
Abstract
Pollution of the environment with aromatic hydrocarbons, such as benzene, toluene, ethylbenzene and xylene (so-called BTEX) is often observed. The cleanup of these toxic compounds has gained much attention in the last decades. In situ bioremediation of aromatic hydrocarbons contaminated soils and groundwater by naturally occurring microorganisms or microorganisms that are introduced is possible. Anaerobic bioremediation is an attractive technology as these compounds are often present in the anoxic zones of the environment. The bottleneck in the application of anaerobic techniques is the lack of knowledge about the anaerobic biodegradation of benzene and the bacteria involved in anaerobic benzene degradation. Here, we review the existing knowledge on the degradation of benzene and other aromatic hydrocarbons by anaerobic bacteria, in particular the physiology and application, including results on the (per)chlorate stimulated degradation of these compounds, which is an interesting new alternative option for bioremediation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
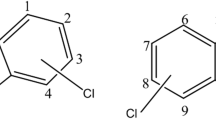
Aromatic compounds are the second most abundant family of organic constituents present in nature after carbohydrates. The most important natural sources of aromatic compounds are poorly biodegradable polymers such as lignin, condensed tannins and humus (Dagley 1985). Since the start of the industrial revolution a wide variety of aromatic compounds has also been introduced into the environment through anthropogenic activity. About 35 million tonnes are produced worldwide annually (http://en.wikipedia.org/wiki/Aromatic). With such huge quantities of these compounds being made, transported and used, it is inevitable that a substantial amount will be lost to the environment. Aromatic compounds are important constituents of crude oil and oil derivatives. Gasoline is rich in monoaromatic hydrocarbons, such as benzene, toluene, ethylbenzene and xylene (BTEX) (Fig. 1). Furthermore, these compounds are used as industrial solvents and they provide the starting materials for the production of pharmaceuticals, agrochemicals, polymers, explosives and many other everyday products (Smith 1990).

Chemical structures of benzene, toluene, ethylbenzene, ortho-xylene, meta-xylene and para-xylene
Accidental spills and industrial discharges have resulted in pollution of the environment with BTEX. Furthermore, gasoline leakage from underground storage tanks has been identified as an important source of groundwater contamination with BTEX. High concentrations of BTEX have been detected in soils, sediments and groundwater. The mobility and toxicity of the BTEX compounds are of major concern (several physical–chemical properties of BTEX compounds are summarized in Table 1). Compared with other oil hydrocarbons, BTEX are relatively water-soluble and therefore a plume of contamination within the groundwater is formed rapidly (Coates et al. 2002; Chakraborty and Coates 2004). Benzene is the most hazardous of the BTEX compounds since it is a known human carcinogen (leukaemogenic potential) (Badham and Winn 2007). Toluene and xylene are not carcinogenic, but toluene can enhance carcinogenesis by other compounds (Dean 1978).
In aquifers contaminated with organic pollutants, generally a sequence of redox zones has developed as a result of organic contamination. Often, near the source of the organic pollutants methanogenic conditions are observed, whereas downstream of the contaminant source zone in the plume sulfate-reducing and iron-reducing conditions may exist. Further downstream and at the fringes of the plume, nitrate and manganese(IV)-reducing conditions prevail (Christensen et al. 2001). As a result, BTEX contamination is often present in the anaerobic zones of the environment (Lovley 1997).
Insight into BTEX degradation has led to the development of biological remediation techniques for BTEX-contaminated sites. All aromatic compounds possess a relative resistance to degradation due to the large (negative) resonance energy. This large resonance energy is caused by the stability of the π-electron cloud (Aihara 1992). Before the 1980s, mainly aerobic BTEX degradation was studied. The last two decades microorganisms that degrade BTEX components in the absence of oxygen were also studied. Many bacteria, especially Pseudomonas species, have been isolated that can use benzene as sole carbon and energy source for aerobic growth. Under aerobic conditions, oxygen does not only serve as a terminal electron acceptor, but it is also used in the initial enzymatic activation of aromatic compounds. Oxygen is incorporated into the aromatic ring and these reactions are catalyzed by mono- or dioxygenases (Gibson and Subramanian 1984). Hence, the biochemical strategy for aromatic hydrocarbon activation under oxic conditions is to introduce a hydroxyl group (monohydroxylation by monooxygenase) or hydroxyl groups (dihydroxylation by dioxygenases) into the aromatic ring. The aerobic degradation of toluene, ethylbenzene and xylene may involve mono- or dioxygenases, but other pathways have also been described (Van Agteren et al. 1998). Aerobic biodegradation of BTEX compounds has been studied since the sixties of the last century and has been reviewed several times (Gibson and Subramanian 1984; Dagley 1985, 1986; Smith 1990). Under anaerobic conditions, oxygen is not available for the initial attack of the ring and therefore other pathways are involved in the BTEX degradation.
In this review the current knowledge on aerobic and anaerobic transformation of BTEX compounds is summarized. The emphasis in this paper lies on the microbial degradation of BTEX compounds under anaerobic conditions. The degradation of BTEX, in particular benzene, under anaerobic conditions will be discussed in more detail hereafter.
2 Anaerobic BTEX degradation
As BTEX compounds are often present in the anoxic zones of the environment, anaerobic bioremediation is an attractive remediation technique. Anaerobic degradation of aromatic hydrocarbons was first described in 1985 (Kuhn et al. 1985). Since then, the degradation pathways of toluene and ethylbenzene by denitrifying and sulfate-reducing microorganisms in particular have been characterized. Anaerobic degradation of BTEX has been reviewed in the past (Schink et al. 1992; Heider et al. 1999; Spormann and Widdel 2000; Phelps and Young 2001; Widdel and Rabus 2001; Chakraborty and Coates 2004; Heider 2007; Foght 2008; Fuchs 2008; Carmona et al. 2009). Aromatic compounds, such as benzene and toluene, are thermodynamically favorable electron donors for growth, because of the high Gibbs free energy change of the oxidation of these compounds with different electron acceptors (see Table 2 for stoichiometric equations and standard free energy changes for benzene oxidations with various electron acceptors). All studies regarding anaerobic BTEX degradation have indicated that anaerobic benzene degradation is most difficult and that toluene is one of the aromatic compounds, which is relatively easy to degrade anaerobically.
2.1 Anaerobic toluene biodegradation
Among the BTEX components, the anaerobic biodegradation of toluene is probably most extensively studied. Toluene can be biodegraded with nitrate, Mn(IV), Fe(III), sulfate or CO2 as terminal electron acceptors (Langenhoff et al. 1997b; Chakraborty and Coates 2004 and others). More recently, it was demonstrated that anaerobic toluene degradation can also be coupled to the reduction of humic substances (Cervantes et al. 2001), chlorine oxyanions, such as perchlorate or chlorate (Chakraborty et al. 2005) or arsenate (Liu et al. 2004). Moreover, toluene can be used as a carbon source by anoxygenic phototrophs (Zengler et al. 1999). Anaerobic toluene degradation has been found in field studies, column studies, enrichment cultures and microcosms and in pure cultures (Phelps and Young 2001). Here, pure cultures studies will be discussed in more detail.
