annual ryegrass
Selection of a somatic embryogenic callus line amenable to Agrobacterium transformation
All ten lines selected based on their regenerative ability as described above were inoculated with EHA105 harbouring pDPI000016 to select a somatic embryogenic callus line amenable to Agrobacterium transformation. Visualisation of fluorescence from the DsRed reporter gene was used to monitor trans-gene expression after co-cultivation with A. tumefaciens. Transient expression of the DsRed gene was first observed 5 days after inoculation (Fig. 2c; Table 1) and stable expression was subsequently observed at 28 day after inoculation (Fig. 2d). Of the ten somatic embryogenic callus genotypes tested, VLR1-46 and VLR1-60 had the highest levels of transient DsRed gene expression with an average of 6.9 ± 0.9 and 7.7 ± 1.1 DsRed foci per callus respectively (Table 1), while only four lines showed stable integration (Table 1). Microscopic evaluation for DsRed revealed that VLR1-60 gave the highest number of putative stable transformants (DsRed observed in 23.3 ± 1.3 % of all somatic embryogenic calli). This observation is consistent with previous studies from other monocotyledenous species that reported A. tumefaciens-mediated transgene integration is genotype dependent (Cheng et al. 2004; Gelvin 2003; Tzfira and Citovsky 2006) and is thought to be a result of variation in the expression of host genes induced by A. tumefaciens infection (Gelvin 2003; Tzfira and Citovsky 2006). The observation that only four genotypes gave rise to stably transformed somatic embryogenic calli suggests that transgene integration rather than gene delivery is critical for successful transformation of L. rigidum.
Hygromycin dose response analysis of somatic embryogenic callus line VLR1-60
Herbicide resistance genes such as phosphinothricin acetyl transferase (bar) and antibiotic resistance genes including hpt and neomycin phosphotransferase (nptII) are widely used as selectable markers for the successful recovery of transgenic plants (Wang and Ge 2005; Tzfira and Citovsky 2006). Hygromycin B concentrations in media of more than 50 mg/L have been used in transformation protocols for some monocotyledonous plants (Dong and Qu 2005; Ge et al. 2006; Li and Qu 2011). However, nearly all L. rigidum calli showed necrosis within 1 week and almost all calli infected with A. tumefaciens died within 2 weeks of inoculation when subcultured onto media containing 50 mg/L hygromycin B. Clearly, VLR1 somatic embryogenic callus is sensitive to hygromycin B. Our results showed that somatic embryogenic callus growth was 81 % of that observed in uninfected VLR1-60 somatic embryogenic calli when hygromycin B concentration was 10 mg/L in the culture medium, and 35.5 % of that observed in uninfected VLR1-60 somatic embryogenic calli when hygromycin B concentration was increased to 25 mg/L (Fig. 3). There were also significant differences in the mean weights of somatic embryogenic calli grown on the relatively high concentrations of hygromycin B of 50, 75, and 100 mg/L, indicating that somatic embryogenic callus growth was inhibited (Fig. 3). In our hands, the good number of recovered transgenic calli were obtained when hygromycin B concentration was increased from 25 to 50 mg/L and then to 75 mg/L with each successive sub-culture.

Dose response plot showing the response of L. rigidum VLR1-60 somatic embryogenic calli to increasing concentrations of hygromycin B. Columns represent the fresh weight (mean ± SE) of somatic embryogenic calli harvested after growth on media for 2 weeks (n = 60)
Recovery of transgenic plants
Co-cultivation periods of 2, 3 and 4 days were compared (Table 2). It was found that co-cultivation for 3 days produced the greatest number of somatic embryogenic callus transiently expressing reporter genes. After 4 days of co-cultivation, the average DsRed foci per callus for the transient expression was not significantly different to calli of a 3 day co-cultivation, but it resulted in a typical detrimental overgrowth of A. tumefaciens. A lower frequency of gene delivery was observed for a 2 day co-cultivation period. The presence of filter paper between the somatic embryogenic calli and media during a 3 day co-cultivation made no difference to the level of transient expression. The average DsRed foci was 6.4 ± 0.9 for calli on filter paper and 7.3 ± 0.8 on co-cultivation medium directly (Table 2).
