
Abstract
Geothermally heated regions of Earth, such as terrestrial volcanic areas (fumaroles, hot springs, and geysers) and deep-sea hydrothermal vents, represent a variety of different environments populated by extremophilic archaeal and bacterial microorganisms. Since most of these microbes thriving in such harsh biotopes, they are often recalcitrant to cultivation; therefore, ecological, physiological and phylogenetic studies of these microbial populations have been hampered for a long time. More recently, culture-independent methodologies coupled with the fast development of next generation sequencing technologies as well as with the continuous advances in computational biology, have allowed the production of large amounts of metagenomic data. Specifically, these approaches have assessed the phylogenetic composition and functional potential of microbial consortia thriving within these habitats, shedding light on how extreme physico-chemical conditions and biological interactions have shaped such microbial communities. Metagenomics allowed to better understand that the exposure to an extreme range of selective pressures in such severe environments, accounts for genomic flexibility and metabolic versatility of microbial and viral communities, and makes extreme- and hyper-thermophiles suitable for bioprospecting purposes, representing an interesting source for novel thermostable proteins that can be potentially used in several industrial processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Background
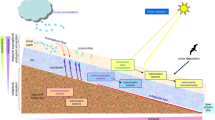
Hot springs populated by extreme thermophiles (Topt: 65–79 °C) and hyperthermophiles (Topt > 80 °C) (DeCastro et al. 2016) are very diverse and some of them show combinations of extreme chemo-physical conditions, such as temperature, acidic or alkaline pHs, high pressure, and high concentrations of salts and heavy metals (López-López et al. 2013). As with all studies of environmental microbiology, our understanding of the composition, functional and physiological dynamics, and evolution of extreme- and hyper-thermophilic microbial consortia has lagged substantially behind. However, recent advances in ‘omics’ technologies, particularly within a system biology context, allowed significant progress in this field. These include the prediction of microbial consortia functionality in situ and the access to enzymes with important potential applications in biotechnology (Cowan et al. 2015). Metagenomics is particularly relevant in geothermal environments since most extremophilic microorganisms are recalcitrant to cultivation-based approaches (Amann et al. 1995; Lorenz et al. 2002). The rapid and substantial cost reduction in next-generation sequencing (NGS) (Fig. 1) has dramatically accelerated the development of sequence-based metagenomics as witnessed by the explosion of metagenome shotgun sequence datasets in the past few years. Metagenomics provides access to the gene composition of microbial communities offering a much deeper description than phylogenetic surveys, which are often based only on the diversity of the 16S rRNA gene. In addition, metagenomic datasets can provide novel, and even unexpected insights into community dynamics. For example, surprisingly, it has been found that both extreme thermophiles and hyperthermophiles showed a statistically significant higher number of clustered regularly interspaced short palindromic repeats (CRISPR) sequences in their genomes than mesophiles, suggesting that viruses/phages may play an important role in shaping composition and function of thermophiles communities as well as in driving their evolution (Cowan et al. 2015). In addition, functional metagenomic strategies, exploiting expression libraries in conventional microbes, are powerful alternatives to conventional genomic approaches for producing novel enzymes for industrial applications.

Evolution of technology sequencing in the past 10 years. The main characteristics of the different techniques, including main pros and cons, are highlighted. NGS, producing millions of shortreads (25–650 bp) is generally less expensive than the Sanger sequencing and shows faster library construction without bias for toxic genes in the cloning host
This review offers an overview on recent developments of metagenomics applied to terrestrial geothermal environments with temperature ≥65 °C. We describe how recent progress in deep sequencing technology led to the expansion of the studies on microbial and phage/viral communities populating these sites (Sect. 2) as well as to the use of CRISPR loci as a metagenomic tool to identify specific hosts for a viral assemblage (Sect. 2.2.1). Then, particular emphasis is given to the most recent literature on the distribution of microbiome and virome communities populating terrestrial geothermal sites worldwide (Sect. 4), and to the exploitation of functional metagenomics for the discovery and production of enzymes for biotechnology (Sect. 5).
2 Microbial and viral metagenomics of geothermal environments
2.1 Microbial metagenomics
The microbiological study of geothermal environments officially started in the early 1970s with the innovative work of Thomas Brock and followed, in the 1980s, by the early microbial studies by using 16S rRNA analysis allowing the identification of Archaea as a kingdom separated from Bacteria [Brock et al. 1971; Woese et al. 1990, which is reviewed in Brock (2001)]. Since then, this approach, overcoming the limitations of thermophilic microorganisms isolation, has continuously revealed novel uncultivated microbial lineages, proving that isolates represent less then 20% of the phylogenetic diversity in Archaea and Bacteria (Reysenbach et al. 1994; Stahl et al. 1984; Wu et al. 2009). Several microbiological surveys of (hyper)thermophilic environments were performed in the last 10 years by using 16S rRNA gene profiling. This led to the first microbial characterization of different geothermal areas, allowing the identification of the dominant phyla/genera among the microbial communities populating these environments and the correlations with geophysical and climatic parameters (Meyer-Dombard et al. 2005; Wang et al. 2014). Despite the easiness of this approach, the assessment of abundance estimation and of microbial diversity from a single 16S rRNA gene is challenging for several reasons (see the section below: Approaches and tools for microbial and viral metagenomics) (Fig. 2).

Schematic flow chart of the analysis approaches of a sample from geothermal sites. 16S rRNA profiling (purple), microbial metagenomics (light blue), viral metagenomics (orange), single-cell genomics (yellow) and functional metagenomics (green). Dashed arrows and box indicate optional steps. (Color figure online)
An alternative approach is the sequence-based metagenomic (SBM) analysis, which has been exploited successfully also on microbiomes populating geothermal sites. This method provides access to the gene composition of a microbial community and to its encoded function, giving a much broader and detailed phylogenetic description than the 16S rRNA profiling (Wu et al. 2014). Indeed, SBM, which is especially valuable for complex communities requiring deeper sequencing, represents the best approach in geothermal hot springs in which, despite the low microbial complexity, the population is not well characterized because of the difficulties in isolating new strains through classical microbiology approaches. As reported by Bahya and co-workers, SBM led for the first time in 2007 to the identification of two different Synechococcus populations inhabiting the microbial mats of the Octopus Spring in the Yellowstone National Park (YNP). This study revealed extensive genome rearrangements and differences related to the assimilation and storage of several elements such as nitrogen, phosphorus and iron suggesting that the two populations have adapted differentially to the fluxes and gradients of chemical elements (Bhaya et al. 2007). In addition, SBM showed correlations between function and phylogeny of unculturable microorganisms allowing the study of evolutionary profiles and the identification of novel candidate phyla (i.e. Geoarchaeota, Lokiarchaeota and Aigarchaeota). These studies contributed to the understanding of the archaea evolution and their metabolic interactions that may not have been addressed with the basic 16S rRNA gene profiling (Kozubal et al. 2013; Spang et al. 2015). At the onset, SBM was generally more expensive than 16S rRNA sequencing, however, the constant reduction of the NGS costs (Fig. 1) made this approach more and more convenient thereby considerably increasing the number of metagenomic projects available and making it a valid support, or even a direct alternative, to the 16S rRNA profiling. In addition, the sequence data banks resulting from SBM studies of geothermal environments are an important repository of genes encoding for novel enzymes with potential biotechnological interest. Therefore, in silico functional screening of metagenomic data banks allows the identification of genes that can be cloned and expressed in mesophilic hosts to produce recombinant enzymes. Alternatively, metagenomic expression libraries can be constructed to perform direct functional screenings of the enzymes of interest (see the section Enzyme discovery below).
2.2 Viral metagenomics
Phages are generally the predominant biological entity in every ecosystem and have the capability to greatly influence the structure, composition and function of their host population(s) (Snyder et al. 2015). This holds true also in geothermal environments, although their density is lower (typically 10–100-fold less viruses than host cells) if compared to mesophilic aquatic systems (López-López et al. 2013). Despite their importance, the knowledge about the diversity and biology of phages on the microbial communities in these ecosystems is still limited (Schoenfeld et al. 2008). Since not obvious common genetic markers exist, phages are still classified according to their host range and morphology, thus making challenging the discovery of genetic variants and novel subtypes. In addition, the rate of lateral gene transfer events within the geothermal environments is exceptionally high, a fact that renders uncertain the resolution of evolutionary histories of the known major viral/phages groups (Diemer and Stedman 2012).
Geothermal environments with temperature >80 °C tend to be dominated by archaea over bacteria and eukaryotes (Bolduc et al. 2015) and therefore, the majority of viruses isolated from two types of habitats are archeoviruses (Snyder et al. 2015). At present, one order and 10 families (Fuselloviridae, Bicaudaviridae, Ampullaviridae, Clavaviridae, Guttaviridae, Lipothrixviridae, Rudiviridae, Globuloviridae, Myoviridae, Siphoviridae) of archaeal viruses have been documented (Fusco et al. 2015a, b; Prangishvili 2013; Snyder et al. 2015; Wang et al. 2015b). Until relatively recent times, the only methodology available to study these viruses was through the cultivation of their hosts (López-López et al. 2013). By systematically applying this approach, our knowledge on viruses populating (hyper)thermal environments over the last 30 years has considerably expanded thanks to the pioneer work of Wolfram Zillig and, subsequently, of several groups in Europe and USA (Bize et al. 2008; Dellas et al. 2013, 2014; Diemer and Stedman 2012; Haring et al. 2005; Peng et al. 2012; Prangishvili et al. 2001, 2006; Prangishvili and Garrett 2004, 2005; Rice et al. 2001, 2004; Snyder et al. 2011; Snyder and Young 2013; Zillig et al. 1996). While enrichment cultures have been invaluable in the study of thermophilic viruses, contextual information, such as relative abundance, diversity, and distribution, was mainly unknown.
Direct SBM analysis of environmental samples together with the development of ad hoc bioinformatics tools (Rampelli et al. 2016; Roux et al. 2014, 2015) had a revolutionary impact on virology of extremophiles providing a better understanding of viral specific role in these environmental niches. In addition, viral metagenomics and genomics of cultured viruses has also revealed that a large proportion of predicted archaeal viral genes are ‘unknown’ or ‘hypothetical’ (Contursi et al. 2014a; Prato et al. 2008) expanding the content of genetic information referred as biological ‘dark matter’ (Martinez-Garcia et al. 2014). It is expected that the annotation of additional sequenced genomes as well as the exploitation of bioinformatics tools based on structural protein homologies will help to disclose this unexplored repository of viral genes (Fig. 2). Interestingly, non-coding nucleic acid sequences also play a critical role in archaeal virus function, which is a virtually underestimated topic in archaeal virology (Contursi et al. 2010).
Despite the promising scientific impact, only few viral metagenomics on geothermal samples have been reported so far (see also the paragraph: Geographical distribution of microbiomes). Some studies were pursued by deep sequencing of environmental samples enriched for virus particles (Bolduc et al. 2012; Garrett et al. 2010; Schoenfeld et al. 2008) whereas others were performed by retrieving viral sequences from whole SBM datasets (Gudbergsdottir et al. 2016; Servín-Garcidueñas et al. 2013a, b).
In addition, recently, the same approach allowed to focus on the study of CRISPR that became one of the most advanced fields in viral metagenomics and that is reviewed below.
2.2.1 CRISPR
CRISPR is a mechanism of acquired immunity playing a role in controlling the equilibrium between prokaryotic populations and their parasites. This system, which is found in the 80% of archaea and 40% of bacteria, recognizes and memorizes short sequences from the genome of the viral or phage invader (Barrangou et al. 2007; Brouns et al. 2008; Fusco et al. 2015a; Marraffini and Sontheimer 2010; van der Oost et al. 2009). The peculiar structure of CRISPR loci (Fig. 3), with alternating spacer and repeat units, results in a computationally identifiable sequence signature. Several bioinformatics tools have been developed to identify CRISPR spacers in bacterial genomes (Biswas et al. 2013; Bland et al. 2007; Edgar and Myers 2005; Grissa et al. 2007; Skennerton et al. 2013), and spacer sequences have also been collected in publicly accessible databases (Grissa et al. 2007; Rousseau et al. 2009).

Overview of the CRISPR-Cas system. Upon infection, CRISPR immune system operates by recognizing foreign genetic elements, such as plasmids and bacteriophages (blue solid line) and cleaving them into short DNA fragments (protospacers). In adaptation, these latter are then inserted, as new spacers, into the sequence array of a CRISPR locus. Such a locus consists of several short palindromic repeats, each approximately 20–50 bp in length, interspersed by spacers (Grissa et al. 2007). This array is typically located adjacent to a leader sequence (black rectangle) and CRISPR-associated cas genes (coloured arrows) (Rath et al. 2015). A newly acquired spacer (blue box highlighted by a black arrow) is generally inserted directionality into the CRISPR array region, which is closest to the leader sequence, thus preserving the history of viral infections. In transcription and processing, the repeats-spacers array is transcribed into a pre-crRNA, which is cleaved at each repeat to yield individual mature CRISPR RNAs (crRNA). These guides a dedicated set of CRISPR-associated (Cas) proteins to their targets during cellular surveillance (Marraffini and Sontheimer 2010; van der Oost et al. 2009). Indeed, upon reinfection, when the DNA or in some cases mRNA of remembered invaders is identified, the CRISPR-Cas system binds to invading phage DNA resulting in the degradation of the phage genome sequence (Stern and Sorek 2011). (Color figure online)
A challenge in the field of archaeal virology is the development of new approaches in order to move rapidly from analysis of SBM to the identification and isolation of the viral nucleic acids present in the environmental samples as well as of their respective hosts. In this regard, analyses of CRISPR spacers across metagenomics data provide high-resolution genetic markers that not only recapitulate the history of infections in the host genomes, but also allow individual phage strains to be tracked by following their presence in the very same host genomes (Vale and Little 2010) (Fig. 2). This has been done either by extracting spacers from sequenced host genomes or by PCR identifying CRISPR spacers from the same sample (Gudbergsdottir et al. 2016). An alternative approach consists in exploiting a microarray platform built up with CRISPR spacer sequences of host metagenomics data to examine temporal changes in viral populations within this environment (Snyder et al. 2010).