2.1.1 Anaerobic toluene-degrading isolates
Several bacteria have been isolated, which couple the degradation of toluene to the reduction of nitrate (Table 3). Most of these bacteria belong either to the Azoarcus or Thauera genus, e.g. Thauera aromatica T1 (Evans et al. 1991), Thauera aromatica K172 (Anders et al. 1995), Azoarcus tolulyticus Tol4 (Fries et al. 1994) and Azoarcus sp. EbN1 (Rabus and Widdel 1995). Also four Magnetospirillum strains, which belong to the Alphaproteobacteria, were described that can degrade toluene with nitrate as the electron acceptor (Shinoda et al. 2005). The toluene-oxidizing nitrate-reducing Thauera and Azoarcus species are facultative anaerobes and are members of the Betaproteobacteria. Most of the these bacteria were isolated from anaerobic sludge or (freshwater) sediments (Anders et al. 1995). Several of the Azoarcus and Thauera species were originally described as Pseudomonas species, but were subsequently reclassified. Probably, the Azoarcus and Thauera species that have these aromatics-degrading capacities will be reclassified in future and separated from the other Azoarcus and Thauera species, which do not have these capacities (Kühner et al. 2005). The genome of strain EbN1 has been sequenced and this bacterium was renamed as Aromatoleum aromaticum (Rabus et al. 2005; Wöhlbrand et al. 2007). Recently, an anaerobic betaproteobacterial strain was described, which uses nitrate, Fe(III) or Mn(IV) as electron acceptor for growth on toluene. This bacterium (strain G5G6) was described as a novel taxon of the Betaproteobacteria, named Georgfuchsia toluolica (Weelink et al. 2009).
One of the first reports about anaerobic toluene degradation dealt with the degradation of toluene coupled to the reduction of Fe(III) by Geobacter metallireducens GS-15 (Lovley and Lonergan 1990; Lovley et al. 1993). This bacterium belongs to the Deltaproteobacteria and can completely oxidize toluene to CO2 coupled to the reduction of Fe(III). More recently, Geobacter grbiciae, Geobacter sp. TMJ1 and Desulfitobacterium aromaticivorans were described which can oxidize toluene with Fe(III) (Coates et al. 2001a; Winderl et al. 2007; Kunapuli et al. 2010). Geobacter metallireducens can also use nitrate, Mn(IV) or humic substances as the electron acceptor (Lovley et al. 1993; Coates et al. 2001a). The betaproteobacterium Georgfuchsia toluolica strain G5G6 also uses nitrate, Fe(III) or Mn(IV) as electron acceptor for growth on toluene (Weelink et al. 2009). In general, Geobacter species are often found to be dominant in the Fe(III)reduction zone of environments contaminated with hydrocarbons (Rooney-Varga et al. 1999; Botton et al. 2007).
Several bacteria have been described capable of degrading toluene with sulfate (Table 3), such as Desulfobacula toluolica and Desulfotignum toluenicum (Rabus et al. 1993; Beller et al. 1996; Harms et al. 1999b; Meckenstock 1999; Morasch et al. 2004; Kuever et al. 2005; Ommedal and Torsvik 2007). These sulfate-reducing bacteria all belong to the Deltaproteobacteria. Toluene degradation with other less common electron acceptors has also been reported. Toluene degradation can be coupled to (per)chlorate respiration by Dechloromonas aromatica RCB (Chakraborty et al. 2005) and to the reduction of arsenate by strain Y5 (Liu et al. 2004). Finally, toluene can also be assimilated as a carbon source by the anoxygenic phototroph, Blastochloris sulfoviridis (Zengler et al. 1999).
2.1.2 Anaerobic toluene degradation pathway
The biochemical pathway of anaerobic toluene degradation has been intensively studied over the last decade (Heider et al. 1999; Spormann and Widdel 2000; Widdel and Rabus 2001). Especially, Azoarcus strain T, Thauera aromatica K172 and Thauera aromatica T1 were studied (Biegert et al. 1996; Beller and Spormann 1997b, 1998; Coschigano et al. 1998; Heider et al. 1998; Leuthner and Heider 1998; Leuthner et al. 1998; Beller and Spormann 1999; Krieger et al. 1999). These studies revealed that the first step in the catabolism of toluene is the addition of toluene to the double bond of fumarate to form benzylsuccinate by benzylsuccinate synthase (bssABC) (Fig. 2) (Leuthner et al. 1998). Benzylsuccinate is then activated to CoA-thioester by a succinyl-CoA-dependent CoA-transferase (bbsEF), and benzylsuccinyl-CoA is then converted to succinyl-CoA and benzoyl-CoA. Benzoyl-CoA reductase (bcrCABD) initiates the degradation of benzoyl-CoA, which is thereafter further oxidized via reductive ring cleavage to carbon dioxide. Benzoyl-CoA has been recognized as central intermediate in the anaerobic degradation of many aromatic compounds (Harwood et al. 1999).

Anaerobic toluene degradation route, according to Kube et al. (2004). BssABC, benzylsuccinate synthase; BbsEF, succinyl-CoA:(R)-benzylsuccinate CoA-transferase; BbsG, (R)-benzylsuccinyl-CoA dehydrogenase; BbsH, phenylitaconyl-CoA hydratase; BbsCD, 2-[hydroxy(phenyl)methyl]-succinyl-CoA dehydrogenase; BbsAB, benzoylsuccinyl-CoA thiolase
Although the bss pathway was first identified in Azoarcus and Thauera species growing under nitrate-reducing conditions, it is now considered as the common mechanism for activation of toluene under various (anaerobic) redox conditions by phylogenetically diverse bacteria. The bss pathway was also found in the toluene-oxidizing Fe(III)-reducing Geobacter metallireducens (Kane et al. 2002), in the sulfate-reducing bacteria, PRTOL1 and Desulfobacula toluolica (Beller and Spormann 1997a; Rabus and Heider 1998) and in the toluene-degrading betaproteobacterium Georgfuchsia toluolica (Weelink et al. 2009). Furthermore, this pathway was demonstrated in the toluene-utilizing phototrophic Blastochloris sulfoviridis (Zengler et al. 1999). Bss genes were also identified in a methanogenic toluene-degrading culture (Washer and Edwards 2007). It is possible that some bacteria use other pathways for the initial toluene conversion, such as a pathway involving direct methyl group hydroxylation to benzyl alcohol or hydroxylation to cresol (Frazer et al. 1995; Langenhoff et al. 1997a). These pathways have not been studied as thoroughly as the bss pathway. Therefore, involvement of fumarate addition cannot be ruled out (Phelps and Young 2001).
