
Abstract
Thrombosis and inflammation are two major factors underlying chronic thromboembolic pulmonary hypertension (CTEPH). Tissue factor (TF), C-reactive protein (CRP), tumor necrosis factor-α (TNF-α) and monocyte chemoattractant protein 1 (MCP-1) may play critical roles in the process of CTEPH thrombosis and pulmonary vascular remodeling. Ten patients with a confirmed diagnosis of CTEPH, 20 patients with acute pulmonary thromboembolism and 15 patients with other types of pulmonary hypertension were enrolled in this study, along with 20 healthy subjects as the control group. The immunoturbidimetric method was used to determine the plasma content of CRP. The plasma levels of TNF-α, MCP-1, and TF antigen were measured by an enzyme-linked immunosorbent assay, and TF activity was measured by the chromogenic substrate method. Percoll density gradient centrifugation was used to separate peripheral blood mononuclear cells from plasma. The level of monocyte TF mRNA was examined by reverse transcriptase-polymerase chain reaction. The correlations between all indices described above were analyzed. In CTEPH patients, the expression of CRP, TNF-α, and MCP-1 was significantly higher than that in controls (P < 0.05). The levels of TF activity, TF antigen, and TF mRNA in monocyte cells were increased in CTEPH patients when compared with control subjects, but only the TF antigen and TF mRNA levels were significantly different (P < 0.05). In CTEPH patients, levels of CRP, MCP-1, and TNF-α significantly correlated with the level of TF antigen in plasma. TF gene expression was increased in patients with CTEPH, suggesting that blood-borne TF mainly comes from mononuclear cells. TF expression significantly correlated with levels of CRP, TNF-α and MCP-1. These factors may play an important role in the development of CTEPH via the inflammation–coagulation–thrombosis cycle.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic thromboembolic pulmonary hypertension (CTEPH) is the consequence of thrombus resolution failure after the establishment of thrombosis within the elastic pulmonary arteries [1]. CTEPH is a common variation of pulmonary hypertension (PH) and is associated with significant morbidity and mortality [2, 3]. Historical data indicate that if left untreated, CTEPH is associated with a poor 5-year survival rate ranging from 10 to 40 % depending on the patient’s pulmonary hemodynamics [3, 4]. The treatment of CTEPH includes pulmonary thrombus endarterectomy (PEA), balloon angioplasty, and medical therapy. The outcome of PEA surgery is favorable,and its mortality rates less than 3–5 % [5]. Patients who are not candidates for surgery may benefit from PH-specific medical therapy [6]. However, the effects of medical therapy are limited. Until recently, the pathophysiology of CTEPH has remained poorly understood [7].
The embolic hypothesis claims that CTEPH is the result of a single or recurrent pulmonary embolism originating from sites of venous thrombosis [8]. Tissue factor (TF) is the initiation factor of the extrinsic coagulation pathway and functions together with factor VII [9]. Studies have shown that increased TF expression plays a critical role in the process of thrombosis. There are some reports of a link between PH and TF expression [10, 11]; however, few of these reports discuss CTEPH and TF expression. In this study, our aim was to detect the mRNA levels of monocyte-derived TF and its role during CTEPH thrombosis and remodeling of the pulmonary vasculature.
The role of inflammation in CTEPH pathogenesis has been emphasized in recent years. Levels of inflammatory markers, such as C-reactive protein (CRP) [7, 12, 13], tumor necrosis factor-α (TNF-α) [14] and monocyte chemoattractant protein1 (MCP-1) [15, 16], may be elevated in the plasma and thrombus tissues of patients with CTEPH, which is in correlation with hemodynamic data. TF is often upregulated upon vascular injury, inflammation (e.g. lipopolysaccharides, interleukins, TNF-α, CRP, MCP-1), hypoxia, and other conditions [17]. However, the relationship between inflammatory markers (e.g. CRP, TNF-α, MCP-1) and TF in CTEPH remains unclear.