3 Approaches and tools for microbial and viral metagenomics
3.1 Preparation of high-molecular weight DNA and metagenomic libraries
The step of metagenomic DNA (mDNA) extraction from samples collected in extreme environments plays a critical role for the whole metagenomic analysis workflow (Fig. 2). To be sure that the genetic information obtained is representative of the whole microbiome, mDNA preparation requires specific protocols to preserve, as much as possible, the quality and the amount of the nucleic acids to guarantee best metagenomic library and the subsequent sequencing approach. mDNA extraction from geothermal environments can be performed by following two general strategies sharing the critical removal of humic acid, a major soil component made of phenolic moieties covalently bound to DNA (Lakay et al. 2007), which inhibits restriction enzymes and PCR amplification (Tebbe and Vahjen 1993). The first method is the “direct mDNA extraction” and consists in cell lysis directly followed by the nucleic acids separation from soil particles within the sample, generally providing quickly high DNA amounts.
By contrast, in the second approach, named “indirect mDNA extraction”, the environmental samples need to be physically and mechanically treated before cell lysis. Although this method requires abundant initial sample and is time-consuming if compared to the “direct mDNA extraction”, it is suitable for in-depth sequencing and creation of fosmid libraries, because of the reduced proportion of eukaryotic sequences present in the sample and increased length of the mDNA chunks (Delmont et al. 2011).
Library production for most sequencing technologies require not only high amounts of mDNA but in some cases also amplification of nucleic acids generally performed by Multiple Displacement Amplification (MDA). This method, used both in metagenomics and single-cell genomics, can amplify femtograms to up to micrograms of DNA (see below).
When only a specific part of the ecological community is the target of analysis, as the viral population, additional steps can be applied (Fig. 2).
Indeed, the relative abundance of viral particles in a sample, compared to that of other organisms such as bacteria or host cells (or their genomes), is a critical factor for the discovery of viruses when using metagenomic analysis. Enrichment methods applied to the detection of viruses in hot springs are methodologically challenging mainly because extremophilic viruses and their microbial hosts are rarely cultivable (Edwards and Rohwer 2005). In addition, the majority of the viral metagenomic reads (50–90%) show no significant similarity to sequences from known organisms. Current approaches for virus isolation and concentration include filtration and/or adsorption to and subsequent elution from positively or negatively charged membrane filters (Katayama et al. 2002) and pelletting of virus particles through ultracentrifugation (Bolduc et al. 2012; Short and Short 2008). The drawbacks include selective adsorption of viruses onto treated filters, limited volume capacity and low or variable recoveries of viruses. An efficient virus purification and concentration method consisting in the combination of tangential flow filtration (TFF) with centrifugal ultrafiltration technology, has been employed in order to obtain high density of viral particles from high turbidity seawaters samples (Sun et al. 2014). Such enrichment method has been applied to recover hot springs viral particles for metagenomic studies (Diemer and Stedman 2012; Schoenfeld et al. 2008). Finally, an efficient and reliable method to concentrate viruses from ecological samples has been developed by using FeCl3 as a low-cost and non-toxic agent that leads to nearly complete recovery (92–95%) (John et al. 2011).
3.2 Next generation sequencing technology
Sanger sequencing, developed almost 40 years ago, is still considered a good method for nucleic acid sequencing, and is characterized by the use of sequencing library with large insert sizes (>30 Kb), long read length (up to 1000 bp) with a relative low error rate. However, over the past 10 years shotgun sequencing, including metagenomic analysis, has gradually shifted from this technology to NGS with faster library construction, non associated to bias for toxic genes for the cloning host (Sorek et al. 2007) and generally less expensive. NGS platforms (Fig. 1) usually produce millions of short sequence reads up to 800 bp. To date, the most used sequencing techniques are Roche 454 and Illumina that have now been extensively applied to metagenomic from geothermal environments (Inskeep et al. 2013b; Menzel et al. 2015) (Table 1).
The Roche 454 system produces an average read length between 600 and 800 bp, reducing significantly the number of reads that are too short to be annotated without assembly (Wommack et al. 2008). The main drawbacks of this method with respect to metagenomic applications are the production of artificial replicate sequences (up to 15% of the resulting sequences), which affect the estimation of both microbial and gene abundances (Gomez-Alvarez et al. 2009) and a high error rate in homopolymer regions (Margulies et al. 2005). Despite these disadvantages, Roche 454 is much cheaper (up to 16,000$ per Gb) than the Sanger sequencing. In addition, also the sample preparation has been optimized requiring nanograms of mDNA for the sequencing of a single-end library (Adey et al. 2010), although pair-end sequencing might still require micrograms quantities.
If compared to the Roche 454 technology, Illumina, being sensibly cheaper with a cost of ~200$ per Gbp, usually reads up to 300 bp (Fig. 1). In addition, although more time consuming, Illumina has limited systematic errors and the quality control allows to detect and eliminate bad reads. Faster analysis can be obtained with the Illumina MiSeq instrument. However, despite there is evidence that the MiSeq offers valuable information for shotgun sequencing and can be used to test-run sequencing libraries before analysis on HiSeq instruments, deeper sequencing is strictly required in order to detect the majority of species in a sample and to perform an high quality assembly with a good abundance estimation of the microorganisms (Clooney et al. 2016).
A detailed comparison about the advantages and limitations of Roche 454 and Illumina platforms was reviewed by Luo et al. (2012). The authors suggest that both NGS technologies are reliable for quantitatively assessing genetic diversity within environmental communities. Moreover, considering the longer and more accurate contigs obtained with Illumina by assembly (despite the substantially shorter read length) and the monetary savings by one fourth of the cost relatively to Roche 454, Illumina method may be a more favourable approach for metagenomic studies (Luo et al. 2012).
3.3 16S rRNA PCR amplification versus sequence-based metagenomics and single cell genomics
Generally, 16S rRNA gene profiling is considered as a first approach in a metagenomic survey and has been applied to the analysis of the different microbial populations since the middle 1990s with recent boosts due to the advances of the NGS sequencing platforms (Fig. 1). The comparison of 16S rRNA sequence profiles across different samples, indeed, can explain how microbial communities are related across different environmental conditions. Typically, this approach involves the amplification and the sequencing by NGS of short hypervariable regions (V1-V9) of the 16S rRNA gene that demonstrate considerable and differential sequence diversity in microorganisms. Although a single hypervariable region is not sufficient to phylogenetically classify microorganisms, the hypervariable regions V2, V3 and V6 show the maximal heterogeneity among the different lineages providing the best discriminating power for the analysis of microbial communities (Chakravorty et al. 2007). Today, NGS can produce large 16S rRNA datasets containing hundreds of thousands of 16S RNAs fragments allowing the survey of several microbial communities simultaneously in different hyperthermophilic environments (Hou et al. 2013; Sahm et al. 2013). Despite the ease with which the 16S rRNA profiling can be made, this approach is known to be limited by the short read lengths obtained, sequencing errors (Quince et al. 2009), differences arising from the different hypervariable regions chosen (Youssef et al. 2009) and problems in the Operational Taxonomic Units (OTUs) assignment (Huse et al. 2010). Moreover, to assess abundance estimation and microbial diversity from the single 16S rRNA gene is challenging for several reasons, e.g.: (1) it may fail to resolve a substantial fraction of the diversity in a community given various biases associated with PCR, (2) sequencing can produce widely varying estimates of diversity because different hypervariable regions have differential power at resolving taxa, (3) sequencing provides just a survey of the taxonomic composition of the microbial community without information about the biological function of the taxa, and (4) sequencing is limited to the analysis of known taxa while novel or highly diverged microorganism or viruses, are difficult to study using this approach. Moreover, given the prevalence of horizontal gene transfer, the inherent difficulties in defining microbial species, and the limited resolution of the 16S rRNA gene among closely related species, 16S rRNA profiling should be evaluated carefully, in particular if applied to the temporal microbial surveys. In this case, by using both 16S amplicon analysis and a metagenomic approach, was observed 1.5- and ~10-times more OTU assigned to phyla and genera respectively with the metagenomic method than the 16S rRNA analysis. This seems masking several levels of intra-genus differentiation and heterogeneity of the microbial population (Poretsky et al. 2014).
SBM is an alternative approach to the study of microbial consortia that avoids the limitation of the 16S rRNA analysis. Reads obtained as described above, align to various genomic locations of the different genomes present in the sample, including viruses. By assembling short reads (e.g. Illumina 100 paired-end) longer genomic contigs are obtained. Then, the contigs can be clustered by “binning”, on the base of their nucleotide composition such as %GC and tetranucleotide frequency (Wu et al. 2014), allowing the taxonomic assignment of the resulting bins by homology searches. The analysis of the contigs provides access to the functional gene composition of microbial communities giving a much broader and detailed description than the phylogenetic surveys based on 16S rRNA profiling. Moreover, by using suitable sequence databases (nucleic and aminoacidic), SBM is useful to obtain genetic information on potentially novel biocatalysts, to reveal correlation between function and phylogeny for uncultured organisms, and to study evolutionary profiles of microbial communities. However, the assembly process is generally affected by the problem that single reads have lower confidence in accuracy (low coverage) than the multiple reads that cover the same segment of genetic information (high coverage). This implies that in a complex microbial community with low coverage, it is unlikely to get many reads covering the same fragment of DNA and affecting the result of the assembling. Nevertheless, without assembly, it is impossible to analyse longer and more complex genetic loci such as CRISPRs (Sun et al. 2016). Despite the clear benefits, metagenomic sequence data are not challenges-free since they are generally complex and large, requiring specific hardware for storing and elaboration to avoid computational issues. In addition environmental metagenomic samples may contain contaminating DNA such as from animals and plants seizing useful reads from the microbial analysis. To determine which reads were generated from a detected contaminant’s genome, especially when the contaminant is abundant or has a large genome, can be problematic. SBM is generally more expensive than 16S rRNA sequencing, especially in complex communities requiring a deepest sequencing. However, the cost reduction of the NGS has dramatically increased the number of metagenomic projects making these approaches a direct substitute of the 16S rRNA profiling. This expansion is reflected in the high number of bioinformatics tools and data resources that have been developed in the last six years and are available for SBM analysis. Many of them work on a command-line environment or are web-based tools, which centralize metagenome data management and analysis, providing an interface ready to use but lacking in customization of the analysis. For a complete and detailed overview of metagenomic tools and strategies see the excellent review of Thomas et al. (2012).
In contrast to the metagenomic approach, single-cell genomics is addressed to the analysis of genomes one cell at a time (Blainey and Quake 2014) (Fig. 2). This approach requires the separation of individual cells from a complex environmental sample (e.g. sediments or microbial mat), cell lysis and the amplification and sequencing of genomic DNA (gDNA). The first step is the isolation of individual cells from the primary samples to obtain a suspension of viable single cells. However, this step can be challenging when the primary sample requires mechanical or enzymatic dissociation (e.g. sediments from a mud pool) keeping the cells viable without biases for specific subpopulations. After lysis of individual cells, the gDNA is amplified by using MDA (Lasken 2012; Zong et al. 2012). Generally, the resulting single amplified genomes (SAG) are screened by 16S rRNA profiling for a preliminary survey and identification of candidate phyla or other taxa. SAGs of interest are then deep sequenced by NGS platforms, assembled, and analyzed. By the analysis of the number of single-copy conserved markers in the assembled sequences, it is possible to evaluate how well a given SAG covers the target microorganism’s genome (Rinke et al. 2013). Although single-cell genomics is a useful tool for the study of unknown and uncultivable microorganisms, in particular from extreme environments such as geothermal areas, this shows several critical issues. For instance, the amplification protocols can introduce chimeric artefacts and a severe bias in genomic coverage. To overcome these problems during the assembling, specific methods have been developed to analyse single-cell genomic datasets combining data from closely related single cells clustered by nucleotide percentage identity (Rinke et al. 2013). The resulting assemblies can often represent nearly complete pangenomes for a given strain or species, allowing the detailed analysis of genes and pathways. Today, single-cell genomics and metagenomics can be considered as complementary approaches because the former is not affected neither by amplification issues nor by problems related to the separation of individual cells from a complex primary sample, while the latter is able to associate directly and unambiguously phylogeny and function (Walker 2014).
A combined approach of these methods, indeed, was recently used to identify two novel candidate phyla, Calescamantes and Candidatus kryptonia, by the analysis of different SAGs and metagenomic databases collected in different high-temperature environments (Eloe-Fadrosh et al. 2016; Kim et al. 2015) proving that, although metagenomics and single-cell genomics are informative of their own, the results of the mixed approach could be greater than the sum of their parts.
4 Geographical distribution of microbiomes
Terrestrial surface hot springs (T > 65 °C), which are spread all over the world, offer a remarkable source of biodiversity. Hereinafter, we report on the state of the art of the metagenomic survey of several hot springs worldwide (Fig. 4). The microbial and viral metagenomic data are summarized in Tables 1 and 2, respectively.