2.2 Anaerobic xylene degradation
Although all three xylene isomers can be biodegraded anaerobically, they have different susceptibilities to anaerobic biodegradation (Foght 2008). Early reports of anaerobic xylene degradation under nitrate-reducing conditions indicated that both p- and m- (but not o-) xylene were degraded (Kuhn et al. 1985), whereas Edwards et al. (1992) found p- and o-xylene degradation (m-xylene was not tested) under sulfate-reducing conditions. Several subsequent studies demonstrated that m-xylene is the most readily degraded isomer in mixed cultures (Beller et al. 1995). In some cases the presence of m-xylene inhibited (Jahn et al. 2005) concomitant o- and p-xylene degradation (Meckenstock et al. 2004a; Morasch et al. 2004).
Degradation of o-xylene was demonstrated under denitrifying and sulfate-reducing conditions (Zeyer et al. 1986; Edwards et al. 1992), and recently the first iron-reducing enrichment cultures were shown to oxidize o-xylene (Jahn et al. 2005) and p-xylene (Botton and Parsons 2006). Although p-xylene is often reported to be recalcitrant under anoxic conditions (Rabus and Widdel 1995), it was degraded by enrichment cultures under denitrifying (Häner et al. 1995; Rotaru et al. 2010), iron-reducing (Botton and Parsons 2007) and sulfate-reducing conditions (Morasch and Meckenstock 2005; Nakagawa et al. 2008). Recently, sulfate-reducing enrichment cultures that degraded m-, o- and p-xylenes, respectively, were described (Herrmann et al. 2009). It is noteworthy that the p-xylene-degrading enrichment cultures described by Häner et al. (1995) and Rotaru et al. (2010) both were very selective; they were unable to grow with benzene, ethylbenzene or o-xylene.
Several pure cultures have been shown to utilize m-xylene for growth under nitrate- (Dolfing et al. 1990; Fries et al. 1994; Rabus and Widdel 1995; Hess et al. 1997) and sulfate-reducing (Harms et al. 1999b) conditions, whereas only two pure cultures of sulfate reducers have been reported to mineralize o-xylene anaerobically, Desulfosarcina sp. strain oXyS1 (Harms et al. 1999b), renamed as Desulfosarcina ovata (Kuever et al. 2005), and Desulfotomaculum sp. strain OX39 (Morasch et al. 2004). Such isolates often show narrow substrate specificity, with mXyS1 degrading only meta- and oXyS1 preferring ortho- substituted aromatics (Harms et al. 1999b). No pure cultures utilizing p-xylene for growth have yet been reported, although Rotaru et al. (2010) described highly enriched Betaproteobacteria growing with p-xylene and nitrate. Phylotypes with 95% 16S rRNA gene sequence identity to Denitratisoma oestradiolicum dominated the enrichment cultures (>91% of all cells) (Rotaru et al. 2010).
The initial steps of anaerobic m-xylene and o-xylene degradation were elucidated in Azoarcus sp. strain T (Beller and Spormann 1997b; Krieger et al. 1999). Research by Krieger et al. (1999) revealed that m-xylene is converted in a series of reactions that are similar to those of fumarate addition in toluene degradation. Homologs corresponding to toluene fumarate addition metabolites have been detected in cultures incubated with xylenes. For example, 4-methylbenzylsuccinate and 4-methylphenylitaconate were extracted from an enrichment culture incubated with p-xylene (Morasch and Meckenstock 2005), and the expected 2-methylbenzylsuccinate homolog was detected in cultures co-metabolizing o-xylene (Beller and Spormann 1997b). In addition, Herrmann et al. (2009) identified the benzylsuccinate synthase (bssA) in several xylene-degrading cultures using bssA-targeted primers.
2.3 Anaerobic ethylbenzene degradation
Anaerobic ethylbenzene degradation has been demonstrated in several nitrate-reducing bacteria and one sulfate-reducing bacterium (Table 3). Although ethylbenzene is chemically very similar to toluene, it is usually degraded via a different pathway. The denitrifying bacterial strains EB1, EbN1 (Aromatoleum aromaticum) and PbN1 degrade ethylbenzene to CO2. These bacteria were used to elucidate the anaerobic ethylbenzene degradation pathway (Ball et al. 1996; Rabus and Heider 1998; Johnson et al. 2001; Rabus et al. 2002). These strains are closely related, and belong to the genus Azoarcus in the Betaproteobacteria. This metabolic pathway (Fig. 3) included an initial step catalyzed by ethylbenzene dehydrogenase, a novel molybdenum/iron-sulfur/heme enzyme (Johnson et al. 2001; Rabus et al. 2002). It oxidizes the methyl group of ethylbenzene independently of oxygen, generating (S)-1-phenylethanol as first intermediate. Further metabolism of (S)-1-phenylethanol proceeds via oxidation to acetophenone, which is then carboxylated at the methyl group forming benzoylacetate. Benzoylacetate is converted via benzoylacetyl-CoA to benzoyl-CoA (Ball et al. 1996), the central intermediate in the anaerobic catabolism of aromatic compounds.

Anaerobic metabolism of ethylbenzene, according to Kühner et al. (2005). ebdABC, ethylbenzene dehydrogenase; ped: (S)-1-phenylethanol dehydrogenase; apc1-5, acetophenone carboxylase; bal, benzoylacetate CoA-ligase
A recent study on anaerobic ethylbenzene degradation under sulfate-reducing conditions resulted in the isolation of a novel organism, strain EbS7 (Kniemeyer et al. 2003). This strain is a member of the Deltaproteobacteria, most closely related to strain mXyS1, which can anaerobically oxidize toluene and m-xylene (Harms et al. 1999b). In contrast to the initial dehydrogenation reaction used by denitrifying ethylbenzene degraders, but similarly to toluene and xylene pathways, activation of ethylbenzene by strain EbS7 is achieved by fumarate addition at the secondary carbon atom of the ethyl group to form 1-phenylethyl-succinate (Kniemeyer et al. 2003).
2.4 Anaerobic benzene degradation
For a long time, benzene was considered to be persistent under anaerobic conditions. Benzene is still considered as the most recalcitrant of all BTEX compounds under anaerobic conditions. A survey of laboratory and field studies with groundwater demonstrated that anaerobic benzene degradation in most cases did not occur (Aronson and Howard 1997). Anaerobic benzene degradation has been observed in some sediment, microcosm and column studies and microbial enrichments. Only recently, isolates capable of anaerobic benzene degradation have been described (Coates et al. 2001b; Kasai et al. 2006). Anaerobic benzene degradation has been extensively reviewed (Lovley 2000; Phelps and Young 2001; Coates et al. 2002; Chakraborty and Coates 2004).