Methods
Study population
All procedures were approved by the Ethics Committee of the First Affiliated Hospital of Fujian Medical University and all patients provided written informed consent. In this study, 10 patients with CTEPH were recruited between June 2013 and October 2014. CTEPH is defined by the following observations after ≥3 months of effective anticoagulation therapy [18]: (1) pulmonary arterial pressure (mPAP) >25 mmHg with a pulmonary capillary wedge pressure ≤15 mmHg; and (2) at least one (segmental) perfusion defect detected by ventilation and perfusion (V/Q) lung scan [19], multidetector computed tomographic (CT) angiography, or pulmonary angiography. If the V/Q scan were the primary mode of diagnosis, the results would need to be interpreted as a high probability of thromboembolic disease in order to be considered a positive test for CTEPH. A negative CT angiogram was not considered sufficient to exclude CTEPH. At the same time, 20 patients with pulmonary thromboembolism (PTE) and 15 patients with non-thromboembolic PH were enrolled in this study. We also enrolled a control group (n = 20) that consisted of healthy blood donors and was defined by the following observations: (1) healthy subjects who had a physical examination in the medical center of the First Affiliated Hospital of Fujian Medical University in the same time period, and (2) the physical examination was normal. The mPAP of CTEPH and non-thromboembolic PH patients was determined by right heart catheterization based on the recent consensus paper [20].
Sample collection
Eight milliliters of blood were collected from each study subject (of note, no drugs that affect coagulation status were taken by subjects before sample collection) in two anticoagulant heparin and sodium citrate (1:9) vacutainers. One vacutainer was centrifuged at 3000 rpm for 15 min within 30 min of collection. The blood plasma was frozen at −80 °C for analysis of levels of CRP, TNF-α, MCP-1, and TF antigen and TF activity within 2 months. Blood monocytes were isolated by the percoll density gradient centrifugation method for measuring TF mRNA levels in monocytes.
Miscellaneous
Plasma CRP levels were measured by the immune turbidimetric assay using the automatic dry biochemical analyzer (Johnson & Johnson, China) according to the manufacturer’s protocol. Levels of TNF-α, MCP-1, and TF antigen were determined by enzyme-linked immunosorbent assay (ELISA) (CUSABIO, China). The activity of TF was measured by the chromogenic substrate method (ab108906, Abcam, England).
Primer design and RT-PCR
Blood monocyte samples were treated with Trizol reagent (Invitrogen, USA) and RNA was extracted in accordance with the manufacturer’s instructions. M-MLV reverse transcriptase was purchased from Thermo Scientific. The purity of the RNA was analyzed by OD260/OD280 ratio measured by ultraviolet spectrophotometry. RNA samples with an OD260/OD280>1.8, indicating high RNA purity, were used for the following experiments. An aliquot of RNA underwent gel electrophoresis at 80–100 mV.
Primer pairs were designed for TF and β-actin (internal control) using their DNA sequences. Primers were synthesized by Shanghai Biotechnology (China). The primers for TF were: forward 5′-TAC TTG GCA CGG GTC TTC TC-3′ and reverse 5′-TGT CCG AGG TTT GTC TCC AG-3′, resulting in a 119 bp product. The primers for β-actin were: forward 5′-CGG GAA ATC GTG CGT GAC-3′ and reverse: 5′-TGG AAG GTG GAC AGC GAG G-3′, resulting in a 434 bp product. Cycling conditions were as follows: 95 °C denaturation for 5 min; 40 cycles of 95 °C for 45 s, 58 °C for 30 s, and 72 °C for 30 s; and a final 10 min 72 °C extension. Products underwent 2 % agarose gel electrophoresis containing 0.5 μg/mL ethidium bromide and a gel imaging system (HBMA-9600, China) was used to visualize and photograph the results with β-actin as the internal reference. TF expression was semi-quantitatively determined by the imaging software.
Statistical analysis
Statistical software SPSS17.0 was used for statistical analysis. Quantitative data were expressed as mean ± standard deviation (SD). Multiple groups were compared by analysis of one-way ANOVA. Coefficients of correlation were calculated by the Spearman rank test. Probability values (two-sided) were considered significant at a value of P < 0.05.
Results
Characteristics of the study population
A total of 65 subjects were included in this study: 10 patients with CTEPH, 20 patients with PTE, 15 patients with non-thromboembolic PH, and a control group of 20 healthy subjects. Non-thromboembolic PH cases included idiopathic PH (n = 5), familial PH (n = 2), and PH associated with respiratory disease (n = 3), connective tissue disease (n = 2) or congenital heart disease (n = 3). Patient demographics and clinical characteristics are reported in Table 1. D-dimmer plasma concentrations were elevated in PTE, non-thromboembolic PH and CTEPH when compared with the control group, but only the difference between the PTE and control groups was statistically significant (P < 0.01). Compared with the control group, fibrinogen plasma concentrations were significantly elevated in the PTE and CTEPH groups at the time of diagnosis (P < 0.01). No significant differences in fibrinogen concentrations were detected between non-thromboembolic PH and control subjects. The above data suggest that the PTE and CTEPH patients are in a hypercoagulable state.