4.1 Yellowstone National Park, USA
The Yellowstone geothermal complex includes more then 10,000 thermal sites such as hot springs, vents, geysers, and mud pools showing broad ranges of pH, temperature and geochemical properties. One of the first detailed environmental and microbiological survey has been reported in 2005 and included three different hot springs in YNP, i.e. the Obsidian Pool (ObP) (80 °C, pH 6.5), the Sylvan Spring (SSp) (81 °C, pH ~ 5.5), and the Bison Pool (BP) (83 °C, pH ~ 8.0) (Meyer-Dombard et al. 2005). The Obsidian and Bison Pools are inhabited, among archaea, mostly by different groups of uncultured crenarchaeota or by members of the family Desulphurococcaceae (in ObP and BP, respectively), while bacteria belong to the genera Thermocrinis, Geothermobacterium and to the phylum Proteobacteria. On the other hand, Hydrogenothermus was the most abundant bacterial genus in SSp, together with a dominance of families Desulphurococcaceae and Thermoproteaceae among archaea (Meyer-Dombard et al. 2005). More recently, Inskeep and co-workers, in an extensive metagenomic survey of the microbial species in the YNP, reported on the identification of the predominant microbial populations, metabolic features, and the relationship between geochemical conditions and gene expression of five geochemically dissimilar high-temperature environments, namely Crater Hills (CH; 75 °C, pH 2.5), Norris Geyser Basin (NGB; 65 °C, pH 3.0), Joseph’s Coat (JCHS; 80 °C, pH 6.1), Calcite (CS; 75 °C, pH 7.8), and Mammoth Hot Springs (MHS; 71 °C, pH 6.6) (Inskeep et al. 2010). Specifically, binning and fragment recruitment approaches revealed that archaea, of the order Sulfolobales (in CH and NGB) and Thermoproteales (in JCHS), mainly dwelt in high-temperature acidic springs. Moreover, the results suggested that the relative abundance of Thermoprotei was modulated by differences in pH and/or concentration of dissolved O2. By contrast, bacteria mainly belonging to the order Aquificales, outnumbered archaea at pH values above 6.0 (CS and MHS). In particular, a predominance of reads showed nucleotide identity with Sulfurihydrogenibium sp. Y03AOP1 in MHS and with Thermus aquaticus and Sulfurihydrogenibium yellowstonensis in CS (Inskeep et al. 2010). To date, the widest investigation of microbial communities in hyperthermophilic environments (known as the YNP metagenome project) spans over 20 different geothermal sites in the YNP, 13 of which showing temperatures above 65 °C. These sites have been pooled in two different ecosystems based on features such as pH, temperature, presence of dissolved sulfide and elemental sulfur that are the main determinants shaping the microbiome within (Inskeep et al. 2013b). The first ecosystem, populated by Aquificales-rich “filamentous-streamer” communities, was identified in six sites: Dragon Spring (DS; 68–72 °C, pH 3.1); 100 Spring Plan (OSP_14; 72–74 °C, pH 3.5), Octopus Spring (OS; 74–76 °C, pH 7.9) and Bechler Spring (BCH; 80–82 °C, pH 7.8) together with MHS and CS described above (Inskeep et al. 2013b; Takacs-Vesbach et al. 2013). Whereas the second one is represented in seven archaeal-dominated sediments: Nymph Lake (NL; 88 °C, pH 4), Monarch Geyser (MG; 78–80 °C, pH 4.0), Cistern Spring (CIS; 78–80 °C, pH 4.4), Washburn Spring (WS; 76 °C, pH 6.4), 100 Spring Plan (OSP_8; 72 °C, pH 3.4), and including CH and JCHS described above (Inskeep et al. 2013a, b). The diversity detected among sites with similar characteristics suggested that additional geochemical and geophysical factors, such as the total dissolved organic carbon (DOC) and the amount of solid-phases of carbon, could play a role in the consortia composition (Inskeep et al. 2013a, b; Takacs-Vesbach et al. 2013). A similar observation but mainly related to the different SO4 2−/Cl− ratio was recently reported in the analysis of three thermal springs sharing pH ~ 4.0 of the YNP: Norris (NOR; 84 °C, pH 4.34), Mary Bay Area (MRY; 80 °C, pH 4.32) and Mud Kettles (MKL, 72 °C pH 4.35), which resulted exclusively populated by bacteria and dominated by microorganisms belonging to the phylum of Cyanobacteria (NOR and MRY) and Aquificales (MKL) (Jiang and Takacs-Vesbach 2017).
A combined approach of metagenomics and single-cell analysis conducted in CH and NL revealed the presence of Nanoarchaeota that, indeed, represent Nanobsidianus stetter cells based on their high 16S rRNA similarity and their overall genome homology.
This is in agreement with previous findings highlighting the wide distribution of Nanobsidianus genus in this kind of YNP geothermal environments (Clingenpeel et al. 2013). Furthermore, single-cell and catalyzed reporter deposition-fluorescence in situ hybridization analysis performed on these environmental YNP samples showed the occurrence of a symbiotic association with extreme thermoacidophilic Crenarchaeota hosts, such as Acidicryptum nanophilium, Acidolobus sp, Vulcanisieta sp. (5%), and Sulfolobus spp.
Genome fragments of Nanobsidianus contain integrated viral sequences. On the other hand, matching viral DNA sequences were found in the viral fractions isolated from the same hot springs, suggesting that Nanobsidianus species can host viruses or support viral replication (Munson-McGee et al. 2015).
Worth of note is also the abundance of archaeal DNA-and RNA-viruses in these environments (Table 2). The first viral metagenomics study (Schoenfeld et al. 2008) lead to the identification of double-stranded DNA viruses in the Octopus (93 °C) and Bear Paw (74 °C) hot springs (Inskeep et al. 2013b). Operons and potentially complete genomes were assembled, thus providing insight to the possibly dominant viral populations within each hot-spring (López-López et al. 2013; Schoenfeld et al. 2008). Viral metagenomes indicated the predominance of a lytic lifestyle as suggested by the significant proportion of lys-like genes encoding for proteins involved in host cell lysis (López-López et al. 2013; Schoenfeld et al. 2008). This evidence is in contrast to the cultured thermophilic crenarchaeal viruses, most of which are non-lytic (Contursi et al. 2006; Prangishvili 2013; Snyder et al. 2015; Wang et al. 2015b). The evidence of the replacement of cellular genes by non-orthologous viral genes (i.e. helicases, DNA polymerases, ribonucleotide reductase, and thymidylate synthase) suggested that viruses might play a critical role in the evolution of DNA and its replication mechanisms (Schoenfeld et al. 2008).
In a further study, Inskeep and co-workers reported the assembly of viral genomes from SBM data collected in several YNP sites (Inskeep et al. 2013a, b). Based on phylogenetic analysis of known viruses, 10 scaffolds from the archaeal-dominated samples were classified as “viral” although the similarity of the scaffolds to known viruses varied considerably. CRISPR regions including both spacer regions and direct repeats were predicted from these assemblies and near perfect alignments were found between CRISPR spacer regions and 8 of the 10 viral-like scaffolds (Inskeep et al. 2010, 2013a).
Novel positive-strand RNA viruses have been also discovered in Nymph Lake hot springs (NL) characterised by high temperature (>80 °C) and low pH < 4 (Bolduc et al. 2012). Three sites (NL10, NL17 and NL18) were selected as putative niches for archaeal RNA viruses based on a viral-fraction-enrichment approach followed by deep sequencing and two genomic fragments of putative archaeal RNA viruses were identified (Bolduc et al. 2012).
An attempt to link these RNA viral genomes to a specific host type was carried out through the analysis of the CRISPR direct repeat (DR) and spacer content present in cellular metagenomics data sets from the same sites (Bolduc et al. 2012). The majority of matching spacer sequences of the RNA metagenome was related to DRs Sulfolobus species (an organismcommonly found in NL10) suggesting that this crenarchaeon, might host not only DNA (Contursi et al. 2014b; Lipps 2006) but also RNA viruses. Intriguingly, the identification of these spacers might indicate that not only DNA viruses but also archaeal RNA viruses elicit CRISPR-mediated immunity.
The genetic diversity of these newly identified putative archaeal RNA viruses was investigated by searching for similarity throughout global metagenomics datasets (Wang et al. 2015a). The authors were able to obtain nine novel partial or nearly complete genomes of novel genogroups or genotypes of the putative RNA viruses previously identified by Bolduc et al. (2012).
Viral sequences were also retrieved from the metagenomic dataset obtained in a recent study on the NL10 (Menzel et al. 2015). Among the viral families, Lipothrixiviridae, is the most abundant and Rudiviridae and Ampullaviridae members were also identified together with viral sequences assigned to Pyrobaculum spherical virus (PSV) and Thermoproteus tenax virus 1 (TTV1) (Haring et al. 2004; Neumann et al. 1989). The high representativeness of archaeal viral families is in agreement with the predominance of archaeal species (58.1% of reads assigned to Archaea) in the NL10 site (Gudbergsdottir et al. 2016).
Despite the fact that the predominance of the same archaeal species concerns the closely located CH1102 site as well (Sulfur Spring, Temp: 79 °C, pH 1.8), a different viral scenario has been detected in this site. Indeed, the virus Sulfolobus Monocaudavirus (SMV1) is the most abundant in CH1102 constituting more than 80% of the identified viral reads (Uldahl et al. 2016). To link the viral genomes and their potential host in the samples, CRISPR loci were identified from the cellular part of the CH1102 metagenome where, interestingly, the number of spacers matching to the novel SMV genomes was generally very high.
By employing a network approach to a time series of viral metagenomics data collected from high temperature Nymph Lake acidic hot springs, Bolduc and co-worker demonstrated the proof-of-concept that the viral assemblage structure and its stability over a 5 year sampling period can be precisely defined (Bolduc et al. 2015). Furthermore, this analysis highlighted the high representativeness of completely novel archaeal viruses, thus demonstrating that the combination of metagenomics dataset with advanced bioinformatics tools is essential to expand our knowledge on the archaeal virosphere.
A metagenomic approach employed to obtain full genome sequences from a hot basic enrichment sample (85 °C and pH 6.0) collected from ObP (Garrett et al. 2010) lead to the identification of two novel genomes HAV1 (linear) and HAV2 (circular) neither of which showing any clear similarity to other known archaeal viruses (Garrett et al. 2010). Extensive genomic differences were detected in multiple variants of a virus HAV1, possibly resulting from CRISPR-Cas-directed interference by unidentified hosts.
4.2 Iceland
Given its location on a divergent tectonic plate boundary (the mid-Atlantic Ridge), Iceland is studded with active volcanic systems. Among these, metagenomics data are available for two distantly located (about 45 km) sites, i.e. Krísuvík (Is3-13) and Grensdalur (Is2-5S). Is3-13 (90 °C, pH 3.5–4.0), belonging to a geothermal complex including solfataras, fumaroles, mud pots and hot springs, had very limited access to organic materials. Instead, Is2-5S (85 °C and pH 5.0) is reached by the flow through streams from other hot springs and is located on a hill plenty of organic materials, such as moss and lichens (Menzel et al. 2015). Genomic DNA, extracted from both sediment and water samples, was sequenced using the Illumina Hiseq and analysed by MEGAN (Huson and Weber 2013). Mapped reads (several millions) were assigned to archaeal microorganisms for 19.7 and 33% in Is3-13 and Is2-5S, respectively. The analysis showed a predominance of Thermoproteales and Sulfolobales in Is3-13 and of Crenarchaeota in Is2-5S, mainly of the Pyrobaculum genus. On the other hand, bacteria were overrepresented by Proteobacteria (in Is3-13), including Gamma- and Beta-proteobacteria, and a large population of Aquificales (Is2-5S) mostly belonging to the species Thermocrinis albus and Sulfurihydrogen ibiumazorense. By comparing their data with those available for other geothermal locations worldwide, authors concluded that the community structure is strongly influenced by environmental parameters rather than geographic distance (Menzel et al. 2015).
The viral community composition and the relative abundance of viruses in IS2-5S and IS3-13 sites are quite different. In both cases, the representativeness of crenarchaeal viral sequences is high despite the predominance of bacterial species. This apparent discrepancy might be due to a compositional bias in the reference database, since most of the thermophilic viruses have been isolated from archaeal hosts (Gudbergsdottir et al. 2016).
The non-crenarchaeal viral order Caudovirales, composed of head to tail viruses infecting members of Bacteria and Euryarchaea is most abundant in Is2-5S (Krupovic et al. 2011). Conversely, the IS3-13 site is mostly populated by Ampullaviridae members and constitutes only a small percentage of all the viral sequences in the former sample. Furthermore, sequences referable to Bicaudoviridae one of the most widely represented crenarchaeal family in hot springs (Wang et al. 2015b), are absent in IS2-5S metagenome (Prangishvili 2013; Snyder et al. 2015). Interestingly, the longest contig in Is3-13 metagenome was assigned to a near complete Acidanius-bottle-shaped (ABV)-like genome (Haring et al. 2005).
Common to both the sites are contigs assigned to the Rudiviridae family (Prangishvili 2013; Snyder et al. 2015; Wang et al. 2015b) and viral sequences assigned to Pyrobaculum spherical virus (PSV) (Haring et al. 2004) and Thermoproteus tenax virus 1 (TTV1) (Neumann et al. 1989) and accordingly the metagenomes also contain sequences assigned to their archaeal Pyrobaculum and Thermoproteus tenax hosts (Menzel et al. 2015).
Unique to IS2-5S site is a 20 kb contig representing an novel incomplete viral genome that, as suggested by CRISPR spacer analysis, is likely to infect Hydrogenobaculum, an host for which no virus has been reported before (Gudbergsdottir et al. 2016; Romano et al. 2013). This is remarkable as a small but significant percentage of cellular reads in the Is2-5S metagenome were assigned to Hydrogenobaculum supporting the CRISPR analysis (Menzel et al. 2015).