A total of five putative transgenic plants (VLR1-60-1, 2, 3, 4 and 5) were recovered from four experiments in which 100 somatic embryogenic calli were inoculated in each experiment (Table 3) following the procedure shown in Fig. 2 (e-l). Similar to the observation of DsRed gene expression in somatic embryogenic calli transformed with pDPI000016 stated previously, histochemical localisation of GUS activity in somatic embryogenic calli transformed with pCAMBIA 1305.2 was monitored at 5 days after inoculation for transient expression and 28 days for stable expression (Fig. 2g, h). Four plants were recovered from two experiments inoculated with A. tumefaciens carrying pCAMBIA 1305.2 and one plant (VLR1-60-5) was recovered from two experiments inoculated with A. tumefaciens carrying pDPI000016. All plants recovered were from somatic embryogenic calli on different plates and were therefore independent events. Across these four experiments, the average transformation efficiency was 1.3 %. The current protocol gave a low efficiency compared to that of other published protocols of Lolium species such as 4.8 % for L. temulentum (Ge et al. 2007) and about 50 % for L. perenne (Zhang et al. 2012). The necrosis of calli of this genotype due to the sensitivity to hygromycin may be the major cause. Further improvement is needed for achieving a high transformation efficiency. The transgenic plants showed normal growth compared to its isogenic control VLR1-60 (Fig. 2l).
Molecular characterization of transgenic annual ryegrass plants
The plant transformed with pDPI000016 showed DsRed fluorescence in all tissues compared to the wild type VLR1-60 control (Fig. 2m–o). Similarly, samples of all four putative transgenic plants transformed with pCAMBIA 1305.2 were analysed with a GUS histochemical assay. All tissues except for the very base of the stem showed blue staining when compared to the wild type VLR1-60 control (Fig. 2p). Four of the five putative transgenic plants (VLR1-60-1, VLR1-60-2, VLR1-60-3 and VLR1-60-5) were analysed for the presence of the transgene, initially by visualization of DsRed fluorescence or GUS activity in transgenic plants (Fig. 2n–p), PCR and subsequently by Southern analysis (Fig. 4). These analyses indicated that all putative events were transgenic and contained either one (Fig. 4a) or three copies of the T-DNA as indicated by the presence of either a single band or a single band plus a doublet on Southern blots (Fig. 4b). The low copy numbers obtained are consistent with the results from other grass species transformed using A. tumefaciens-mediated gene delivery (Bettany et al. 2003; Luo et al. 2004; Dong and Qu 2005; Han et al. 2005; Wang and Ge 2005; Bajaj et al. 2006; Ge et al. 2006; Gao et al. 2008).

Southern hybridisation analysis of L. rigidum VLR1-60 plants. All DNA was digested to completion with HindIII. M = 1 KB Molecular Marker. a Analysis of plants transformed with pCAMBIA1305.2. Lanes contain 10 μg DNA purified from wild type (VLR1-60) and putative transgenic (VLR1-60-1, 2 and 3) plants. The membrane was probed with a DIG-labelled probe specific to uidA, to detect the presence of the T-DNA. b Analysis of plants transformed with pDPI000016. Lanes contain 20 ng purified plasmid DNA (pDPI000016), or 10 μg DNA purified from wild type (VLR1-60) or putative transgenic (VLR1-60-5) plant. The membrane was hybridised with a DIG-labelled probe specific to DsRed to detect the presence of the T-DNA
Conclusion
A highly regenerable single genotype somatic embryogenic callus line of L. rigidum, named VLR1-60 was selected and maintained in vitro. A robust selection regime for this genotype that utilises hygromycin B has been established. Confirmation of the transgenic nature of the regenerated plants by molecular analysis indicated that the protocol presented here is reliable. To our knowledge, this is the first report of A. tumefaciens-mediated genetic transformation of L. rigidum. This protocol enables future functional genomic studies of this economically important grass species.