CRP, MCP-1, and TNF-α levels in CTEPH
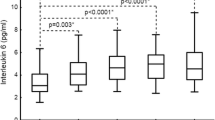
As shown in Fig. 1, CRP and TNF-α levels increased significantly in all groups (CTEPH, PTE, and non-thromboembolic PH) compared with the control group (P < 0.05). In the CTEPH and PTE groups, the increase in the level of MCP-1 was significant (P < 0.01). In the CTEPH group, the mPAP ranged from 62 to 132 mmHg, with a mean ± SD of 79.7 ± 21.19 mmHg. Moreover, in CTEPH patients, both CRP (r = 0.92, P < 0.01) and MCP-1 (r = 0.66, P < 0.05) levels significantly correlated with pulmonary artery systolic pressure, while TNF-α levels did not (r = 0.49, P > 0.05) (Fig. 2), suggesting that there is a link between CRP and MCP-1 and severity of disease in CTEPH.

CRP, MCP-1 and TNF-α levels in CTEPH. a Plasma level of CRP in CTEPH (26.44 ± 5.18 mg/L, n = 10), PTE (33.83 ± 21.47 mg/L, n = 20), and non-thromboembolic PH (8.35 ± 5.03 mg/L, n = 15) patients and in control subjects (2.42 ± 1.71 mg/L, n = 20). b Plasma level of MCP-1 in CTEPH (45.49 ± 16.52 pg/mL, n = 10), PTE (69.37 ± 27.58 pg/mL, n = 20), and non-thromboembolic PH (34.20 ± 25.55 pg/mL, n = 15) patients and in control subjects (22.27 ± 8.59 pg/mL, n = 20). c Plasma level of TNF-α in CTEPH (32.34 ± 4.53 pg/mL, n = 10), PTE (31.26 ± 6.62 pg/mL, n = 20), non-thromboembolic PH (23.23 ± 8.44 pg/mL, n = 15) patients and in control subjects (12.12 ± 7.40 pg/mL, n = 20). Plasma concentrations are expressed as mean ± SD. ** P value <0.01 and n.s indicates not significant (P > 0.05) versus control group. # P < 0.05 versus PTE group and ▲ P < 0.01 versus non-thromboembolic PH group

Correlation between mPAP and CRP, MCP-1, and TNF-α in CTEPH patients. a Correlation between mPAP and plasma level of CRP. b Correlation between mPAP and plasma level of MCP-1. c Correlation between mPAP and plasma level of TNF-α
TF levels in CTEPH patients
In this study, the activity of plasma TF increased in all groups (CTEPH, PTE, and non-thromboembolic PH) when compared with the control group, but only the PTE group resulted in a significant difference (P < 0.05). In CTEPH and PTE groups, the level of the TF antigen increased significantly (P < 0.01). TF mRNA levels in monocyte cells were significantly different in both CTEPH and PTE groups compared with the control group (P < 0.01). There also was a significant difference in TF mRNA levels between CTEPH and PTE groups compared to non-thromboembolic PH patients (P < 0.01; Fig. 3). Moreover, in the CTEPH and PTE groups, TF antigen levels significantly correlated with monocyte TF mRNA levels (r = 0.83, P < 0.01; r = 0.91, P < 0.01), suggesting monocyte TF may play a key role in CTEPH thrombosis and remodeling of the pulmonary vasculature (Fig. 4). Finally, in CTEPH patients, CRP (r = 0.73, P < 0.05), MCP-1 (r = 0.70, P < 0.05) and TNF-α (r = 0.66, P < 0.05) levels significantly correlated with plasma levels of TF antigen (Fig. 5), suggesting that these factors may promote the expression of TF antigen in monocytes and exacerbate thrombosis, thereby affecting the pathological process of CTEPH.