4.3 Kamchatka Peninsula, Russia
In the Kamchatka peninsula, also known as the land of fire, an extended volcanic region of approximately 472,300 km2, three different areas, Uzon (81 °C, pH 7.2–7.4), Kam37 (85 °C, pH 5.5) and Mutnovsky (70 °C, pH 3.5–4.0), were surveyed to study the diversity of their microbial communities (Chernyh et al. 2015; Eme et al. 2013; Merkel et al. 2017; Wemheuer et al. 2013). This analysis, besides showing that uncultivated members of the Aquificales, Euryarchaeota, Crenarchaeota, and a Miscellaneous Crenarchaeotic Group were dominating in Kam37, led also to the discovery of two ancient (hyper)thermophilic archaeal lineages, namely Hot Thaumarchaeota-related Clade 1 and Hot Thaumarchaeota-related Clade 2. Thaumarcheota, along with Proteobacteria and Thermotogae, thrive in the Uzon and Mutnovsky sites as well. Interestingly enough, these results further confirm the previous assumption that comparable environmental conditions result in similar microbial communities as in the case of the ObP in YNP and the Uzon Caldera hot spring sharing geochemical features as well as microbial community structures (Meyer-Dombard et al. 2005; Simon et al. 2009). A recent microbial census by Merkel and co-workers of several hot springs with temperature >65 °C spread across Uzon and Mutnovsky (Sery: 80 °C, pH 6.1; Thermofilny: 67 °C, pH 6.1; Bourlyashchy 82 °C, pH 7.0; Izvilist: 77 °C pH 5.9; 3423: 72 °C pH 5.0; 3462: 72 °C, pH 5.1; 3460: 68 °C, pH 6.1; 3401: 90 °C pH 3.5; 3404: 70 °C pH 6.0) revealed that as observed in other thermal habitat (i.e. YNP), bacteria belonging to the genus Sulfurihydrogenibium are the most abuntant and widely distributed group of lithoautotrophic prokaryotes in these environments as also previously reported for Bourlyashchy hot springs, the hottest thermal pool of Uzon (Chernyh et al. 2015). These microorganisms represent the only dominating representatives of Aquificae in the springs analysed together with other lithoautotrophic bacteria such as Caldimicrobium and Thermocrinis. This indicates that reduced sulfur compounds such as dissolved hydrogen sulfide, are the primary energy source for lithoautotrophic carbon assimilation. Because of the simultaneous presence of both aerobic (i.e. Sulfurihydrogenibium) and anaerobic (i.e. Caldimicrobium) microorganisms in these hot springs, the authors suggest that the aerobic sulfur oxidation, anaerobic hydrogen oxidation, and the reduction of the sulfur compounds are the main energy-giving processes in these sites (Merkel et al. 2017).
4.4 Furnas Valley, Azores
The Furnas Valley (Island of São Miguel) is the main geothermal area of the Azores archipelago. Unlike YNP and Iceland, here the largest spring is alkaline and located on a height, whereas smaller ones are in the valley and are more acidic (Brock and Brock 1967). Recently, nine sites showing a wide range of physico-chemical characteristics (51–92 °C; pH 2.5–8.0) were explored, i.e. AI-AIV near Caldeira Do Esgucho, BI-BIII near Caldeirão and CI and CII near Caldeira Asmodeu (Sahm et al. 2013). In this study several approaches were used to assess the prokaryotic diversity, i.e. fluorescence in situ hybridization (FISH), analysis of 16S rRNA and denaturing gradient gel electrophoresis (DGGE). The AI site (51 °C, pH 3.0) was found to be populated by few euryarchaeota, mainly belonging to the genus Thermoplasma, whereas among bacteria dominated Proteobacteria (80%), especially genera known for their acidophilic (Acidicaldus) and chemolithoautotrophic (Acidithiobacillus) lifestyles. By contrast, in the AIV site (92 °C, pH 8.0) an even distribution of archaea (35%) and bacteria (40%) was detected. In particular, phyla Thermotogales (genus Fervidobacterium), Firmicutes (genus Caldicellulosiruptor) and Dictyoglomi (genus Dictyoglomus) were abundant among bacteria, whereas the archaeal population was almost exclusively composed by Crenarchaeota belonging to the Desulfurococcaceae and Thermoproteaceae families (Sahm et al. 2013). A general observations was that, once again, the pH was the predominant parameter, influencing microbial complexity in different areas surveyed, with the highest bacterial diversity detected at sites where temperatures and pHs ranged 55–85 °C and 7.0–8.0, respectively. Intriguingly, unlike other hot spring-environments where Aquificales are dominant, here heterotrophic bacteria prevail. To explain this, authors suggested that a 20-400-fold higher DOC in the Furnas spring could be a reason for the abundance of heterotrophic bacteria (Sahm et al. 2013).
4.5 Sungai Klah, Malaysia
The Sungai Klah (SK) hot spring in Malaysia is surrounded by a wooded area, which makes it continuously fed by plant-derived material that results in a higher degree of total organic carbon (TOC) if compared with the other 60 geothermal sites present in Malaysia. Moreover, three additional key factors were found to be characteristic of SK: (1) temperature exceeds 100 °C in many spring pools along the main stream, (2) it fluctuates between 50 and 110 °C throughout the stream and, (3) the pH is not uniform and spans from 7.0 to 9.0 along the stream. In general, SK is a shallow and fast-flowing stream with temperatures of 75–85 °C and pH 8.0. Samples retrieved from this area were studied through 16S rRNA gene profiling (Chan et al. 2015). This approach led to the identification of 83 phyla among which the predominant were Firmicutes, Proteobacteria, Chloroflexi, Bacteroidetes, Euryarchaeota and Crenarchaeota. Interestingly, by studying sequence affiliations the authors could highlight a relationship between the population diversity and the geochemical parameters within the hot spring. Moreover, it was shown that microbial communities were able to survive by exploiting different symbiotic strategies to prosper under multiple environmental stresses (Chan et al. 2015).
4.6 Tengchong, China
Located on the northeastern edge of Tibet–Yunnan geothermal zone between the Eurasian and Indian plates, Tengchong (China) is one of the most active geothermal areas in the world with Rehai (Hot Sea) and Ruidian geothermal fields characterized by the most intense geothermal activity. While mainly large pools with neutral pH (e.g. Gongxiaoshe and Jinze) are located in Ruidian (Wang et al. 2014), Rehai hosts several types of hot springs showing a wide range of physico-chemical conditions, such as temperatures from 58 to 97 °C and pH values between 1.8 and 9.3. Examples are: (1) small source, high discharge springs (Gumingquan and Jiemeiquan); (2) small, shallow acidic mud pools featured by a decreasing temperature gradient (Diretiyanqu); (3) shallow acidic pools like Zhenzhuquan; and (4) shallow springs with multiple geothermal sources such as Shuirebaozha (Hou et al. 2013). Besides few metagenomic studies mainly focused on Crenarchaeota (Song et al. 2010) or ammonia oxidizing archaea (Jiang et al. 2010), the study by Hou et al. (2013) is the first report on a wide-ranging survey of the microbial community in 16 different hot springs of Tengchong, aiming at shedding light on the relationship between the diversity of the thermophilic microbial communities and local geochemical conditions. In particular, predicted number of OTU as well as Shannon and equitability indexes based on 16S rRNA gene sequence data, were used to highlight correlations between microbial diversity and environmental geochemistry. Analysis of these indexes using Mantel test revealed higher microbial richness, equitability, and diversity in Ruidian than in Rehai. Authors concluded that this is mainly due to differences in the pH, temperatures and TOC between the two springs (Hou et al. 2013). The effect of seasonal changes on the microbial diversity in Tengchong hot springs, which are located in a subtropical area with heavy temporal monsoon rain fall, has been studied by Briggs et al. (2014) and Wang et al. (2014). Specifically, they compared the samples collected between June and August (rainy season) with those of Hou and colleagues sampled in January during the dry season. By doing so, they revealed that Ruidian sediments contained more diverse microbial lineages than Rehai sediments thanks to the neutral pH and moderate temperatures, and that neutral springs contained similar microbial lineages in January and June while in August a single dominant lineage of Thermus emerged. Once again, the pH turned out to be the primary factor influencing the microbial community, followed by temperature and DOC (Wang et al. 2014). Overall, both studies indicated that temperature, pH, and other geochemical conditions play a key role in shaping the microbial community structure in Tengchong hot springs over the seasons.
4.7 Taupo Volcanic Zone, New Zealand
In the central area of the New Zealand’s Northern Island, a group of high temperature geothermal systems take the name of the Taupo Volcanic Zone (TVZ). Within this area, the Waiotapu region is characterized by a large number of springs exhibiting elevated arsenic concentrations (Mountain et al. 2003). With its 65-meter diameter, the Champagne Pool (CP) represents the largest geothermal feature of Waiotapu with arsenic concentrations ranging from 2.9 to 4.2 mg/L (Hedenquist and Henley 1985). The inner rim of CP is characterized by subaqueous orange amorphous As-S precipitate responsible of the characteristic orange color (Jones et al. 2001). Whereas convection stabilizes the water temperature around 75 °C, the surrounding silica terrace (Artist’s Palette) shows lower temperatures (~45 °C). In order to understand the evolution of arsenic resistance in sulfidic geothermal systems, Hug and co-workers studied the microbial contributions to coupled arsenic and sulfur cycling at CP. To this aim, the authors sampled four different CP areas with distinctive physical and chemical features: (1) the inner pool (CPp), (2) the inner rim (CPr), (3) the outflow channel (CPc), and (4) the outer silica terrace (AP). The concentration of the total dissolved arsenic was measured and was found to be 3.0, 2.9, 3.6, and 4.2 mg/L at sites CPp, CPr, CPc and AP, respectively. Moreover, total dissolved sulfur concentrations were rather even in all the sites analysed, i.e. 91–105 mg/L (Hug et al. 2014). It was previously reported that the combination between dissolved toxic metal(loid)s and high temperature in hot springs represents a strong selective pressure on the inhabiting microbial communities (Hirner et al. 1998). Indeed, sulfide ions commonly used as electron donors/acceptors by microorganisms under geothermal conditions, are highly reactive with arsenic (Macur et al. 2013). Therefore, microbially-mediated sulfur cycling could exert an indirect, but profound, influence on arsenic speciation, by affecting the concentration of thioarsenate species (Stauder et al. 2005). To shed light on the potential microbial contributions to arsenic speciation in CP, and to characterize the microbial diversity, total genomic DNA was extracted from sediments and analyzed by deep sequencing (Hug et al. 2014). This analysis revealed that sequences assigned to the Archaea domain were mostly belonging to genera Sulfolobus, Thermofilum, Pyrobaculum, Desulfurococcus, Thermococcus, and Staphylothermus, and that the percentage of total Archaea was of 28, 21, 12 and 2% at CPp, CPc, CPr and AP, respectively. On the other hand, most abundant sequences belonging to Bacteria in all sites were closely related to the genus Sulfurihydrogenibium. According to these authors, the combination of sulfide dehydrogenase and sulfur oxygenase–reductase encoding genes detected as major sulfur oxidation genes at CPp, suggests a two-step sulfide oxidation process to sulfite and thiosulfate, also producing sulfide. Moreover, biogenic sulfide produced would then be available to transform arsenite to monothioarsenate. Interestingly enough, the whole metagenomic analysis allowed to unravel the impact on sulfur speciation by genes underpinning sulfur redox transformations, thus highlighting a microbial role in sulfur-dependent transformation of arsenite to thioarsenate (Hug et al. 2014).
4.8 Phlegraean Fields, Italy
An extended area to the west of Naples, South Italy, known as Phlegraean Fields, comprises 24 craters and volcanic features, mostly lying underwater. In the middle of this area is located the Solfatara volcano, which is one of the youngest volcanoes formed within this active volcanic field (Isaia et al. 2009; Orsi et al. 1996; Petrosino et al. 2012; Rosi and Santacroce 1984). The site with the most intense geothermal activity is the Pisciarelli area that, despite its limited extension (about 800 m2), is featured by over 20 physically and chemically different springs and mud holes. Besides sulfide, arsenic is one of the most prominent heavy metals detectable in this high-temperature environment (Huber et al. 2000), thus suggesting the presence of hyperthermophilic microorganisms able to use these compounds for their metabolism, similarly to what was shown by Hug et al. (2014) in the Champagne Pool (New Zealand). Recently, Solfatara volcano (It6) and Pisciarelli hot spring (It3) were analysed by Menzel and colleagues with the aim of defining the biodiversity, genome contents and inferred functions of bacterial and archaeal communities (Menzel et al. 2015). It6 sample (76 °C, pH 3.0, water/sediment) is subjected to IlluminaHiseq sequencing and whereas those from the It3 site (86 °C, pH 5.5, water/sediment) is sequenced using the Roche/454 Titanium (Menzel et al. 2015). This study showed that in It6 78.6% of the mapped reads were assigned to Bacteria (phyla Proteobacteria and Thermoprotei), while only 17.6% to Archaea (phyla Crenarchaeota and Euryarchaeota). Conversely, It3 was mainly populated by Archaea (96.6%), including species such as Acidianus, Sulfolobus and Pyrobaculum (Menzel et al. 2015). When these data were compared with those from previous studies (Inskeep et al. 2013a; Sahm et al. 2013; Urbieta et al. 2014; Wemheuer et al. 2013) the authors conclude that environmental chemico-physical parameters are the major determinants in shaping the structure and composition of the microbial community (Menzel et al. 2015).
It3 and It6 metagenomes were screened for viral sequences as well (Table 2). A common feature to these two sites is the predominance of the Lipothrixviridae (Prangishvili 2013; Snyder et al. 2015; Wang et al. 2015b) viral sequences, representing the 43.5 and 81.7%, respectively (Gudbergsdottir et al. 2016). Despite the high abundance of contigs assigned to this family, the absence of a complete lipothrixviral genome in the metagenomes could indicate the presence of multiple related but not identical genomes of similar abundance.