2.4.1 Anaerobic benzene degradation under different redox conditions
Anaerobic benzene degradation with nitrate has been demonstrated in microcosms (Major et al. 1988), in enrichment cultures (Burland and Edwards 1999; Ulrich and Edwards 2003) and in pure cultures (Coates et al. 2001b; Kasai et al. 2006). So far anaerobic benzene degradation with Fe(III) as the electron acceptor has not been observed in pure cultures, but only in microcosms (Lovley et al. 1994, 1996a; Anderson et al. 1998; Anderson and Lovley 1999; Botton and Parsons 2006) and in enrichment cultures (Rooney-Varga et al. 1999; Botton and Parsons 2007; Kunapuli et al. 2007). Moreover, anaerobic benzene degradation with Fe(III) as the electron acceptor was stimulated by humic substances, which serve as electron shuttle compounds between Fe(III)-reducing bacteria and insoluble Fe(III) oxides (Lovley et al. 1996b). With sulfate as the electron acceptor, anaerobic benzene degradation has been found in a column study (Vogt et al. 2007), in microcosms (Edwards and Grbic-Galic 1992; Lovley et al. 1995; Coates et al. 1996a, b; Kazumi et al. 1997; Weiner et al. 1998; Anderson and Lovley 2000), and in enrichments (Phelps et al. 1996, 1998; Ulrich and Edwards 2003; Musat and Widdel 2007; Herrmann et al. 2008; Kleinsteuber et al. 2008; Laban et al. 2009). Under methanogenic conditions, anaerobic benzene degradation has been demonstrated in microcosms (Kazumi et al. 1997; Weiner and Lovley 1998) and in enrichments (Vogel and Grbic-Galic 1986; Grbic-Galic and Vogel 1987; Ulrich and Edwards 2003; Chang et al. 2005). Furthermore, benzene was degraded with Mn(IV) as the electron acceptor in microcosms and sediment columns (Villatoro-Monzon et al. 2003, 2008). Benzene degradation with (per)chlorate as the electron acceptor was shown in pure culture (Coates et al. 2001b; Weelink et al. 2008) and in column studies (Tan et al. 2006).
2.4.2 Ecology of anaerobic benzene-degrading bacteria
Little is known about the microorganisms responsible for anaerobic benzene degradation. Most information has been obtained by using molecular approaches. It was only recently that pure cultures of two Dechloromonas strains (RCB and JJ) and two Azoarcus strains (DN11 and AN9) were isolated, which are capable to degrade benzene anaerobically (Coates et al. 2001b; Kasai et al. 2006). Dechloromonas strain RCB (Dechloromonas aromatica RCB) and strain JJ are phylogenetically closely related (98.1% 16S rRNA sequence similarity) and both are members of the newly described Dechloromonas genus in the Betaproteobacteria (Achenbach et al. 2001; Coates et al. 2001b). Strain RCB was isolated from river sediment with 4-chlorobenzoate as the electron donor and chlorate as the electron acceptor. Strain JJ was isolated from lake sediment with anthrahydroquinone 2,6-disulphonate (AQDS, a humic substance analogue) as the electron donor and nitrate as the electron acceptor. Both strains are also able to oxidize benzene coupled to the reduction of nitrate. Benzene (160 μM) was completely degraded to CO2 within 5 days (Coates et al. 2001b). So far, subsequent research was carried out with strain RCB rather than with strain JJ. In addition to benzene, strain RCB is able to use toluene, ethylbenzene and m- and o-xylene as the electron donor with nitrate as the electron acceptor. Moreover, in addition to nitrate, strain RCB could degrade benzene with perchlorate, chlorate and oxygen (Chakraborty et al. 2005). The Dechloromonas species, together with the closely related Azospira (previously named Dechlorosoma) species, are considered to represent the predominant (per)chlorate-reducing bacteria in the environment (Coates et al. 1999b) and they may be important in the nitrate-dependent anaerobic degradation of benzene in the environment (Chakraborty and Coates 2004).
Two Azoarcus strains (DN11 and AN9) were isolated, which degrade benzene with nitrate as the electron acceptor (Kasai et al. 2006). Subsequently, the degradative capacities of strain DN11 and the potential for its application in bioaugmentation were investigated. Strain DN11 could grow on benzene, toluene, m-xylene and benzoate as the sole carbon and energy source under nitrate-reducing conditions, but o- and p-xylene were only cometabolically transformed in the presence of toluene. Phenol was not utilized under anaerobic conditions. Furthermore, strain DN11 could significantly enhance the benzene degradation after addition of the strain to laboratory batches containing benzene-contaminated groundwater (Kasai et al. 2007).
Under Fe(III)-reducing conditions, Geobacter species have often been associated with anaerobic benzene degradation. Benzene-degrading Fe(III)-reducing sediments and enrichments were dominated by bacteria of the family Geobacteraceae (Anderson et al. 1998; Rooney-Varga et al. 1999; Botton et al. 2007). However, several species in the genus Geobacter have the ability to anaerobically degrade toluene and other aromatic compounds (Lovley et al. 2004), but none of the Geobacter species tested degrades benzene. Recently, a detailed functional and phylogenetic characterization of a benzene-degrading iron-reducing enrichment using stable isotope probing (SIP) was presented. The authors obtained indications that benzene degradation in the enrichment involved an unusual syntrophy, in which members of the genus Thermincola within the family Peptococcaceae primarily oxidize benzene and partially share electrons from benzene with members of the Desulfobulbaceae as syntrophic partners (Kunapuli et al. 2007). Syntrophic BTX degradation was also suggested to occur in other iron-reducing enrichments (Botton et al. 2007) and in benzene-degrading sulfate-reducing consortia (Kleinsteuber et al. 2008; Herrmann et al. 2010).