TF level in CTEPH patients. a Plasma level of TF activity in CTEPH (24.35 ± 6.10 pM/mL, n = 10), PTE (29.35 ± 8.26 pM/mL, n = 20), and non-thromboembolic PH (24.41 ± 5.20 pM/mL, n = 15) patients and in control subjects (20.24 ± 3.29 pM/mL, n = 20). b Plasma level of TF antigen in CTEPH (45.68 ± 12.06 pg/mL, n = 10), PTE (54.68 ± 15.98 pg/mL, n = 20), and non-thromboembolic PH (29.11 ± 5.55 pg/mL, n = 15) patients and in control subjects (27.92 ± 3.33 pg/mL, n = 20). c TF mRNA expression in monocytes in CTEPH (0.33 ± 0.019, n = 10), PTE (0.39 ± 0.027, n = 20), and non-thromboembolic PH (0.26 ± 0.01, n = 15) patients and in control subjects (0.25 ± 0.01, n = 20). d RT-PCR products of TF from monocytes by gel electrophoresis. Plasma concentrations are expressed as mean ± SD. **P value <0.01, *P < 0.05 and n.s indicates not significant (P > 0.05) versus control group. △ P < 0.01 versus PE group and ▲ P < 0.01 versus non-thromboembolic PH group

Correlation between TF antigen and monocyte TF mRNA levels. a Correlation between TF antigen and monocyte TF mRNA in patients with CTEPH. b Correlation between TF antigen and monocyte TF mRNA in patients with PTE

Correlation between TF antigen and CRP, MCP-1, and TNF-α levels in patients with CTEPH. a Correlation between TF antigen and plasma level of CRP. b Correlation between TF antigen and plasma level of MCP-1. c Correlation between TF antigen and plasma level of TNF-α
Discussion
TF hypercoagulability leading to thrombosis
TF is also known as factor III and it can initiate extrinsic coagulation by binding to and activating coagulation factor VIIa. The resultant TF-VIIa complex then activates factor X (factor Xa), triggering the generation of thrombin, which converts soluble fibrinogen into insoluble fibrin [21, 22]. Under normal circumstances, blood mononuclear cells and endothelial cells do not express TF [23]. It has been shown that the level of circulating TF in the blood of healthy donors is extremely low and does not appear to contribute to hemostasis [24, 25]. This suggests that increased TF levels and activity in the blood may trigger thrombosis [26].
Some reports have suggested that an underlying hypercoagulable state may be responsible for the development of CTEPH. We found increased TF activity in PTE patients, but not in CTEPH or PH patients, manifesting the hypothesis that thrombosis is associated with TF activity. However, TF activity does not always correlate with TF gene expression because TF activity may be affected by the change in expression of tissue factor pathway inhibitor (TFPI) or other anticoagulants or by anticoagulant therapy. In the present study, we have demonstrated the higher plasma level of TF antigen and activity in CTEPH patients compared with healthy subjects. It is also interesting that only the level of TF antigen in plasma is significantly increased while the level of TF activity is not. Burley argued that anticoagulants (e.g. low molecular weight heparin, heparin, TFPI, antithrombin III) can reduce TF expression and activity [17]. As we know, many patients with CTEPH often undergo anticoagulation therapy before diagnosis. Our results also illustrate this point as the expression of the TF antigen and TF mRNA levels increased in CTEPH patients but with no corresponding increase in TF activity, likely due to the impact of anticoagulant therapy.
Tissue factor closely related with monocytes
Monocytes were the first cell type identified that can synthesize and express TF. TF is also present in other cell types that associate with monocytes. Our study showed that the levels of monocyte TF mRNA in circulating blood increased in all groups (only CTEPH and PTE groups showed a significant difference) when compared to the control group, with a concurrent increase in the levels of TF antigen and activity. This suggests that TF expression in monocytes can play an important role in the pathogenesis of PTE and CTEPH. Furthermore, we have found that the expression of the TF antigen significantly correlates with monocyte TF mRNA levels in both CTEPH and PTE patients, suggesting that levels of TF antigen are closely related to TF mRNA levels in monocytes under pathological conditions. Therefore, the results indicate that mononuclear cells are the source of blood-borne TF, which plays a key role in the process of physiological hemostasis and thrombosis [27].
In this study, we observed that the increase in expression of TF in monocytes can result in the formation and maintenance of thrombus and may play an important role in the development of PTE and CTEPH. Zhang et al. have shown TF involvement in the pathophysiological process of PTE, and that high levels of TF expression in pulmonary arterial tissue adjacent to emboli could lead to an increase in local coagulation activity [9]. Kooiman et al. have also suggested that it is indispensable to assess the relationship between TF and acute/chronic thromboembolic disease [28].