Acidianus two-tailed virus (ATV)-like sequences are also abundant in these two italian metagenomes in agreement with the fact that ATV, the single member of Bicaudaviridae, was originally isolated around 10 years ago from the It6 site (Prangishvili 2013; Snyder et al. 2015; Wang et al. 2015b). However, no full genome of an ATV-like virus was recovered (Gudbergsdottir et al. 2016). Conversely, a long Acidanius-bottle-shaped (ABV)-like contig was identified in the It6 metagenome indicating that this linear genome, belonging to a novel representative of the Ampullaviridae family, was complete as judged by the presence of inverted terminal repeats. In addition, another long ARV-like contig identified in the same metagenome (Gudbergsdottir et al. 2016) was assigned to a new member of the Rudiviridae family (Prangishvili 2013; Snyder et al. 2015; Wang et al. 2015b).
The It3 metagenome contains also a ≈20 kbp contig assigned to HAV2 (see above) (Garrett et al. 2010). However, since only one ORF showed similarity (38% a.a. identity) to an HAV2 gene the authors raised the possibility that this contig was part of a novel viral genome (Gudbergsdottir et al. 2016). The CRISPR analysis revealed that four spacers in the It3 metagenome matched to this novel genome originating from the hyperthermophilic crenarchaeon Pyrobaculum and indicating that the contig is an extracellular sequence of either viral or plasmid origin (Gudbergsdottir et al. 2016).
4.9 Lassen Volcanic National Park, USA
Viral metagenomic was carried out on samples from the Boiling Springs Lake (BSL), an acidic, high temperature lake (temperature ranging between 52 and 95 °C with a pH of approximately 2.5) located in Lassen Volcanic National Park, USA (Table 2; Diemer and Stedman 2012). The study revealed the presence of a unique circular, putatively single-stranded DNA virus, named RDHV (RNA-DNA hybrid virus; Diemer and Stedman 2012). Indeed, this viral genome harboured genes homologous to both ssRNA and ssDNA viruses with the ORFs arranged in an uncommon orientation. Intriguingly, the hybrid nature of this virus was explained with the occurrence of an interviral RNA-DNA recombination event in which a DNA circovirus-like progenitor acquired a capsid protein gene from a ssRNA virus via reverse transcription and recombination (Faurez et al. 2009). Mining environmental sequence databases for genetic similar configurations allowed the identification of three candidate BSL RDHV-like genomes, thus indicating that BSL RDHV is not endemic to Boiling Springs Lake. Such recombination events, although occur infrequently, might constitute one of the driving force for the evolution of novel viruses originating through genetic exchange between distinct virus lineages (Diemer and Stedman 2012).
4.10 Los Azufres National Park, Mexico
To date, the only microbial survey carried out in the Los Azufres National Park (Mexico) was reported by Brito and co-workers (Brito et al. 2014) analyzing five different samples among which AM1 (87 °C; pH 3.4) is the only one with temperature >65 °C. AM1, collected from the main geyser present in the “Los Azufres spa” showed high concentration of metals such as Zn and Mn and of heavy metals (Hg, Pb and Fe) up 1000 fold the EPA and WHO drinking water standards. The analysis by T-RFLP and 16S of the sites revealed an overall low bacterial diversity and that in particular AM1 was dominated exclusively by a microorganism related to Lysobacter spp. before identified in different extreme environments. This result suggests that the bacterial community of this site, if compared with the other in the area at lower temperature, was mainly influenced by the concentration of Zn, Mn and temperature (Brito et al. 2014).
A study performed by Servín-Garcidueñas et al. (2013a, b) identified the consensus sequence of a novel archaeal rudivirus (SMR1) as well as of a new member of the family Fuselloviridae (SMF1) by metagenomic reads assembly. Despite the large geographical distance from the locations of other sequenced rudiviruses and fuselloviruses, SMR1 and SMF1 retained a core set of conserved genes specific to Rudiviridae and Fuselloviridae, respectively. These genes were inferred to be important for the viral life cycle and their occurrence on the genomes of viruses geographically separated supported the hypothesis of exchange of genetic material over intercontinental distances (Servín-Garcidueñas et al. 2013a, b ).
5 Enzyme discovery
One of the driving interests toward metagenomic of geothermal environments is the discovery and exploitation of a rich pool of uncharacterised metabolic pathways as well as of novel thermostable enzymes (thermozymes) with biochemical characteristics evolved to accommodate the unique environments that the microbes reside in Bartolucci et al. (2013). Specifically, thermozymes exhibit an intrinsic stability to common protein chemical–physical denaturants and therefore are of great interest in biotechnological applications representing a valuable alternative to the available enzymes from mesophiles (Cobucci-Ponzano et al. 2015; Sharma et al. 2012).
5.1 Sequence-based function prediction
SBM enables the construction of data banks of all the genes present in a geothermal sample. In silico screening for sequence similarity or the presence of conserved motifs followed by amplified through PCR-approaches can allow the production of enzymes of potential applicative interest. Some frequently used databases for functional annotation are the SEED annotation system, the KEGG orthology (KO) database or the Pfam database (Finn et al. 2016; Kanehisa et al. 2014; Overbeek et al. 2014). Then, identified genes can be cloned and expressed in conventional hosts from native or inducible promoters and recombinant enzymes can be purified and characterized in detail. This approach increased enormously the number of genes to analyse if compared to genomic screening requiring the isolation of microbial strains, and it is especially convenient for extremophilic microorganisms whose isolation is particularly challenging. On the other hand, also this approach shows limitations. Firstly, the databases may be subjected to phylogenetic biases, as some communities are more accurately annotated than others; secondly, since prediction of genes function is based on sequence similarity to not many already characterized genes and pathways in the public databases, currently, about 50% of genes in genomes are defined as “hypothetical” or proteins of unknown function. Thirdly, the presence of a gene on a metagenome does not mean that it is expressed. To increase the probability of finding active functional genes involved in a substrate uptake and transformation, some studies use activity-based screenings on expression libraries (functional metagenomics) or a substrate-induced enrichment of the community before the mDNA extraction (see below).
5.2 Functional metagenomics
A powerful alternative or a complement, to SBM is functional metagenomics that relies on the construction of metagenomic libraries by cloning environmental DNA into expression vectors and propagating them in the appropriate hosts, followed by activity-based screening (Fig. 2). Depending on the size of the insert, functional metagenomics can be explored using vectors carrying short (<10 kb) or long size inserts (200 kb). Bigger inserts have the potential to carry entire gene-clusters as well as their own promoter sequences, allowing the expression of more enzymes. By using an appropriate screening method, genes expressing a particular enzymatic activity can be identified and their products characterized. Lambda phage-based expression vectors offer the possibility of screening for particular enzymatic activities directly on phage plaques. Indeed E. coli cells are lysed at the end of the infection cycle and the translated metagenomic proteins are released into the extracellular matrix. The main advantages of this approach are: (1) isolation of entire genes, (2) direct identification of enzymes fully active in recombinant form, and (3) identification of novel enzymatic activities, whose functions would not be predicted from the available sequence databases (López-López et al. 2014).
The description of the thermozymes isolated through functional metagenomics goes beyond the aims of this manuscript. One of the main limitations of this approach is the expression of heterologous genes in E. coli (Gabor et al. 2004). Although the commonly used E.coli strains have relaxed requirements for promoter recognition and translation initiation, in this host proteins may not fold correctly and many genes from extremophilic environmental samples are not translated efficiently, especially those belonging to archaea, which might show a codon usage bias (Prato et al. 2008). Alternative hosts and broad-host-range vectors may be required to overcome these limitations (Angelov et al. 2009; Cheng et al. 2014). Recently, Leis and co-workers identified novel esterases from hot springs in the Azores based by complementing the growth of a custom, esterase-deficient strain of Thermus thermophilus. By using this method, they uncover several enzymes from underrepresented species of archaeal origin leading to the identification of new biocatalysts that do not share any known sequence signatures at all (Leis et al. 2015).
5.3 Extremophilic microbiome enrichments
Direct selection of the enzymes by functional metagenomics is not always the best approach and adaptation of entire extremophilic microbiomes on specific substrates is an interesting alternative choice. Often, complex substrates, recalcitrant to enzymatic conversion, require the combined action of many catalytic activities and accessory proteins that, in nature, are provided by a complex microbiome. This is particularly true for the conversion of plant lignocellulosic material including cellulose and hemicelluloses (xylans, xyloglucans, pectins, etc.) that are the two most abundant polymers on Earth (global cellulose production estimates range between 9 × 1012 and 1.5 × 1012 tons/year (Ha et al. 2011; Pinkert et al. 2009) and are remarkably stable to spontaneous hydrolysis (half-life of the glycosidic bond is 4.7 × 106 years, Wolfenden et al. 1998). Thus, carbohydrate active enzymes (cazymes) from (hyper)thermophiles have interesting biotechnological potential (Cobucci-Ponzano et al. 2006, 2010a, b). In fact, their impressive stability (Ausili et al. 2004) at the conditions at which plant lignocellulose is pretreated in second generation biorefineries (steam-explosion at extremes of temperatures and pHs), make them the ideal catalysts for the hydrolysis of (hemi)cellulose into fermentable sugars for the production of bioethanol and plastic precursors (Castiglia et al. 2016; Cobucci-Ponzano et al. 2013, 2015; Iacono et al. 2016; Aulitto et al. 2017).
Functional metagenomics through selections at high temperatures (60–75 °C) allowed the identification of interesting thermophilic cazymes, including β-glucosidase, cellulase, β-xylosidases/α-arabinofuranosidase, endoxylanase, α-fucosidase, and acetyl xylan esterase from samples collected in environmental niches, like guts of animals and insects, and compost, swine waste, and thermophilic methanogenic digesters (Allgaier et al. 2010; Dougherty et al. 2012; Liang et al. 2010; Wang 2009; Wang et al. 2015c). However, direct selection of microbiomes from extreme environments on plant biomass is a useful alternative. Gladden and collaborators adapted samples from composts to grow on switchgrass by multiple passages at 60 °C. The enrichments produced a reduction in microbiome diversity showing by 16S rRNA the presence of thermophilic Gram-positive bacteria Firmicutes, Bacteriodetes, Chloroflexi, and, remarkably, an uncultivated lineage related to the Gemmatimonadetes phylum (Gladden et al. 2011). In addition, supernatants of the enriched cultures showed endoglucanase and xylanase activities more stable than those commercially available and exploited in biorefineries. With a similar approach, enrichments of a sample from a 94 °C geothermal pool in Nevada (USA) on Miscanthus and cellulose Avicel in strictly anaerobic conditions at 90 °C were performed (Graham et al. 2011). Three 16S rRNA genes were identified corresponding to the archaea Ignisphaera aggregans, Pyrobaculum islandicum, and Thermophilum pendens, with the Ignisphaera-like strain dominating the microbiome. In addition, CMC active enzymes were detected in the enriched cultures and the annotation analysis of the metagenomic data bank allowed to identify and characterize a novel GH5 endoglucanase. This study showed the first microbiome of archaea able to deconstruct lignocellulose at 90 °C and a novel uncommon cellulase showing promising application in biorefineries. Finally, a metagenomic analysis of an environmental sample dominated by Firmicutes and collected in hot springs (50-70 °C) in Xiamen-China has been reported very recently. Upon enrichment on sugarcane bagasse, a novel cellulase, XM70-Cel9, was identified and biochemically characterized displaying relevant biotechnological properties i.e. optimal temperature of 70 °C and good pH tolerance (Zhao et al. 2017).
6 Conclusions
The interest in the diversity, ecology, physiology and biochemistry of extreme- and hyper-thermophilic microorganisms has increased enormously during the past few decades. An unanticipated phylogenetic and physiological diversity of thermophiles has been revealed through metagenomic studies conducted in different thermophilic biotopes. Figure 5 shows the principal component analysis of the geothermal sites herein described taking into account pH, temperature and dominant phyla. The analysis clearly indicates a direct correlation between the predominance of archaeal phyla (i.e. Crenarchaeota) in higher temperature (~75–90 °C) and low pH (2.6–5.5) sites and of bacterial phyla (i.e. Aquificae) under lower temperature (~65 to 80 °C) and high pH (5.5–9.4) conditions. This observation is in perfect agreement with the studies reported by Inskeep et al. (2013b) in the YNP metagenomic project.

Principal component representation of the geothermal sites described in this review (see Table 1) considering temperature, pH and dominant phyla as clustering parameters. The two main clusters of geothermal sites, A and B, reflect the correlation between the dominant phyla and the environmental parameters (pH and T) confirming the presence of dominant archaeal and bacterial phyla in high temperature/low pH and low temperature/high pH geothermal environments, respectively. 1–20 Yellowstone National Park (red), 21–22 Iceland (light blue), 23–33 Kamchatka (orange), 34–35 Furnas Valley (purple), 36 Malasya (grey), 37–50 Rehai and Ruidia (pink); 51–54 Taupo (green) and 55–56 Phlegraean fields (green); 57 Los Azufres (black). (Color figure online)
It is expected that the combination of DNA metagenomic studies together with metatranscriptomic and metaproteomic approaches will allow us to understand the functional dynamics of microbial communities as well as to advance the prediction of the in situ microbial activities and productivity of microbial consortia.
Besides the bacterial and archaeal communities, viral metagenomic studies have provided information concerning viral biogeography, diversity and community structure of hot environments, and new viral types contributing to the discovery of potential archaeal RNA viruses. Viral metagenomics analysis has also revealed the presence of high percentage of unknown sequences demonstrating the vast novelty of genetic information to be still obtained from viruses.
Microbial and viral metagenomics open up the roads to analyze and screen the genetically and metabolically rich microbial thermophilic communities in their entirety. By further developing high-throughput screening methods and chromogenic substrates-based tests for the detection of thermozymes it is possible to foresee a quantum leap in the discovery of novel biocatalysts to be successfully exploited in sustainable processes for industrial applications.