References
Altpeter F (2006) Perennial ryegrass (Lolium perenne L.). In: Wang K (ed) Methods in molecular biology, vol 344: Agrobacterium protocols. Humana Press Inc, Totowa, pp 55–64
Anson S, Delgado I, Muñoz F (1997) Evaluation of the productivity of Lolium rigidum. Pastos 27:165–176
Bajaj S, Ran Y, Phillips J, Kulrajathevan G, Pal S, Cohen D, Elborough K, Puthigae S (2006) A high throughput Agrobacterium tumefaciens-mediated transformation method for functional genomics of perennial ryegrass (Lolium perenne L.). Plant Cell Rep 25:651–659
Bertani G (1951) Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol 62:293–300
Bettany AJE, Dalton SJ, Timms E, Manderyck B, Dhanoa MS, Morris P (2003) Agrobacterium tumefaciens-mediated transformation of Festuca arundinacea (Schreb.) and Lolium multiflorum (Lam.). Plant Cell Rep 21:437–444
Bhalla PL, Swoboda I, Singh MB (1999) Antisense mediated silencing of a gene encoding a major ryegrass pollen allergen. Proc Natl Acad Sci USA 96:11676–11680
Broster JC, Pratley JE (2006) A decade of monitoring herbicide resistance in Lolium rigidum in Australia. Aust J Exp Agr 46:1151–1160
Burnet MWM, Holtum JAM, Powles SB (1994) Resistance to nine herbicide classes in a Lolium rigidum biotype. Weed Sci 42:369–377
Cao MX, Huang JQ, He YL, Liu SJ, Wang CL, Jiang WZ, Wei ZM (2006) Transformation of recalcitrant turfgrass cultivars through improvement of tissue culture and selection regime. Plant Cell Tiss Organ Cult 85:307–331
Chenault K, Melcher U (1993) Plant gene register-cauliflower mosaic virus lsolate CMV- 1. Plant Physiol 101:1395–1396
Cheng M, Lowe B, Spencer T, Ye X, Armstrong C (2004) Factors influencing Agrobacterium-mediated transformation of monocotyledonous species. In Vitro Cell Dev Biol Plant 40:31–45
Christiansen P, Henrik CA, Didion T, Folling M, Nielsen KK (2005) A rapid and efficient transformation protocol for the grass Brachypodium distachyon. Plant Cell Rep 23:751–758
Dalton SJ, Bettany AJE, Timms E, Morris P (1999) Co-transformed, diploid Lolium perenne (perennial ryegrass), Lolium multiflorum (Italian ryegrass) plants produced by microprojectile bombardment. Plant Cell Rep 18:721–726
Dong S, Qu R (2005) High efficiency transformation of tall fescue with Agrobacterium tumefaciens. Plant Sci 168:1453–1458
Gao CX, Long D, Lenk I, Nielsen KK (2008) Comparative analysis of transgenic tall fescue (Festuca arundinacea Schreb.) plants obtained by Agrobacterium-mediated transformation and particle bombardment. Plant Cell Rep 27:1601–1609
Gao CX, Liu JX, Nielsen KK (2009) Agrobacterium-mediated transformation of meadow fescue (Festuca pratensis Huds.). Plant Cell Rep 28:1431–1437
Ge Y, Norton T, Wang Z (2006) Transgenic zoysiagrass (Zoysia japonica) plants obtained by Agrobacterium-mediated transformation. Plant Cell Rep 25:792–798
Ge Y, Cheng X, Hopkins A (2007) Generation of transgenic Lolium temulentum plants by Agrobacterium tumefaciens-mediated transformation. Plant Cell Rep 26:783–789
Gelvin SB (2003) Agrobacterium-mediated plant transformation: the biology behind the “Gene-Jockeying” tool. Microbiol Mol Biol Rev 67:16–37
Goggin DE, Powles SB, Steadman KJ (2012) Understanding Lolium rigidum seeds: the key to managing a problem weed? Agronomy 2:222–239
Hajdukiewicz P, Svab Z, Maliga P (1994) The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol Biol 25:989–994
Han N, Chen D, Bian H, Deng M, Zhu M (2005) Production of transgenic creeping bentgrass Agrostis stolonifera var. palustris plants by Agrobacterium tumefaciens-mediated transformation using hygromycin selection. Plant Cell, Tissue Organ Cult 81:131–138
Hayashi H, Czaja I, Lubenow H, Schell J, Walden R (1992) Activation of a plant gene by T-DNA tagging: auxin-independent growth in vitro. Science 258:1350–1352
Heap J, Knight R (1982) A population of ryegrass tolerant to the herbicide diclofop-methyl. J Aust Inst Agr Sci 48:156–157
Hisano H, Kanazawa A, Kawakami A, Yoshida M, Shimamoto Y, Yamada T (2004) Transgenic perennial ryegrass plants expressing wheat fructosyltransferase genes accumulate increased amounts of fructan and acquire increased tolerance on a cellular level to freezing. Plan Sci 167:861–868
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep 5:387–405