Coagulation–inflammation–thrombosis circuit
Our observations raise the following question: why are levels of TF elevated in CTEPH? As mentioned above, TF may participate in the pathogenesis of CTEPH by triggering the extrinsic coagulation pathway. The level of TF in monocytes may increase in various pathological conditions such as infection, inflammation, thrombosis, and cancer [17, 29, 30]. In this study, we found that the expression of inflammatory factors (e.g. TNF-α, CRP, MCP-1) increased and significantly correlated with TF antigen content in CTEPH. This implies that the expression of TF may be upregulated by these inflammatory factors and it may be of clinical value to detect the levels of these factors in CTEPH patients to provide further information on their condition and prognosis.
Generally, CRP, TNF-α and MCP-1 may be involved in the process of CTEPH pathogenesis by mediating the inflammatory process, which induces the expression of TF in monocytes and aids in the formation of blood clots. An increase in monocyte TF could be disastrous because inflammatory cells, which are regulated by a variety of inflammatory mediators, can not only enhance the expression and activity of TF but can also lead to the injury of vascular endothelial cells. With injury to vascular endothelial cells, increasing amounts of TF are exposed to the blood, which activates the extrinsic coagulation pathway and causes thrombosis formation and deposits in the blood vessel walls. As a consequence, blood vessels become narrow and produce large amounts of inflammatory mediators, resulting in further inflammation. Inflammatory cytokines can induce TF expression and activate the coagulation system, giving rise to the formation of the inflammation-coagulation-thrombosis cycle. There is mounting evidence supporting the existence of such a positive feedback/reversible loop in a complete coagulation-inflammation cycle [31]. For example, TNF-α upregulates the expression of TF in acute respiratory distress syndrome (ARDS) [32], and CRP drastically increases TF expression [33]. Long pentraxin-3, an acute inflammatory molecule, upregulates TF expression in lung injuries [34]. Conversely, guggulsterone (an anti-inflammatory phytosterol) inhibits TF expression and arterial thrombosis [35], adding further proof of inflammation-triggered coagulation. Furthermore, the TF hypercoagulable state results in progressive inflammation as a result of continuous refueling of the coagulation-inflammation cycle. Thrombosis and inflammation are two major pathogeneses of CTEPH, between which there is cross-talk and cooperativity [36]. The inflammation-thrombosis connection provides an alternative pathway through which blood coagulation can indirectly contribute to thrombosis through inflammation, and both processes together promote CTEPH pulmonary vascular remodeling [37]. Currently, few reports have focused on the role of monocyte TF and CTEPH. Our study involved a small number of patients and did not examine the mechanism of TF in CTEPH nor did we compare the change in inflammatory factors before and after treatment. Therefore, further studies with larger patient cohorts that elucidate the molecular mechanism are needed.
Conclusions
In summary, TF expression was increased in the plasma of patients with CTEPH, partly due to an increase in monocyte TF mRNA levels. Monocyte TF may play an important role during the CTEPH thrombotic process. At the same time, the inflammatory factors CRP, TNF-α and MCP-1 increased in the plasma of patients with CTEPH and correlated with mPAP, indicating that they are involved in the pathogenesis of CTEPH and determine disease severity. Moreover, high expression of TF correlated with expression of the inflammatory factors CRP, TNF-α and MCP-1 in patients with CTEPH. TF, CRP, TNF-α, and MCP-1 may not be attractive molecules to test for screening of CTEPH but may have value in determination of prognosis, which was not evaluated in our study.