References
Adey A, Morrison HG, Asan Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, Shendure J (2010) Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. doi:10.1186/gb-2010-11-12-r119
Allgaier M, Reddy A, Park JI, Ivanova N, Dhaeseleer P, Lowry S, Sapra R, Hazen TC, Simmons BA, VanderGheynst JS, Hugenholtz P (2010) Targeted discovery of glycoside hydrolases from a switchgrass-adapted compost community. PLoS ONE 5:9
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Angelov A, Mientus M, Liebl S, Liebl W (2009) A two-host fosmid system for functional screening of (meta)genomic libraries from extreme thermophiles. Syst Appl Microbiol 32:177–185. doi:10.1016/j.syapm.2008.01.003
Aulitto M, Fusco S, Fiorentino G, Limauro D, Pedone E, Bartolucci S, Contursi P (2017) Thermus thermophilus as source of thermozymes for biotechnological applications: homologous expression and biochemical characterization of an α-galactosidase. Microb Cell Fact 16(1):28. doi:10.1186/s12934-017-0638-4
Ausili A, Di Lauro B, Cobucci-Ponzano B, Bertoli E, Scire A, Rossi M, Tanfani F, Moracci M (2004) Two-dimensional IR correlation spectroscopy of mutants of the beta-glycosidase from the hyperthermophilic archaeon Sultolobus soltataricus identifies the mechanism of quaternary structure stabilization and unravels the sequence of thermal unfolding events. Biochem J 384:69–78. doi:10.1042/Bj20040646
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi:10.1126/science.1138140
Bartolucci S, Contursi P, Fiorentino G, Limauro D, Pedone E (2013) Responding to toxic compounds: a genomic and functional overview of Archaea. Front Biosci Landmark 18:165–189. doi:10.2741/4094
Beam JP, Jay ZJ, Ma Kozubal, Inskeep WP (2014) Niche specialization of novel thaumarchaeota to oxic and hypoxic acidic geothermal springs of Yellowstone National Park. ISME J 8:938–951. doi:10.1038/ismej.2013.193
Bhaya D, Grossman AR, Steunou AS, Khuri N, Cohan FM, Hamamura N, Melendrez MC, Bateson MM, Ward DM, Heidelberg JF (2007) Population level functional diversity in a microbial community revealed by comparative genomic and metagenomic analyses. ISME J 1:703–713. doi:10.1038/ismej.2007.46
Biswas A, Gagnon JN, Brouns SJJ, Fineran PC, Brown CM (2013) CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol 10:817–827. doi:10.4161/rna.24046
Bize A, Peng X, Prokofeva M, MacLellan K, Lucas S, Forterre P, Garrett RA, Bonch-Osmolovskaya EA, Prangishvili D (2008) Viruses in acidic geothermal environments of the Kamchatka Peninsula. Res Microbiol 159:358–366. doi:10.1016/j.resmic.2008.04.009
Blainey PC, Quake SR (2014) Dissecting genomic diversity, one cell at a time. Nat Methods 11:19–21
Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P (2007) CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. doi:10.1186/1471-2105-8-209
Bolduc B, Shaughnessy DP, Wolf YI, Koonin EV, Roberto FF, Young M (2012) Identification of novel positive-strand RNA viruses by metagenomic analysis of archaea-dominated Yellowstone hot springs. J Virol 86:5562–5573. doi:10.1128/JVI.07196-11
Bolduc B, Wirth JF, Mazurie A, Young MJ (2015) Viral assemblage composition in Yellowstone acidic hot springs assessed by network analysis. ISME J 9:2162–2177. doi:10.1038/ismej.2015.28
Briggs BR, Brodie EL, Tom LM, Dong H, Jiang H, Huang Q, Wang S, Hou W, Wu G, Huang L, Hedlund BP, Zhang C, Dijkstra P, Hungate BA (2014) Seasonal patterns in microbial communities inhabiting the hot springs of Tengchong, Yunnan Province, China. Environ Microbiol 16:1579–1591. doi:10.1111/1462-2920.12311
Brito EM, Villegas-Negrete N, Sotelo-Gonzalez IA, Caretta CA, Goni-Urriza M, Gassie C, Hakil F, Colin Y, Duran R, Gutierrez-Corona F, Pinon-Castillo HA, Cuevas-Rodriguez G, Malm O, Torres JP, Fahy A, Reyna-Lopez GE, Guyoneaud R (2014) Microbial diversity in Los Azufres geothermal field (Michoacan, Mexico) and isolation of representative sulfate and sulfur reducers. Extremophiles 18:385–398. doi:10.1007/s00792-013-0624-7
Brock T (2001) The origins of research on thermophiles. In: Reysenbach A-L, Voytek M, Mancinelli R (eds) Thermophiles biodiversity, ecology and evolution. Springer, Boston, pp 1–9
Brock TD, Brock ML (1967) The hot springs of the Furnas Valley, Azores. Internationale Revue der gesamten Hydrobiologie und Hydrographie 52:545–558. doi:10.1002/iroh.19670520405
Brock TD, Brock ML, Bott TL, Edwards MR (1971) Microbial life at 90 °C: the sulfur bacteria of Boulder Spring. J Bacteriol 107:303–314
Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, van der Oost J (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. doi:10.1126/science.1159689
Castiglia D, Sannino L, Marcolongo L, Ionata E, Tamburino R, De Stradis A, Cobucci-Ponzano B, Moracci M, La Cara F, Scotti N (2016) High-level expression of thermostable cellulolytic enzymes in tobacco transplastomic plants and their use in hydrolysis of an industrially pretreated Arundo donax L. biomass. Biotechnol Biofuels. doi:10.1186/S13068-016-0569-Z
Chakravorty S, Helb D, Burday M, Connell N, Alland D (2007) A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods 69:330–339. doi:10.1016/j.mimet.2007.02.005
Chan CS, Chan K-G, Tay Y-L, Chua Y-H, Goh KM (2015) Diversity of thermophiles in a Malaysian hot spring determined using 16S rRNA and shotgun metagenome sequencing. Front Microbiol 6:177. doi:10.3389/fmicb.2015.00177
Cheng J, Pinnell L, Engel K, Neufeld JD, Charles TC (2014) Versatile broad-host-range cosmids for construction of high quality metagenomic libraries. J Microbiol Methods 99:27–34. doi:10.1016/j.mimet.2014.01.015
Chernyh NA, Mardanov AV, Gumerov VM, Miroshnichenko ML, Lebedinsky AV, Merkel AY, Crowe D, Pimenov NV, Rusanov II, Ravin NV, Moran MA, Bonch-Osmolovskaya EA (2015) Microbial life in Bourlyashchy, the hottest thermal pool of Uzon Caldera, Kamchatka. Extremophiles 19:1157–1171. doi:10.1007/s00792-015-0787-5
Clingenpeel S, Kan J, Macur RE, Woyke T, Lovalvo D, Varley J, Inskeep WP, Nealson K, McDermott TR (2013) Yellowstone lake nanoarchaeota. Front Microbiol 4:274. doi:10.3389/fmicb.2013.00274
Clooney AG, Fouhy F, Sleator RD, O’Driscoll A, Stanton C, Cotter PD, Claesson MJ (2016) Comparing apples and oranges?: next generation sequencing and its impact on microbiome analysis. PLoS ONE 11:1–16. doi:10.1371/journal.pone.0148028
Cobucci-Ponzano B, Conte F, Benelli D, Londei P, Flagiello A, Monti M, Pucci P, Rossi M, Moracci M (2006) The gene of an archaeal alpha-l-fucosidase is expressed by translational frameshifting. Nucl Acids Res 34:4258–4268. doi:10.1093/Nar/Gkl574
Cobucci-Ponzano B, Aurilia V, Riccio G, Henrissat B, Coutinho PM, Strazzulli A, Padula A, Corsaro MM, Pieretti G, Pocsfalvi G, Fiume I, Cannio R, Rossi M, Moracci M (2010a) A New Archaeal beta-Glycosidase from Sulfolobus solfataricus seeding a novel retaining beta-glycan-specific glycoside hydrolase family along with the human non-lysosomal glucosylceramidase gba2. J Biol Chem 285:20691–20703. doi:10.1074/jbc.M109.086470
Cobucci-Ponzano B, Conte F, Strazzulli A, Capasso C, Fiume I, Pocsfalvi G, Rossi M, Moracci M (2010b) The molecular characterization of a novel GH38 alpha-mannosidase from the crenarchaeon Sulfolobus solfataricus revealed its ability in de-mannosylating glycoproteins. Biochimie 92:1895–1907. doi:10.1016/j.biochi.2010.07.016
Cobucci-Ponzano B, Ionata E, La Cara F, Morana A, Ferrara MC, Maurelli L, Strazzulli A, Giglio R, Moracci M (2013) Extremophilic (hemi)cellulolytic microorganisms and enzymes. In: Faraco V (ed) Lignocellulose conversion enzymatic and microbial tools for bioethanol production. Springer, Berlin, pp 111–130. doi:10.1007/978-3-642-37861-4
Cobucci-Ponzano B, Strazzulli A, Iacono R, Masturzo G, Giglio R, Rossi M, Moracci M (2015) Novel thermophilic hemicellulases for the conversion of lignocellulose for second generation biorefineries. Enzyme Microb Technol. doi:10.1016/j.enzmictec.2015.06.014
Contursi P, Cannio R, She QX (2010) Transcription termination in the plasmid/virus hybrid pSSVx from Sulfolobus islandicus. Extremophiles 14:453–463. doi:10.1007/s00792-010-0325-4
Contursi P, Farina B, Pirone L, Fusco S, Russo L, Bartolucci S, Fattorusso R, Pedone E (2014a) Structural and functional studies of Stf76 from the Sulfolobus islandicus plasmid-virus pSSVx: a novel peculiar member of the winged helix-turn-helix transcription factor family. Nucleic Acids Res 42:5993–6011. doi:10.1093/nar/gku215
Contursi P, Fusco S, Cannio R, She QX (2014b) Molecular biology of fuselloviruses and their satellites. Extremophiles 18:473–489. doi:10.1007/s00792-014-0634-0
Cowan D, Ramond J-B, Makhalanyane T, De Maayer P (2015) Metagenomics of extreme environments. Curr Opin Microbiol 25:97–102. doi:10.1016/j.mib.2015.05.005
DeCastro ME, Rodriguez-Belmonte E, Gonzalez-Siso MI (2016) Metagenomics of thermophiles with a focus on discovery of novel thermozymes. Front Microbiol 7:1521. doi:10.3389/fmicb.2016.01521
Dellas N, Lawrence MC, Young JM (2013) A survey of protein structures from archaeal viruses. Life. doi:10.3390/life3010118
Dellas N, Snyder JC, Bolduc B, Young MJ (2014) Archaeal viruses: diversity, replication, and structure. Ann Rev Virol 1:399. doi:10.1146/annurev-virology-031413-085357
Delmont TO, Robe P, Clark I, Simonet P, Vogel TM (2011) Metagenomic comparison of direct and indirect soil DNA extraction approaches. J Microbiol Methods 86:397–400. doi:10.1016/j.mimet.2011.06.013
Diemer GS, Stedman KM (2012) A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol Direct. doi:10.1186/1745-6150-7-13
Dougherty MJ, D’haeseleer P, Hazen TC, Simmons BA, Adams PD, Hadi MZ (2012) Glycoside hydrolases from a targeted compost metagenome, activity-screening and functional characterization. BMC Biotechnol. doi:10.1186/1472-6750-12-38
Edgar RC, Myers EW (2005) PILER: identification and classification of genomic repeats. Bioinformatics 21:I152–I158. doi:10.1093/bioinformatics/bti1003
Edwards RA Rohwer F (2005) Opinion: Viral metagenomics. Nat Rev Microbiol 3(6):504–510
Eloe-Fadrosh EA, Paez-Espino D, Jarett J, Dunfield PF, Hedlund BP, Dekas AE, Grasby SE, Brady AL, Dong H, Briggs BR, Li WJ, Goudeau D, Malmstrom R, Pati A, Pett-Ridge J, Rubin EM, Woyke T, Kyrpides NC, Ivanova NN (2016) Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs. Nat Commun 7:10476. doi:10.1038/ncomms10476
Eme L, Reigstad LJ, Spang A, Lanzen A, Weinmaier T, Rattei T, Schleper C, Brochier-Armanet C (2013) Metagenomics of Kamchatkan hot spring filaments reveal two new major (hyper)thermophilic lineages related to Thaumarchaeota. Res Microbiol 164:425–438. doi:10.1016/j.resmic.2013.02.006
Faurez F, Dory D, Grasland B, Jestin A (2009) Replication of porcine circoviruses. Virol J. doi:10.1186/1743-422x-6-60
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. doi:10.1093/nar/gkv1344
Fusco S, Aulitto M, Bartolucci S, Contursi P (2015a) A standardized protocol for the UV induction of Sulfolobus spindle-shaped virus 1. Extremophiles 19:539–546. doi:10.1007/s00792-014-0717-y
Fusco S, Liguori R, Limauro D, Bartolucci S, She QX, Contursi P (2015b) Transcriptome analysis of Sulfolobus solfataricus infected with two related fuselloviruses reveals novel insights into the regulation of CRISPR-Cas system. Biochimie 118:322–332. doi:10.1016/j.biochi.2015.04.006
Gabor EM, Alkema WB, Janssen DB (2004) Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ Microbiol 6:879–886. doi:10.1111/j.1462-2920.2004.00640.x
Garrett RA, Prangishvili D, Sa Shah, Reuter M, Stetter KO, Peng X (2010) Metagenomic analyses of novel viruses and plasmids from a cultured environmental sample of hyperthermophilic neutrophiles. Environ Microbiol 12:2918–2930. doi:10.1111/j.1462-2920.2010.02266.x
Gladden JM, Allgaier M, Miller CS, Hazen TC, Vandergheynst JS, Hugenholtz P, Ba Simmons, Singer SW (2011) Glycoside hydrolase activities of thermophilic bacterial consortia adapted to switchgrass. Appl Environ Microbiol 77:5804–5812. doi:10.1128/AEM.00032-11