Kloot PM (1983) The genus Lolium in Australia. Aust J Bot 31:421–435
Li R, Qu R (2011) High throughput Agrobacterium-mediated switchgrass transformation. Biomass Bioenerg 35:1046–1054
Lorraine-Colwill DF, Powles SB, Hawkes TR, Preston C (2001) Inheritance of evolved glyphosate resistance in Lolium rigidum (Gaud.). Theor Appl Genet 102:545–550
Luo H, Hu Q, Nelson K, Longo C, Kausch AP, Chandlee JM, Wipff JK, Fricker CR (2004) Agrobacterium tumefaciens-mediated creeping bentgrass (Agrostis stolonifera L.) transformation using phosphinothricin selection results in a high frequency of single-copy transgene integration. Plant Cell Rep 22:645–652
Maguire TL, Collins GG, Sedgley M (1994) A modified CTAB DNA extraction procedure for plants belonging to the family proteaceae. Plant Mol Biol Rep 12:106–109
McElroy D, Zhang W, Cao J, Wu R (1990) Isolation of an efficient actin promoter for use in rice transformation. Plant Cell 2:163–171
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant 15:473–497
Neve P, Powles SB (2005) Recurrent selection with reduced herbicide rates results in the rapid evolution of herbicide resistance in Lolium rigidum. Theor Appl Genet 110:1154–1166
Powles SB, Yu Q (2010) Evolution in action: plants resistance to herbicides. Annu Rev Plant Biol 61:317–347
San Emeterio L, Arroyo A, Canals RM (2004) Allelopathic potential of Lolium rigidum Gaud. on the early growth of three associated pasture species. Grass Forage Sci 59:107–112
Sato H, Takamizo T (2006) Agrobacterium tumefaciens-mediated transformation of forage-type perennial ryegrass (Lolium perenne L.). Grassland Sci 52:92–98
Spangenberg G, Wang ZY, Wu XL, Nagel J, Potrykus I (1995) Transgenic perennial ryegrass (Lolium perenne) plants from microprojectile bombardment of embryogenic suspension cells. Plant Sci 108:209–217
Toki S, Takamatsu S, Nojiri C, Ooba S, Anzai H, Iwata M, Christensen AH, Quail PH, Uchimiya H (1992) Expression of a maize ubiquitin gene promoter-bar chimeric gene in transgenic rice plants. Plant Physiol 100:1503–1507
Travella S, Ross SM, Harden J, Everett C, Snape JW, Harwood WA (2005) A comparison of transgenic barley lines produced by particle bombardment and Agrobacterium-mediated techniques. Plant Cell Rep 23:780–789
Tu Y, Rochfort S, Liu Z, Ran Y, Griffith M, Badenhorst P, Louie G, Bowman M, Smith K, Noel J, Mouradov A, Spangenberg G (2010) Functional analysis of caffeic acid O-methyltransferase and cinnamoyl-CoA. -reductase genes from perennial reygrass (Lolium perenne). Plant Cell 22:3357–3373
Tzfira T, Citovsky V (2006) Agrobacterium-mediated genetic transformation of plants: biology and biotechnology. Curr Opin Biotechnol 17:147–154
Wang ZY, Ge Y (2005) Agrobacterium-mediated high efficiency transformation of tall fescue (Festuca arundinacea). J Plant Physiol 162:103–113
Wang ZY, Ge Y (2006) Recent advances in genetic transformation of forage and turf grasses. In Vitro Cell Dev Biol Plant 42:1–18
Waterhouse PM, Helliwell CA (2003) Exploring plant genomes by RNA-induced gene silencing. Nat Rev Genet 4:26–35
Wu YY, Chen QJ, Chen M, Chen J, Wang XC (2005) Salt-tolerant transgenic perennial ryegrass (Lolium perenne L.) obtained by Agrobacterium tumefaciens-mediated transformation of the vacuolar Na +/H + antiporter gene. Plant Sci 169:65–73
Ye XD, Wang ZY, Wu XL, Potrykus I, Spangenberg G (1997) Transgenic Italian ryegrass (Lolium multiflorum) plants from microprojectile bombardment of embryogenic suspension cells. Plant Cell Rep 16:379–384
Yu Q, Cairns A, Powles SB (2004) Paraquat resistance in a population of Lolium rigidum. Funct Plant Biol 31:247–254
Zhang WJ, Dewey RE, Boss W, Phillippy BQ, Qu R (2012) Enhanced Agrobacterium-mediated transformation efficiencies in monocot cells is associated with attenuated defence responses. Plant Mol Biol 81:273–286
Acknowledgments
We would like to thank Dr Margaret Buchanan, Dr Stephen Panter and Dr Matthew Hayes for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Ran, Y., Patron, N., Yu, Q. et al. Agrobacterium-mediated transformation of Lolium rigidum Gaud.. Plant Cell Tiss Organ Cult 118, 67–75 (2014). https://doi.org/10.1007/s11240-014-0462-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-014-0462-z