Phelps et al. (1998) described the molecular characterization of a benzene-degrading sulfate-reducing enrichment. This enrichment received benzene as the only carbon and energy source for a period of 3 years, but repeated attempts to isolate the responsible bacteria failed. Molecular characterization revealed a diverse collection of phylotypes; 16S rRNA genes belonging to Proteobacteria, Cytophageles and Gram-positive phyla as well as one deeply branching phylotype not closely related to any known bacterium were detected (Phelps et al. 1998). Four clones fell within the Deltaproteobacteria, in the family Desulfobacteriaceae and one of these clones was closely related to a known aromatic hydrocarbon degrader, Desulfobacula toluolica. The large diversity of bacteria maintained over such a long period of time suggests that a consortium of bacteria may be needed to degrade benzene anaerobically in this enrichment. Members of the family Desulfobacteriaceae have also been found in a methanogenic benzene-degrading enrichment culture. This enrichment was dominated (33% of the total population) by a clone belonging to the family Desulfobacteriaceae (Ulrich and Edwards 2003). In another study, DGGE (Denaturing Gradient Gel Electrophoresis) analysis demonstrated that a bacterium related to Desulfobacterium sp. clone OR-M2 dominated a methanogenic benzene-degrading enrichment (Da Silva and Alvarez 2007). It was not proven that this Desulfobacterium was a benzene degrader, but it was suggested that it either initiates benzene degradation or is a critical (commensal) partner. Musat and Widdel (2007) demonstrated anaerobic benzene degradation by a marine sulfate-reducing enrichment culture. The dominant phylotype in this enrichment was closely related to a clade of Deltaproteobacteria that includes sulfate-reducing bacteria able to degrade naphthalene and other aromatic hydrocarbons. Cell hybridization with specifically designed 16S rRNA-targeted fluorescent oligonucleotide probes showed that the retrieved phylotype accounted for more than 85% of the cells detectable via DAPI staining in the enrichment culture (Musat and Widdel 2007). Liou et al. (2008) used laboratory incubations of coal-tar waste-contaminated sediment microbial communities under relatively controlled physiological conditions (anaerobic with or without sulfate or nitrate additions versus aerobic) to interpret results of a field-based SIP assay. By using this SIP approach the authors were able to associate sets of active benzene-degrading taxa with consumption of particular electron acceptors (nitrate, sulfate or mixed aerobic/anaerobic metabolism) and then qualitatively compare these taxa with those retrieved from an uncontrolled in situ field experiment. 13C-DNA clone libraries revealed a broad diversity of taxa involved in benzene metabolism and distinct libraries for each biodegradation treatment. Perhaps most importantly, in the field SIP experiment the clone libraries produced were dominated by Pelomonas (Betaproteobacteria) sequences similar to those found in the anaerobic benzene laboratory experiment (Liou et al. 2008).
Recently, benzene-degrading sulfate-reducing consortia were enriched from a benzene-contaminated aquifer (Zeitz field site, Germany). Benzene-mineralizing organisms were enriched under sulfate-reducing conditions: (1) using in situ microcosms, filled with different solids (sand, lava and Amberlite XAD-7), placed for 9 weeks within a groundwater monitoring well located downstream from the source zone of the plume, which were subsequently incubated in the laboratory (Herrmann et al. 2008) and (2) in columns percolated with benzene-containing anoxic groundwater, filled with either sand or pumice (Vogt et al. 2007). In control microcosms without filling material, benzene was initially degraded, but the benzene-degrading capacity could not be sustained. Herrmann et al. (2008) suggested that it could be favorable to use solids for the in situ enrichment of anaerobic benzene-degrading bacteria, a strategy that might be useful for the cultivation of bacteria that are considered to be hardly or not, cultivable. From the sand-filled columns, described by Vogt et al. (2007), Kleinsteuber et al. (2008) established stable benzene-degrading sulfate-reducing enrichment culture (named ZzBs1-4). It was composed of Delta- and Epsilonproteobacteria, Clostridia, Chloroflexi, Actinobacteria and Bacteroidetes. The most prominent phylotype of the consortium was related to the genus Sulfurovum, followed by Desulfovibrio sp. and the Cryptanaerobacter/Pelotomaculum phylotype. The Cryptanaerobacter/Pelotomaculum phylotype appeared to be specific for benzene as a growth substrate and might play a key role in benzene degradation in the consortium. Based on the possible functions of the community members, a functional model for syntrophic benzene degradation under sulfate-reducing conditions was proposed (Kleinsteuber et al. 2008) that was later confirmed by the DNA-SIP experiment described by Herrmann et al. (2010). In this enrichment two phyloptypes were dominant: a member of the Cryptanaerobacter/Pelotomaculum group within the Peptococcaceae, and a phylotype belonging to the Epsilonproteobacteria. Herrmann et al. (2010) hypothesized that the Cryptanaerobacter/Pelotomaculum phylotype was responsible for the first steps of benzene degradation. It was proposed that they form metabolites like hydrogen, acetate or other low-molecular mass fermentation products that are used by the other bacterial partners (such as the Epsilonproteobacteria phyloptype) and the Methanosaetaceae-like Archaea. This would imply that benzene is mineralized by a consortium consisting of syntrophs, hydrogenotrophic sulfate reducers and to a minor extent of aceticlastic methanogens. The function of the Epsilonproteobacteria phyloptype could not be elucidated completely, which means that sofar it is not clear how this bacterium degrades the fermentation products (Herrmann et al. 2010). In another study, different molecular biology techniques showed 95% dominance of a phylotype that is affiliated to the Gram-positive bacterial genus Pelotomaculum, also found in a benzene-degrading sulfate-reducing enrichment culture (Laban et al. 2009). Since there was only one dominant organism present, Laban et al. (2009) proposed that, unlike the Cryptanaerobacter/Pelotomaculum phylotype described by Kleinsteuber et al. (2008) and Herrmann et al. (2010), bacteria with Pelotomaculum related 16S rRNA gene sequences oxidize benzene directly with sulfate as electron acceptor. In another study, members of the family Desulfobacteraceae have been identified as significant bacteria in a sulfate-reducing benzene-degrading enrichment culture, described first by Phelps et al. (1998), using SIP (Oka et al. 2008).
2.4.3 Anaerobic benzene degradation pathway
Several mechanisms are known to cleave the aromatic ring anaerobically for aromatic compounds with functional groups such as carboxyl or hydroxyl groups. Anaerobic degradation of benzene, however, is more difficult due to the stability of benzene. The mechanisms of activation and further degradation of benzene are still unknown. The possible initial steps are hydroxylation, carboxylation and methylation, and subsequent transformation to the central aromatic intermediate benzoyl-CoA (Fig. 4), which is further degraded to CO2. Below, the different pathways that have been proposed will be discussed in more detail.

Possible mechanisms of the initial steps of benzene degradation under anaerobic conditions: a benzene hydroxylation, b benzene methylation, c benzene carboxlation
-
A.
Benzene hydroxylation
Studies of Grbic-Galic and Vogel demonstrated that benzene could be degraded under methanogenic conditions (Grbic-Galic 1986; Vogel and Grbic-Galic 1986; Grbic-Galic and Vogel 1987). Phenol, cyclohexanone, and propionate were detected as putative intermediates, suggesting initial hydroxylation of benzene to phenol and subsequent ring reduction of phenol to cyclohexanone. Experiments with 18O-labeled water suggested that the oxygen incorporated into the aromatic ring was derived from water. Weiner and Lovley (1998) found phenol, propionate, and acetate as intermediates in a benzene-degrading methanogenic enrichment using 14C-labeled benzene. Furthermore, benzoate, phenol, o-hydroxybenzoate, and acetate were detected in a benzene-degrading enrichment derived from a mixed inoculum of cow dung and anaerobic digester sludge (Chaudhuri and Wiesmann 1995). In another study using 13C-labeled benzene, phenol was detected as intermediate in sulfate-reducing, Fe(III)-reducing and methanogenic enrichments, and benzoate was detected as intermediate in the sulfate-reducing enrichment (Caldwell and Suflita 2000). Mass spectral results indicated that the carboxyl group of the produced benzoate was 13C-labeled during 13C-benzene degradation.