Gomez-Alvarez V, Teal TK, Schmidt TM (2009) Systematic artifacts in metagenomes from complex microbial communities. ISME J 3:1314–1317. doi:10.1038/ismej.2009.72
Graham JE, Clark ME, Nadler DC, Huffer S, Chokhawala HA, Rowland SE, Blanch HW, Clark DS, Robb FT (2011) Identification and characterization of a multidomain hyperthermophilic cellulase from an archaeal enrichment. Nat Commun 2:375. doi:10.1038/ncomms1373
Grissa I, Vergnaud G, Pourcel C (2007) The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. doi:10.1186/1471-2105-8-172
Gudbergsdottir SR, Menzel P, Krogh A, Young M, Peng X (2016) Novel viral genomes identified from six metagenomes reveal wide distribution of archaeal viruses and high viral diversity in terrestrial hot springs. Environ Microbiol 18:863–874. doi:10.1111/1462-2920.13079
Ha SH, Mai NL, An G, Koo Y-M (2011) Microwave-assisted pretreatment of cellulose in ionic liquid for accelerated enzymatic hydrolysis. Biores Technol 102:1214–1219. doi:10.1016/j.biortech.2010.07.108
Haring M, Peng X, Brugger K, Rachel R, Stetter KO, Garrett RA, Prangishvili D (2004) Morphology and genome organization of the virus PSV of the hyperthermophilic archaeal genera Pyrobaculum and Thermoproteus: a novel virus family, the Globuloviridae. Virology 323:233–242. doi:10.1016/j.virol.2004.03.002
Haring M, Rachel R, Peng X, Garrett RA, Prangishvili D (2005) Viral diversity in hot springs of Pozzuoli, Italy, and characterization of a unique archaeal virus, acidianus bottle-shaped virus, from a new family, the Ampullaviridae. J Virol 79:9904–9911. doi:10.1128/JVI.79.15.9904-9911.2005
Hedenquist JW, Henley RW (1985) Hydrothermal eruptions in the Waiotapu geothermal system, New Zealand; their origin, associated breccias, and relation to precious metal mineralization. Econ Geol 80:1640–1668. doi:10.2113/gsecongeo.80.6.1640
Hirner AV, Feldmann J, Krupp E, Grumping R, Goguel R, Cullen WR (1998) Metal(loid)organic compounds in geothermal gases and waters. Org Geochem 29:1765–1778. doi:10.1016/S0146-6380(98)00153-3
Hou W, Wang S, Dong H, Jiang H, Briggs BR, Peacock JP, Huang Q, Huang L, Wu G, Zhi X, Li W, Ja Dodsworth, Hedlund BP, Zhang C, Hartnett HE, Dijkstra P, Ba Hungate (2013) A comprehensive census of microbial diversity in hot springs of Tengchong, Yunnan Province China using 16S rRNA gene pyrosequencing. PLoS ONE. doi:10.1371/journal.pone.0053350
Huber R, Huber H, Stetter KO (2000) Towards the ecology of hyperthermophiles: biotopes, new isolation strategies and novel metabolic properties. FEMS Microbiol Rev 24:615–623
Hug K, Wa Maher, Stott MB, Krikowa F, Foster S, Moreau JW (2014) Microbial contributions to coupled arsenic and sulfur cycling in the acid-sulfide hot spring Champagne Pool, New Zealand. Front Microbiol 5:1–14. doi:10.3389/fmicb.2014.00569
Huse SM, Welch DM, Morrison HG, Sogin ML (2010) Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12:1889–1898. doi:10.1111/j.1462-2920.2010.02193.x
Huson DH, Weber N (2013) Microbial community analysis using MEGAN. Methods Enzymol 531:465–485. doi:10.1016/B978-0-12-407863-5.00021-6
Iacono R, Cobucci-Ponzano B, Strazzulli A, Giglio R, Maurelli L, Moracci M (2016) (Hyper)thermophilic biocatalysts for second generation biorefineries. Chem Today 34:34–37
Inskeep WP, Rusch DB, Jay ZJ, Herrgard MJ, Ma Kozubal, Richardson TH, Macur RE, Hamamura N, Jennings RDM, Fouke BW, Reysenbach AL, Roberto F, Young M, Schwartz A, Boyd ES, Badger JH, Mathur EJ, Ortmann AC, Bateson M, Geesey G, Frazier M (2010) Metagenomes from high-temperature chemotrophic systems reveal geochemical controls on microbial community structure and function. PLoS ONE. doi:10.1371/journal.pone.0009773
Inskeep WP, Jay ZJ, Herrgard MJ, Kozubal MA, Rusch DB, Tringe SG, Macur RE, Jennings RD, Boyd ES, Spear JR, Roberto FF (2013a) Phylogenetic and functional analysis of metagenome sequence from high-temperature archaeal habitats demonstrate linkages between metabolic potential and geochemistry. Front Microbiol. doi:10.3389/Fmicb.2013.00095
Inskeep WP, Jay ZJ, Tringe SG, Herrgard MJ, Rusch DB, Co YMPS (2013b) The YNP metagenome project: environmental parameters responsible for microbial distribution in the Yellowstone geothermal ecosystem. Front Microbiol. doi:10.3389/Fmicb.2013.00067
Isaia R, Marianelli P, Sbrana A (2009) Caldera unrest prior to intense volcanism in Campi Flegrei (Italy) at 4.0 ka B.P.: implications for caldera dynamics and future eruptive scenarios. Geophys Res Lett 36:L21303. doi:10.1029/2009GL040513
Jiang X, Takacs-Vesbach CD (2017) Microbial community analysis of pH 4 thermal springs in Yellowstone National Park. Extremophiles 21:135–152. doi:10.1007/s00792-016-0889-8
Jiang H, Huang Q, Dong H, Wang P, Wang F, Li W, Zhang C (2010) RNA-based investigation of ammonia-oxidizing archaea in hot springs of Yunnan Province, China. Appl Environ Microbiol 76:4538–4541. doi:10.1128/AEM.00143-10
John SG, Mendez CB, Deng L, Poulos B, Kauffman AK, Kern S, Brum J, Polz MF, Boyle EA, Sullivan MB (2011) A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ Microbiol Rep 3:195–202. doi:10.1111/j.1758-2229.2010.00208.x
Jones B, Renaut R, Rosen M (2001) Biogenicity of gold- and silver-bearing siliceous sinters forming in hot (75 °C) anaerobic spring-waters of Champagne Pool, Waiotapu, North Island, New Zealand. J Geol Soc Lond 158:895–911. doi:10.1144/0016-764900-131
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42:D199–D205. doi:10.1093/nar/gkt1076
Katayama H, Shimasaki A, Ohgaki S (2002) Development of a virus concentration method and its application to detection of enterovirus and norwalk virus from coastal seawater. Appl Environ Microbiol 68:1033–1039
Kim Y-M, Nowack S, Olsen MT, Becraft ED, Wood JM, Thiel V, Klapper I, Kühl M, Fredrickson JK, Da Bryant, Ward DM, Metz TO (2015) Diel metabolomics analysis of a hot spring chlorophototrophic microbial mat leads to new hypotheses of community member metabolisms. Front Microbiol. doi:10.3389/fmicb.2015.00209
Kozubal MA, Romine M, Jennings Rd, Jay ZJ, Tringe SG, Rusch DB, Beam JP, McCue LA, Inskeep WP (2013) Geoarchaeota: a new candidate phylum in the Archaea from high-temperature acidic iron mats in Yellowstone National Park. ISME J 7:622–634. doi:10.1038/ismej.2012.132
Krupovic M, Prangishvili D, Hendrix RW, Bamford DH (2011) Genomics of bacterial and archaeal viruses: dynamics within the prokaryotic virosphere. Microbiol Mol Biol Rev 75:610. doi:10.1128/MMBR.00011-11
Lakay FM, Botha A, Prior BA (2007) Comparative analysis of environmental DNA extraction and purification methods from different humic acid-rich soils. J Appl Microbiol 102:265–273. doi:10.1111/j.1365-2672.2006.03052.x
Lasken RS (2012) Genomic sequencing of uncultured microorganisms from single cells. Nat Rev Microbiol 10:631–640. doi:10.1038/nrmicro2857
Leis B, Angelov A, Mientus M, Li HJ, Pham VTT, Lauinger B, Bongen P, Pietruszka J, Goncalves LG, Santos H, Liebl W (2015) Identification of novel esterase-active enzymes from hot environments by use of the host bacterium Thermus thermophilus. Front Microbiol. doi:10.3389/Frricb.2015.00275
Liang Y, Yesuf J, Feng Z (2010) Toward plant cell wall degradation under thermophilic condition: a unique microbial community developed originally from swine waste. Appl Biochem Biotechnol 161:147–156. doi:10.1007/s12010-009-8780-z
Lipps G (2006) Plasmids and viruses of the thermoacidophilic crenarchaeote Sulfolobus. Extremophiles 10:17–28. doi:10.1007/s00792-005-0492-x
López-López O, Cerdán EM, González-Siso IM (2013) Hot spring metagenomics. Life. doi:10.3390/life3020308
López-López O, Cerdán ME, González Siso MI (2014) New extremophilic lipases and esterases from metagenomics. Curr Protein Pept Sci 15:445–455
Lorenz P, Liebeton K, Niehaus F, Eck J (2002) Screening for novel enzymes for biocatalytic processes: accessing the metagenome as a resource of novel functional sequence space. Curr Opin Biotechnol 13:572–577
Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT (2012) Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 7:e30087. doi:10.1371/journal.pone.0030087
Macur RE, Jay ZJ, Taylor WP, Kozubal MA, Kocar BD, Inskeep WP (2013) Microbial community structure and sulfur biogeochemistry in mildly-acidic sulfidic geothermal springs in Yellowstone National Park. Geobiology 11:86–99. doi:10.1111/gbi.12015
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen ZT, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu PG, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. doi:10.1038/nature03959
Marraffini LA, Sontheimer EJ (2010) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11:181–190. doi:10.1038/nrg2749
Martinez-Garcia M, Santos F, Moreno-Paz M, Parro V, Anton J (2014) Unveiling viral-host interactions within the ‘microbial dark matter’. Nat Commun. doi:10.1038/Ncomms5542
Menzel P, Gudbergsdóttir SR, Rike AG, Lin L, Zhang Q, Contursi P, Moracci M, Kristjansson JK, Bolduc B, Gavrilov S, Ravin N, Mardanov A, Bonch-Osmolovskaya E, Young M, Krogh A, Peng X (2015) Comparative metagenomics of eight geographically remote terrestrial hot springs. Microb Ecol. doi:10.1007/s00248-015-0576-9
Merkel AY, Pimenov NV, Rusanov II, Slobodkin AI, Slobodkina GB, Tarnovetckii IY, Frolov EN, Dubin AV, Perevalova AA, Bonch-Osmolovskaya EA (2017) Microbial diversity and autotrophic activity in Kamchatka hot springs. Extremophiles 21:307–317. doi:10.1007/s00792-016-0903-1
Meyer-Dombard D, Shock E, Amend J (2005) Archaeal and bacterial communities in geochemically diverse hot springs of Yellowstone National Park, USA. Geobiology 3:211–227. doi:10.1111/j.1472-4669.2005.00052.x
Mountain B, Benning L, Boerema J (2003) Experimental studies on New Zealand hot spring sinters: rates of growth and textural development. Can J Earth Sci 40:1643–1667. doi:10.1139/E03-068
Munson-McGee JH, Field EK, Bateson M, Rooney C, Stepanauskas R, Young MJ (2015) Nanoarchaeota, their sulfolobales host, and Nanoarchaeota virus distribution across Yellowstone National Park Hot Springs. Appl Environ Microbiol 81:7860–7868. doi:10.1128/AEM.01539-15
Neumann H, Schwass V, Eckerskorn C, Zillig W (1989) Identification and characterization of the genes encoding 3 structural proteins of the thermoproteus-tenax virus Ttv1. Mol Gen Genet 217:105–110. doi:10.1007/Bf00330948
Orsi G, De Vita S, di Vito M (1996) The restless, resurgent Campi Flegrei nested caldera (Italy): constraints on its evolution and configuration. J Volcanol Geotherm Res 74:179–214. doi:10.1016/S0377-0273(96)00063-7
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia FF, Stevens R (2014) The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42:D206–D214. doi:10.1093/nar/gkt1226
Peng X, Garrett RA, She QX (2012) Archaeal viruses-novel, diverse and enigmatic. Sci China Life Sci 55:422–433. doi:10.1007/s11427-012-4325-8
Petrosino S, Damiano N, Cusano P, Di Vito MA, de Vita S (2012) Subsurface structure of the Solfatara volcano (Campi Flegrei caldera, Italy) as deduced from joint seismic-noise array, volcanological and morphostructural analysis. Geochem Geophys Geosyst. doi:10.1029/2011gc004030
Pinkert A, Marsh KN, Pang SS, Staiger MP (2009) Ionic liquids and their interaction with cellulose. Chem Rev 109:6712–6728. doi:10.1021/cr9001947
Poretsky R, Rodriguez-R LM, Luo C, Tsementzi D, Konstantinidis KT (2014) Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE. doi:10.1371/journal.pone.0093827
Prangishvili D (2013) The wonderful world of archaeal viruses. Annu Rev Microbiol 67:565–585. doi:10.1146/annurev-micro-092412-155633
Prangishvili D, Garrett RA (2004) Exceptionally diverse morphotypes and genomes of crenarchaeal hyperthermophilic viruses. Biochem Soc Trans 32:204–208
Prangishvili D, Garrett RA (2005) Viruses of hyperthermophilic Crenarchaea. Trends Microbiol 13:535–542. doi:10.1016/j.tim.2005.08.013
Prangishvili D, Stedman K, Zillig W (2001) Viruses of the extremely thermophilic archaeon Sulfolobus. Trends Microbiol 9:39–43. doi:10.1016/S0966-842x(00)01910-7