References
Toshner M, Pepke-Zaba J (2014) Chronic thromboembolic pulmonary hypertension: time for research in pathophysiology to catch up with developments in treatment. F1000Prime Rep 6:38. doi:10.12703/P6-38
Lang IM (2004) Chronic thromboembolic pulmonary hypertension-not so rare after all. N Engl J Med 350(22):2236–2238. doi:10.1056/NEJMp048088
Olsson KM, Meyer B, Hinrichs J, Vogel-Claussen J, Hoeper MM, Cebotari S (2014) Chronic thromboembolic pulmonary hypertension. Dtsch Arztebl Int 111(50):856–862. doi:10.3238/arztebl.2014.0856
Riedel M, Stanek V, Widimsky J, Prerovsky I (1982) Long-term follow-up of patients with pulmonary thromboembolism. Late prognosis and evolution of hemodynamic and respiratory data. Chest 81(2):151–158. doi:10.1378/chest.81.2.151
Mercier O, Fadel E, Mussot S, Fabre D, Ladurie FL, Angel C et al (2014) Surgical treatment of chronic thromboembolic pulmonary. Presse Med 43(9):994–1007. doi:10.1016/j.lpm.2014.07.007
Suntharalingam J, Treacy CM, Doughty NJ, Goldsmith K, Soon E, Toshner MR et al (2008) Long-term use of sildenafil in inoperable chronic thromboembolic pulmonary hypertension. Chest 134(2):229–236. doi:10.1378/chest.07-2681
Wynants M, Quarck R, Ronisz A, Alfaro-Moreno E, Van Raemdonck D, Meyns B et al (2012) Effects of C-reactive protein on human pulmonary vascular cells in chronic thromboembolic pulmonary hypertension. Eur Respir J 40(4):886–894. doi:10.1183/09031936.00197511
Peacock A, Simonneau G, Rubin L (2006) Controversies, uncertainties and future research on the treatment of chronic thromboembolic pulmonary hypertension. Proc Am Thorac Soc 3(7):608–614
Zhang JX, Chen YL, Zhou YL, Guo QY, Wang XP (2014) Expression of tissue factor in rabbit pulmonary artery in an acute pulmonary embolism model. World J Emerg Med 5(2):144–147. doi:10.5847/wjem.j.1920-8642.2014.02.012
White RJ, Meoli DF, Swarthout RF, Kallop DY, Galaria II, Harvey JL (2007) Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 293(3):L583–L590. doi:10.1152/ajplung.00321.2006
White TA, Witt TA, Pan S, Mueske CS, Kleppe LS, Holroyd EW et al (2010) Tissue factor pathway inhibitor overexpression inhibits hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 43(1):35–45. doi:10.1165/rcmb.2009-0144OC
Quarck R, Nawrot T, Meyns B, Delcroix M (2009) C-reactive protein: a new predictor of adverse outcome in pulmonary arterial hypertension. J Am Coll Cardiol 53(14):1211–1218. doi:10.1016/j.jacc.2008.12.038
Quarck R, Wynants M, Verbeken E, Meyns B, Delcroix M (2015) Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur Respir J 46(2):431–443. doi:10.1183/09031936.00009914
Langer F, Schramm R, Bauer M, Tscholl D, Kunihara T, Schäfers HJ (2004) Cytokine response to pulmonary thromboendarterectomy. Chest 126(1):135–141. doi:10.1378/chest.126.1.135
Kimura H, Okada O, Tanabe N, Tanaka Y, Terai M, Takiguchi Y et al (2001) Plasma monocyte chemoattractant protein-1 and pulmonary vascular resistance in chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 164(2):319–324. doi:10.1164/ajrccm.164.2.2006154
Zabini D, Heinemann A, Foris V, Nagaraj C, Nierlich P, Bálint Z et al (2014) Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur Respir J 44(4):951–962. doi:10.1183/09031936.00145013
Chu Arthur J (2011) Tissue factor, blood coagulation, and beyond: an overview. Int J Inflamm 2011:367284. doi:10.4061/2011/367284
Lang IM, Pesavento R, Bonderman D, Yuan JX (2013) Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J 41(2):462–468. doi:10.1183/09031936.00049312
Dartevelle P, Fadel E, Mussot S, Chapelier A, Hervé P, de Perrot M et al (2004) Chronic thromboembolic pulmonary hypertension. Eur Respir J 23(4):637–648. doi:10.1183/09031936.04.00079704
Kim NH, Delcroix M, Jenkins DP, Channick R, Dartevelle P, Jansa P et al (2013) Chronic thromboembolic pulmonary hypertension. J Am Coll Cardiol 62(25 Suppl):D92–D99. doi:10.1016/j.jacc.2013.10.024
Altman R, Scazziota AS, Herrera MDL, Gonzalez C (2006) Thrombin generation by activated factor VII on platelet activated by different agonists. Extending the cell-based model of hemostasis. Thromb J 4:5. doi:10.1186/1477-9560-4-5