It is possible, but difficult to hydrate molecules with double bonds that lack an adjacent carbon with a functional group, such as benzene (Coates et al. 2002). It is not known which enzymes are involved in this reaction, but a recently purified molybdenum-containing, iron-sulfur protein that adds a hydroxyl group to ethylbenzene may provide a model for hydroxylation of benzene (Johnson et al. 2001). In most prokaryotic systems studied, these oxomolybdenum enzymes catalyse the transfer of an oxygen atom from water to the substrate (Coates et al. 2002). Hydroxylation of the aromatic ring can also occur by a non-enzymatic mechanism involving highly reactive hydroxyl radicals (HO·), which can be formed by a Fenton-like reduction of hydrogen peroxide (H2O2) in the presence of ferrous iron complexes. These radicals can be incorporated into the aromatic ring. However, it is not clear how hydrogen peroxide could be formed anaerobically though such a mechanism may be possible at interfaces between aerobic and anaerobic zones (Coates et al. 2002). The conversion of phenol to benzoate probably involves the carboxylation of phenol to form p-hydroxybenzoate (4-hydroxybenzoate). Several pure cultures degrade phenol anaerobically via initial carboxylation and this has been observed under nitrate-reducing, sulfate-reducing and methanogenic conditions (Heider and Fuchs 1997). Anaerobic phenol degradation in Thauera aromatica, for instance, starts with the phosphorylation to form phenylphosphate, which is then carboxylated by phenylphosphate carboxylase, forming p-hydroxybenzoate (Schühle and Fuchs 2004). The p-hydroxybenzoate is further activated by a specific CoA ligase and the hydroxyl group is reductively removed (Heider and Fuchs 1997). However, in studies dealing with anaerobic benzene degradation, p-hydroxybenzoate has never been detected as intermediate. Evidence for benzene degradation via phenol formation and subsequent conversion to benzoate was found in a benzene-degrading methanogenic enrichment culture (Ulrich et al. 2005) and in a benzene-degrading iron-reducing enrichment (Botton and Parsons 2007).
In Dechloromonas aromatica RCB using nitrate as terminal electron acceptor initial hydroxylation of benzene to phenol and subsequent carboxylation to p-hydroxybenzoate, and loss of the hydroxyl group to form benzoate (or the CoA derivative, benzoyl-CoA) was found (Chakraborty and Coates 2005) (Fig. 4, pathway A). GC–MS analysis revealed the transient formation of phenol and benzoate (Chakraborty and Coates 2005). To ensure that the benzene degradation with nitrate as the electron acceptor in strain RCB was not due to a mechanism involving molecular oxygen, chemical reductants (0.5 mM sodium dithionite or 0.1 mM sodium ascorbate) were added to active benzene-degrading cultures to remove any traces of oxygen. In the presence of the reductants, 14C-benzene was still rapidly oxidized to 14CO2. Formation of phenol was detected in cultures amended with sodium dithionite, however, no phenol was detected in benzene-degrading cultures of strain RCB with oxygen as the electron acceptor. Although the benzene metabolism was not inhibited, addition of dithionite (0.5 mM) retarded the benzene degradation rate and subsequent phenol formation. Higher concentrations of dithionite (>1 mM) completely inhibited benzene degradation. However, even at a dithionite concentration of 0.5 mM, the phenol degradation rates remained unaffected. It was suggested that dithionite inhibited the initial hydroxylation step of benzene degradation in strain RCB rather than the subsequent oxidation of phenol to CO2 (Chakraborty and Coates 2005). To investigate the origin of the hydroxyl group of phenol, experiments with 18O-labeled H2O were performed. However, hardly any incorporation of 18O label into the hydroxyl group of phenol was found in benzene-degrading culture of strain RCB and it was concluded that H2O is not the source of the hydroxyl group. Unfortunately, it was not investigated whether the hydroxyl group of phenol could originate from nitrate, for instance using 18O-labeled NO3 −. On the other hand, it was investigated whether highly reactive hydroxyl free radicals (HO·) were involved in the hydroxylation of benzene to phenol in strain RCB. They found that hydroxyl free radical scavengers, such as sodium iodide (0.5 mM), propyl iodide (0.5 mM), 5,5-dimethyl-1-pyrroline-N-oxide, or mannitol (10 mM), inhibited benzene degradation with nitrate as the electron acceptor. These results suggested that hydroxyl free radicals play an important role in the benzene ring hydroxylation. Since phenol and benzoate have been detected in several studies as intermediates of anaerobic benzene degradation under different redox conditions, Chakraborty et al. (2005) suggested that a single universal pathway may exist for anaerobic benzene degradation and that it is similar to the pathway in strain RCB (Fig. 4, pathway A).
The genome of Dechloromonas aromatica strain RCB has been sequenced in 2005 (GenBank, http://www.ncbi.nlm.nih.gov/, accession no. CP000089) and has recently been published (Salinero et al. 2009). Dechloromonas aromatica has a single circular DNA chromosome with a length of 4,501,104 bp and 4,204 predicted protein coding genes. A function can be assigned to 69% of the protein coding genes. Several putative genes can be found in the genome sequence that could be involved in benzene metabolism. Recently, the genome of Dechloromonas aromatica was analyzed. Analysis indicated that gene families that constitute metabolic cycles presumed to create Dechloromonas aromatica’s environmental ‘foot-print’ indicate a high level of diversification between its predicted capabilities and those of its close relatives, Aromatoleum aromaticum strain EbN1 and Azoarcus BH72. For example, the benzylsuccinate synthase (bssABC) genes (responsible for fumarate addition to toluene) and genes involved in the central benzoyl-CoA pathway for monoaromatics are missing in the Dechloromonas aromatica genome. However, many enzymes responsible for aerobic aromatic degradation were found inside the genome (Salinero et al. 2009). As D. aromatica was described to degrade benzene anaerobically with nitrate, it cannot be excluded that during nitrate reduction oxygen is formed which initiates oxygenase-dependent benzene degradation as described below for chlorate. Oxygen formation linked to denitrification was recently shown for anaerobic methane-degrading culture (Ettwig et al. 2010). If a comparable oxygenic mechanism would exist under nitrate-reducing conditions in strain RCB, the addition of chemical reductants such as dithionite or ascorbate to the medium had no effect and thus oxidation by oxygenases can not be excluded.