Prangishvili D, Forterre P, Garrett RA (2006) Viruses of the Archaea: a unifying view. Nat Rev Microbiol 4:837–848. doi:10.1038/nrmicro1527
Prato S, Vitale RM, Contursi P, Lipps G, Saviano M, Rossi M, Bartolucci S (2008) Molecular modeling and functional characterization of the monomeric primase-polymerase domain from the Sulfolobus solfataricus plasmid pIT3. FEBS J 275:4389–4402. doi:10.1111/j.1742-4658.2008.06585.x
Pride DT, Schoenfeld T (2008) Genome signature analysis of thermal virus metagenomes reveals Archaea and thermophilic signatures. BMC Genom 9:420. doi:10.1186/1471-2164-9-420
Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IM, Read LF, Sloan WT (2009) Accurate determination of microbial diversity from 454 pyrosequencing data. Nat Methods 6:639–641. doi:10.1038/NMETH.1361
Rampelli S, Soverini M, Turroni S, Quercia S, Biagi E, Brigidi P, Candela M (2016) ViromeScan: a new tool for metagenomic viral community profiling. BMC Genom 17:165. doi:10.1186/s12864-016-2446-3
Rath D, Amlinger L, Rath A, Lundgren M (2015) The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie 117:119–128. doi:10.1016/j.biochi.2015.03.025
Reysenbach AL, Wickham GS, Pace NR (1994) Phylogenetic analysis of the hyperthermophilic pink filament community in Octopus Spring, Yellowstone National Park. Appl Environ Microbiol 60:2113–2119
Rice G, Stedman K, Snyder J, Wiedenheft B, Willits D, Brumfield S, McDermott T, Young MJ (2001) Viruses from extreme thermal environments. Proc Natl Acad Sci USA 98:13341–13345. doi:10.1073/pnas.231170198
Rice G, Tang L, Stedman K, Roberto F, Spuhler J, Gillitzer E, Johnson JE, Douglas T, Young M (2004) The structure of a thermophilic archaeal virus shows a double-stranded DNA viral capsid type that spans all domains of life. Proc Natl Acad Sci USA 101:7716–7720. doi:10.1073/pnas.0401773101
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F, Darling A, Malfatti S, Swan BK, Ea Gies, Ja Dodsworth, Hedlund BP, Tsiamis G, Sievert SM, Liu W-T, Ja Eisen, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437. doi:10.1038/nature12352
Romano C, D’Imperio S, Woyke T, Mavromatis K, Lasken R, Shock EL, McDermott TR (2013) Comparative genomic analysis of phylogenetically closely related Hydrogenobaculum sp. isolates from Yellowstone National Park. Appl Environ Microbiol 79:2932–2943. doi:10.1128/AEM.03591-12
Rosi M, Santacroce R (1984) Volcanic hazard assessment in the Phlegraean Fields: a contribution based on stratigraphic and historical data. Bull Volcanol 47:359–370. doi:10.1007/BF01961567
Rousseau C, Gonnet M, Le Romancer M, Nicolas J (2009) CRISPI: a CRISPR interactive database. Bioinformatics 25:3317–3318. doi:10.1093/bioinformatics/btp586
Roux S, Tournayre J, Mahul A, Debroas D, Enault F (2014) Metavir 2: new tools for viral metagenome comparison and assembled virome analysis. BMC Bioinform 15:76. doi:10.1186/1471-2105-15-76
Roux S, Hallam SJ, Woyke T, Sullivan MB (2015) Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife. doi:10.7554/eLife.08490
Sahm K, John P, Nacke H, Wemheuer B, Grote R, Daniel R, Antranikian G (2013) High abundance of heterotrophic prokaryotes in hydrothermal springs of the Azores as revealed by a network of 16S rRNA gene-based methods. Extremophiles 17:649–662. doi:10.1007/s00792-013-0548-2
Schoenfeld T, Patterson M, Richardson PM, Wommack KE, Young M, Mead D (2008) Assembly of viral metagenomes from yellowstone hot springs. Appl Environ Microbiol 74:4164–4174. doi:10.1128/AEM.02598-07
Servín-Garcidueñas LE, Peng X, Garrett RA, Martinez-Romero E (2013a) Genome sequence of a novel archaeal fusellovirus assembled from the metagenome of a mexican hot spring. Genome Announc 1:e0016413. doi:10.1128/genomeA.00164-13
Servín-Garcidueñas LE, Peng X, Garrett RA, Martínez-Romero E (2013b) Genome sequence of a novel archaeal rudivirus recovered from a mexican hot spring. Genome Announc 1:e00040-12
Sharma A, Kawarabayasi Y, Satyanarayana T (2012) Acidophilic bacteria and archaea: acid stable biocatalysts and their potential applications. Extremophiles 16:1–19. doi:10.1007/s00792-011-0402-3
Short SM, Short CM (2008) Diversity of algal viruses in various North American freshwater environments. Aquat Microb Ecol 51:13–21. doi:10.3354/ame01183
Simon C, Wiezer A, Strittmatter AW, Daniel R (2009) Phylogenetic diversity and metabolic potential revealed in a glacier ice metagenome. Appl Environ Microbiol 75:7519–7526. doi:10.1128/AEM.00946-09
Skennerton CT, Imelfort M, Tyson GW (2013) Crass: identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res. doi:10.1093/nar/gkt183
Snyder JC, Young MJ (2013) Lytic viruses infecting organisms from the three domains of life. Biochem Soc Trans 41:309–313. doi:10.1042/BST20120326
Snyder JC, Bateson MM, Lavin M, Young MJ (2010) Use of cellular CRISPR (clusters of regularly interspaced short palindromic repeats) spacer-based microarrays for detection of viruses in environmental samples. Appl Environ Microbiol 76:7251–7258. doi:10.1128/AEM.01109-10
Snyder JC, Brumfield SK, Peng N, She QX, Young MJ (2011) Sulfolobus turreted icosahedral virus c92 protein responsible for the formation of pyramid-like cellular lysis structures. J Virol 85:6287–6292. doi:10.1128/JVI.00379-11
Snyder JC, Bolduc B, Young MJ (2015) 40 Years of archaeal virology: expanding viral diversity. Virology 479:369–378. doi:10.1016/j.virol.2015.03.031
Song Z-Q, Chen J-Q, Jiang H-C, Zhou E-M, Tang S-K, Zhi X-Y, Zhang L-X, Zhang C-LL, Li W-J (2010) Diversity of Crenarchaeota in terrestrial hot springs in Tengchong, China. Extremophiles 14:287–296. doi:10.1007/s00792-010-0307-6
Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, Rubin EM (2007) Genome-wide experimental determination of barriers to horizontal gene transfer. Science. doi:10.1126/science.1147112
Spang A, Saw JH, Jorgensen SL, Zaremba-Niedzwiedzka K, Martijn J, Lind AE, van Eijk R, Schleper C, Guy L, Ettema TJG (2015) Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521:173. doi:10.1038/nature14447
Stahl DA, Lane DJ, Olsen GJ, Pace NR (1984) Analysis of hydrothermal vent-associated symbionts by ribosomal RNA sequences. Science 224:409–411. doi:10.1126/science.224.4647.409
Stauder S, Raue B, Sacher F (2005) Thioarsenates in sulfidic waters. Environ Sci Technol 39:5933–5939
Stern A, Sorek R (2011) The phage-host arms race: shaping the evolution of microbes. BioEssays 33:43–51. doi:10.1002/bies.201000071
Sun G, Xiao J, Wang H, Gong C, Pan Y, Yan S, Wang Y (2014) Efficient purification and concentration of viruses from a large body of high turbidity seawater. MethodsX 1:197–206. doi:10.1016/j.mex.2014.09.001
Sun CL, Thomas BC, Barrangou R, Banfield JF (2016) Metagenomic reconstructions of bacterial CRISPR loci constrain population histories. ISME J 10:858–870. doi:10.1038/ismej.2015.162
Takacs-Vesbach C, Inskeep WP, Jay ZJ, Herrgard MJ, Rusch DB, Tringe SG, Kozubal MA, Hamamura N, Macur RE, Fouke BW, Reysenbach AL, McDermott TR, Jennings RD, Hengartner NW, Xie G (2013) Metagenome sequence analysis of filamentous microbial communities obtained from geochemically distinct geothermal channels reveals specialization of three Aquificales lineages. Front Microbiol. doi:10.3389/Fmicb.2013.00084
Tebbe CC, Vahjen W (1993) Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and a yeast. Appl Environ Microbiol 59:2657–2665
Thomas T, Gilbert J, Meyer F (2012) Metagenomics—a guide from sampling to data analysis. Microb Inform Exp 2:3. doi:10.1186/2042-5783-2-3
Uldahl KB, Jensen SB, Bhoobalan-Chitty Y, Martinez-Alvarez L, Papathanasiou P, Peng X (2016) Life cycle characterization of Sulfolobus monocaudavirus 1, an extremophilic spindle-shaped virus with extracellular tail development. J Virol 90:5693–5699. doi:10.1128/JVI.00075-16
Urbieta MS, Toril EG, Giaveno MA, Bazan AA, Donati ER (2014) Archaeal and bacterial diversity in five different hydrothermal ponds in the Copahue region in Argentina. Syst Appl Microbiol 37:429–441. doi:10.1016/j.syapm.2014.05.012
Vale PF, Little TJ (2010) CRISPR-mediated phage resistance and the ghost of coevolution past. Proc R Soc B Biol Sci 277:2097–2103. doi:10.1098/rspb.2010.0055
van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJJ (2009) CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci 34:401–407. doi:10.1016/j.tibs.2009.05.002
Walker A (2014) Adding genomic ‘foliage’ to the tree of life. Nat Rev Microbiol 12:78. doi:10.1038/nrmicro3203
Wang LX (2009) Expanding the repertoire of glycosynthases. Chem Biol 16:1026–1027. doi:10.1016/j.chembiol.2009.10.003
Wang S, Dong H, Hou W, Jiang H, Huang Q, Briggs BR, Huang L (2014) Greater temporal changes of sediment microbial community than its waterborne counterpart in Tengchong hot springs, Yunnan Province, China. Sci Rep UK 4:7479. doi:10.1038/srep07479
Wang HM, Yu YX, Liu TG, Pan YJ, Yan SL, Wang YJ (2015a) Diversity of putative archaeal RNA viruses in metagenomic datasets of a yellowstone acidic hot spring. Springerplus. doi:10.1186/S40064-015-0973-Z
Wang HN, Peng N, Shah SA, Huang L, She QX (2015b) Archaeal extrachromosomal genetic elements. Microbiol Mol Biol R 79:117–152. doi:10.1128/MMBR.00042-14
Wang M, Lai G-L, Nie Y, Geng S, Liu L, Zhu B, Shi Z, Wu X-L (2015c) Synergistic function of four novel thermostable glycoside hydrolases from a long-term enriched thermophilic methanogenic digester. Front Microbiol 6:1–10. doi:10.3389/fmicb.2015.00509
Wemheuer B, Taube R, Akyol P, Wemheuer F, Daniel R (2013) Microbial diversity and biochemical potential encoded by thermal spring metagenomes derived from the Kamchatka Peninsula. Archaea (Vancouver, BC) 2013:136714. doi:10.1155/2013/136714
Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 87:4576–4579
Wolfenden R, Lu XD, Young G (1998) Spontaneous hydrolysis of glycosides. J Am Chem Soc 120:6814–6815. doi:10.1021/Ja9813055
Wommack KE, Bhavsar J, Ravel J (2008) Metagenomics: read length matters. Appl Environ Microbiol. doi:10.1128/AEM.02181-07
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, Hooper SD, Pati A, Lykidis A, Spring S, Anderson IJ, D’Haeseleer P, Zemla A, Singer M, Lapidus A, Nolan M, Copeland A, Han C, Chen F, Cheng JF, Lucas S, Kerfeld C, Lang E, Gronow S, Chain P, Bruce D, Rubin EM, Kyrpides NC, Klenk HP, Eisen JA (2009) A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462:1056–1060. doi:10.1038/nature08656
Wu Y-W, Tang Y-H, Tringe SG, Ba Simmons, Singer SW (2014) MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation–maximization algorithm. Microbiome 2:26. doi:10.1186/2049-2618-2-26
Youssef N, Sheik CS, Krumholz LR, Najar FZ, Ba Roe, Elshahed MS (2009) Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl Environ Microbiol 75:5227–5236. doi:10.1128/AEM.00592-09
Zhao C, Chu Y, Li Y, Yang C, Chen Y, Wang X, Liu B (2017) High-throughput pyrosequencing used for the discovery of a novel cellulase from a thermophilic cellulose-degrading microbial consortium. Biotechnol Lett 39:123–131. doi:10.1007/s10529-016-2224-y
Zillig W, Prangishvilli D, Schleper C, Elferink M, Holz I, Albers S, Janekovic D, Gotz D (1996) Viruses, plasmids and other genetic elements of thermophilic and hyperthermophilic Archaea. FEMS Microbiol Rev 18:225–236. doi:10.1111/J.1574-6976.1996.Tb00239.X
Zong C, Lu S, Chapman AR, Xie XS (2012) Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338:1622–1626. doi:10.1126/science.1229164
Funding
This work was supported by a Grant from BIOPOLIS: PON03PE_00107_1 CUP: E48C14000030005 and by the project “Esobiologia e ambienti estremi: dalla Chimica delle Molecole alla Biologia degli Estremofili—ECMB” No. 2014-026-R.0 of the Italian Space Agency.
Author information
Authors and Affiliations
Contributions
AS, SF, BCP, MM and PC wrote, reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Strazzulli, A., Fusco, S., Cobucci-Ponzano, B. et al. Metagenomics of microbial and viral life in terrestrial geothermal environments. Rev Environ Sci Biotechnol 16, 425–454 (2017). https://doi.org/10.1007/s11157-017-9435-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11157-017-9435-0