Shetty S, Bhandary YP, Shetty SK, Velusamy T, Shetty P, Bdeir K et al (2010) Induction of tissue factor by urokinase in lung epithelial cells and in the lungs. Am J Respir Crit Care Med 181(12):1355–1366. doi:10.1164/rccm.200901-0015OC
Owens AP, Mackman N (2010) Tissue factor and thrombosis: the clotstarts here. Thromb Haemost 104(3):432–439. doi:10.1160/TH09-11-0771
Darbousset R, Thomas GM, Mezouar S, Frère C, Bonier R, Mackman N et al (2012) Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 120(10):2133–2143. doi:10.1182/blood-2012-06-437772
Rauch U, Nemerson Y (2000) Circulating tissue factor and thrombosis. Curr Opin Hematol 7(5):273–277
Hron G, Kollars M, Weber H, Sagaster V, Quehenberger P, Eichinger S et al (2007) Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost 97(1):119–123. doi:10.1160/TH06-03-0141
Østerud B, Bjørklid E (2006) Sources of tissue factor. Semin Thromb Hemost 32(1):11–23. doi:10.1055/s-2006-933336
Kooiman J, den Exter PL, Kilicsoy I, Cannegieter SC, Eikenboom J, Huisman MV et al (2015) Association between micro particle-tissue factor activity, factor VIII activity and recurrent VTE in patients with acute pulmonary Embolism. J Thromb Thrombolysis. doi:10.1007/s11239-015-1180-z
Zwicker JI, Trenor CC 3rd, Furie BC, Furie B (2011) Tissue factor bearing microparticles and thrombus formation. Arterioscler Thromb Vasc Biol 31(4):728–733. doi:10.1161/ATVBAHA.109.200964
de Meis E, Azambuja D, Ayres-Silva JP, Zamboni M, Pinheiro VR, Levy RA et al (2010) Increased expression of tissue factor and protease-activated receptor-1 does not correlate with thrombosis in human lung adenocarcinoma. Braz J Med Biol Res 43(4):403–408. doi:10.1590/S0100-879X2010007500017
Chu AJ (2006) Tissue factor upregulation drives a thrombosis-inflammation circuit in relation to cardiovascular complications. Cell Biochem Funct 24(2):173–192. doi:10.1002/cbf.1200
Kambas K, Markiewski MM, Pneumatikos IA, Rafail SS, Theodorou V, Konstantonis D et al (2008) C5a and TNF-α up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J Immunol 180(11):7368–7375
Wu J, Stevenson MJ, Brown JM, Grunz EA, Strawn TL, Fay WP (2008) C-reactive protein enhances tissue factor expression by vascular smooth muscle cells: mechanisms and in vivo significance. Arterioscler Thromb Vasc Biol 28(4):698–704. doi:10.1161/ATVBAHA.107.160903
Napoleone E, di Santo A, Peri G, Mantovani A, de Gaetano G, Donati MB et al (2004) The long pentraxin PTX3 up-regulates tissue factor in activated monocytes:another link between inflammation and clotting activation. J Leukoc Biol 76(1):203–209. doi:10.1189/jlb.1003528
Gebhard C, Stämpfli SF, Gebhard CE, Akhmedov A, Breitenstein A, Camici GG et al (2009) Guggulsterone, an anti-inflammatory phytosterol, inhibits tissue factor and arterial thrombosis. Basic Res Cardiol 104(3):285–294. doi:10.1007/s00395-008-0757-5
Libby P, Simon DI (2001) Inflammation and thrombosis: the clot thickens. Circulation 103(13):1718–1720
Strukova S (2006) Blood coagulation-dependent inflammation. Coagulation-dependent inflammation and inflammation-dependent thrombosis. Front Biosci 11:59–80
Acknowledgments
This research was supported by Programs of National Natural Science Foundation of China (81570264), Young and Middle-Aged Talent Cultivation of Fujian Provincial Health System (2015-ZQN-ZD-18) and Fujian Provincial Medical Innovation Subject (2014-CXB-12). We are also very grateful to Changsheng Xu for expert technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they do not have any competing or financial interests.
Additional information
Minxia Yang and Chaosheng Deng have contributed equally to this paper.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yang, M., Deng, C., Wu, D. et al. The role of mononuclear cell tissue factor and inflammatory cytokines in patients with chronic thromboembolic pulmonary hypertension. J Thromb Thrombolysis 42, 38–45 (2016). https://doi.org/10.1007/s11239-015-1323-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-015-1323-2