Azoarcus strain DN11 could not grow on phenol, indicating that this strain uses a different anaerobic benzene degradative pathway than Dechloromonas strain RCB (Kasai et al. 2007). The benzene-degrading sulfate-reducing enrichment described by Musat and Widdel (2007) did not show metabolic activity towards phenol or toluene. Based on this observation the authors suggested that benzene degradation by the enrichment does not proceed via anaerobic hydroxylation to phenol or methylation to toluene (Musat and Widdel 2007). Recently, in a study with 13C6-labeled benzene as the growth substrate for a benzene-degrading iron-reducing enrichment culture, it was demonstrated that caution should be exercised in interpreting hydroxylated benzene derivatives as metabolic intermediates of anaerobic benzene degradation (Kunapuli et al. 2008). Phenol was identified as an intermediate, however, it was clearly shown that phenol was formed abiotically by autoxidation of benzene during the sampling and analysis procedure as a result of exposure to air. This results in the production of hydroxyl radicals which readily react with aromatic compounds such as benzene producing phenol and hydroxylation products of phenol. The authors suggested that autoxidation during sampling could also be a possible reason for non-detection of label in the hydroxyl group of phenol when the anaerobic benzene-degrading, denitrifying Dechloromonas strain RCB was incubated with labeled H 182 O (Chakraborty and Coates 2005). Chakraborty and Coates (2005) ruled out water or air as possible sources of the hydroxyl group, as only 1% label incorporation was observed when the culture was incubated with H 182 O and also phenol production occurred when dithionite was added to scavenge any oxygen present. As water is the only reasonable oxygen source for microorganisms in the absence of molecular oxygen, the hydroxyl oxygen had to derive from air, perhaps by autoxidation during sampling (Kunapuli et al. 2008).
-
B.
Benzene methylation
Benzene is susceptible to substitution by strong electrophiles as in Friedel–Crafts alkylation. The electrophile could be the H3C+ unit derived from S-adenosylmethionine. Biologically mediated methylation of benzene to toluene has previously been observed with human bone marrow incubated with S-adenosylmethionine (Flesher and Myers 1991). Methylation of benzene to toluene is energetically favorable using S-adenosylmethionine as the methyl donor (Coates et al. 2002). Methylation has also been proposed for the anaerobic activation of another unsubstituted aromatic hydrocarbon, naphthalene. This reaction has been found in a sulfate-reducing enrichment culture and the methyl group might possibly be generated from bicarbonate via a reverse CO-dehydrogenase pathway (Safinowski and Meckenstock 2005).
The first direct evidence for benzene methylation was recently found by Ulrich et al. (2005). In a nitrate-reducing and methanogenic enrichment culture [ring-13C]-toluene was detected by GC/MS when 13C-benzene was added to these cultures. The nature of the methyl group and the subsequent toluene degradation pathway are not known, but degradation via the benzylsuccinate synthase pathway was hypothesized (Ulrich et al. 2005).
-
C.
Benzene carboxylation
Carboxylation of benzene to form benzoate (or benzoyl-CoA) has been reported (Caldwell and Suflita 2000; Phelps et al. 2001). In a sulfate-reducing enrichment, deuterated benzoate (D5) was detected when deuterated benzene (D6) was added to the enrichment, but phenol was not detected (Phelps et al. 2001). The carboxyl group of benzoate was not labeled when 13C-bicarbonate or 13C-acetic acid were added to the enrichment. This is in contrast to the proposed pathway for anaerobic naphthalene and phenanthrene metabolism, where carboxylation of the aromatic ring by carbon dioxide (or bicarbonate) is the initial activation step (Zhang and Young 1997; Meckenstock et al. 2000). The lack of incorporation of 13C-labeled bicarbonate into the aromatic ring of benzene (Phelps et al. 2001) and the suggestion that the carboxyl group of benzoate may be derived from benzene itself (Caldwell and Suflita 2000) could argue against a direct carboxylation mechanism. However, Kunapuli et al. (2008) demonstrated that in a benzene-degrading iron-reducing enrichment culture the carboxyl group of benzoate derived from the bicarbonate buffer, indicating a direct carboxylation of benzene. With 13C6-labeled benzene as the growth substrate, C7-labeled benzoate appeared, indicating that the carboxyl group of benzoate derived from CO2 that was produced from mineralization of labeled benzene. This was confirmed by growing the culture in 13C-bicarbonate-buffered medium with unlabeled benzene as the substrate, as the label appeared in the carboxyl group of benzoate produced. When using unlabeled buffer and high concentrations of 13C-labeled benzene, a significant portion of the bicarbonate buffer became labeled by the release of 13HCO3 from complete benzene oxidation. As the carboxyl group stems from the bicarbonate buffer, this eventually leads to the generation of 13C7-benzoate. One could speculate that this finding might also explain why other authors could detect 13C-carboxy-labeled benzoate when applying labeled benzene together with unlabeled bicarbonate buffer (Caldwell and Suflita 2000). Recently, benzoate was found as intermediate compound in a benzene-degrading sulfate-reducing enrichment, supporting a direct carboxylation of benzene as the initial activation mechanism. However, additional reactions leading to the formation of benzoate could not be excluded definitely (Laban et al. 2009).
2.4.4 Characterizing and assessing anaerobic benzene degradation: isotopic fractionation
Biodegradation is the most important process leading to a decrease in BTEX concentrations in soil and groundwater. Therefore, the evaluation of in situ BTEX biodegradation is essential for the implementation of groundwater management strategies such as natural attenuation (NA). Traditional methods used to confirm bioremediation in the field included monitoring decreases in contaminant concentrations and electron acceptors, and increases in microbial (by)products (such as carbon dioxide). This is difficult for benzene because of e.g. high background HCO3 − concentrations. However, the challenge is to prove biodegradation in the field, since other processes such as volatilization, dispersion, and sorption can cause loss of the contaminant, and accurate mass balances are difficult to obtain (Mancini et al. 2003). In recent years, stable isotope fractionation analysis has gained attention as a tool for characterizing and assessing in situ biodegradation of organic pollutants in contaminated aquifers (Meckenstock et al. 2004b; Fischer et al. 2007). This concept relies on the fractionation of stable isotopes during the microbial degradation, leading to an enrichment of heavier isotopes in the residual fraction of a pollutant. Isotopes of elements such as carbon (12C and 13C) and hydrogen (1H and 2H) react at slightly different rates during mass-differentiating reactions. During biodegradation, bonds containing the lighter isotopes are preferentially broken, causing the remaining contaminant to be enriched in the heavier isotopes compared to the original isotopic value. A large isotopic fractionation effect (primary isotope effect) can be observed if a bond (e.g. C–H bond) containing the element of interest is broken or formed in the rate-limiting step. Stable isotope fractionation has the ability to identify biodegradation of aromatic hydrocarbons in the field and to distinguish contaminant mass loss due to biodegradation versus that due to (abiotic) physicochemical processes.
In studies concerning stable isotope fractionation of benzene, carbon (12C and 13C) and hydrogen (1H and 2H) isotope ratios are expressed in the delta notation (δ13C and δ2H) in per mil (‰) units according to the following equation:
In this equation, R sample and R standard are the 13C/12C or 2H/1H-ratio of the sample and an international standard, respectively. Vienna Pee Dee Belemnite (VPDB) was used as the standard for the analysis of carbon isotope signature and Vienna Standard Mean Ocean Water (VSMOW) was used as the standard for the detection of hydrogen isotope ratios. For the description of isotope fractionation of biochemical reactions the Rayleigh equation can be used:
where R t , C t and R 0, C 0 are the stable isotope ratios and concentrations of a compound at a given point in time and at the beginning of a transformation reaction, respectively. The enrichment factor ε [‰] provides the link between the changes in stable isotope ratios (R t /R 0) and the changes in the concentrations (C t /C 0) (Fischer et al. 2007). In recently published laboratory experiments, carbon and hydrogen isotope discrimination were determined for aerobic and anaerobic benzene biodegradation (Hunkeler et al. 2001; Mancini et al. 2002; Mancini et al. 2003; Fischer et al. 2008; Mancini et al. 2008; Fischer et al. 2009) (see Table 4).
The biodegradation pathway of the aerobic cultures used in the study by Fischer et al. (2008) was known, i.e. mono- or dihydroxylation, and therefore a relation between enrichment factors and biodegradation pathway could be deduced. Their results indicate that carbon enrichments factors for dihydroxylation of benzene are significantly smaller compared to monohydroxylation and anaerobic degradation. No systematic differences are given for carbon enrichments factors of monohydroxylation and anaerobic degradation. The carbon enrichment factors for monooxygenase reaction are higher as benzene dihydroxylation, which can be expected when a cleavage of a C–H bond is involved to some extent as revealed in several studies (Hanzlik et al. 1984; Mitchell et al. 2003). Hydrogen enrichment factors for benzene monohydroxylation are higher compared to dihydroxylation. Hydrogen enrichment factors for benzene degradation under anaerobic (nitrate-reducing, sulfate-reducing and methanogenic) conditions were significantly higher than for aerobic benzene degradation. The variability in the hydrogen isotope fractionation for the benzene biodegradation can be explained by different initial reaction mechanisms. Carboxylation, methylation or hydroxylation of benzene might cause the cleavage of a C–H bond under anaerobic condition. In contrast, aerobic benzene biodegradation should only lead to secondary hydrogen isotope effects if a dihydroxylation reaction occurs, because no hydrogen bond is broken in the first irreversible reaction step of the transformation pathway (Hunkeler et al. 2001; Mancini et al. 2003, 2008) or low isotope effect can be expected when an epoxylation is the initial reaction step (Mitchell et al. 2003). Similarly, the transformation of benzene by mammalian and methane monooxygenases exhibited a small or even negligible hydrogen isotope effect which was explained by the absence of C–H bond cleavage in the initial enzymatic degradation step (Hunkeler et al. 2001).
Dual parameter plots of carbon and hydrogen isotopic data (∆δ2H/∆δ13C) demonstrated that these plots from each culture were linear, suggesting a consistent reaction mechanism as degradation proceeded. Methanogenic and sulfate-reducing cultures showed consistently higher slopes compared to nitrate-reducing cultures providing evidence for different initial reaction mechanisms (Fischer et al. 2008; Mancini et al. 2008).
3 BTEX biodegradation with (per)chlorate
From information above it is quite clear that major differences exist between BTEX degradation under aerobic and anaerobic conditions. Bacteria that degrade BTEX in the absence of oxygen do not have the ability to employ degradation pathways with mono- or dioxygenases. Therefore, all the pathways in anaerobic bacteria are oxygenase-independent. However, the situation changes when the bacteria are able to produce oxygen during anaerobic respiration. This is the case for bacteria that respire with perchlorate (ClO4 −) and chlorate (ClO3 −).
3.1 (Per)chlorate reducing bacteria
It has been known for more than 50 years that microorganisms can reduce oxyanions of chlorine such as perchlorate and chlorate under anaerobic conditions. The high reduction potential of perchlorate (ClO4 −/Cl− E 0 = 1.287 V) and chlorate (ClO3 −/Cl− E 0 = 1.03 V) makes them ideal electron acceptors for microbial metabolism. When the reduction of (per)chlorate is coupled to electron-transport phosphorylation, microorganisms are able to grow by (per)chlorate reduction. Until 1996, little was known about the biochemical pathway of (per)chlorate reduction. Many denitrifying bacteria are able to reduce (per)chlorate, but in general, this reduction is not coupled to growth (Coates and Achenbach 2004). These denitrifying bacteria most probably cannot grow on chlorate because of the accumulation of toxic chlorite during (per)chlorate reduction (Fig. 5). Therefore, bacteria are able to grow by dissimilatory (per)chlorate reduction when: (1) these oxyanions can be used as electron acceptor and (2) the toxic intermediate chlorite is converted into chloride and oxygen.

(Per)chlorate reduction scheme including involved enzymes
Many dissimilatory (per)chlorate reducing bacteria have been isolated and described. A comprehensive overview is given in literature (Coates and Achenbach 2004). (Per)chlorate reducing bacteria are phylogenetically diverse and can be found in the Alpha-, Beta-, Gamma- and Epsilonproteobacteria. Almost all (per)chlorate-reducing species in the Betaproteobacteria belong to either the Dechloromonas or Azospira (formerly Dechlorosoma) genus. (Per)chlorate reducing bacteria have been isolated from a broad diversity of environments, including both pristine and contaminated soils and sediments (Coates et al. 1999b). This was remarkable, because of the limited natural occurrence of (per)chlorate. However, these bacteria have diverse metabolic capabilities and this could account for their presence in environments in which (per)chlorate was not found. Phenotypic characterization revealed that the (per)chlorate reducing bacteria exhibited a broad range of metabolic capabilities, including the oxidation of hydrogen, simple organic acids and alcohols, aromatic hydrocarbons, hexoses, reduced humic substances, both soluble and insoluble ferrous iron, and hydrogen sulfide. All of the known (per)chlorate reducing bacteria are facultatively anaerobic or microaerophilic microorganisms, which is understandable because oxygen is produced during (per)chlorate reduction. Some, but not all, (per)chlorate reducing bacteria can use nitrate as terminal electron acceptor for growth. So far, all microorganisms capable of perchlorate reduction can also use chlorate as the electron acceptor. However, microorganisms capable of chlorate reduction are not always capable of using perchlorate as the electron acceptor (Coates and Achenbach 2004).
3.2 Potential of (per)chlorate-reducing bacteria in bioremediation
(Per)chlorate reducing bacteria can be used for the treatment of perchlorate- and chlorate-containing waste streams and groundwater (Logan 1998; Urbansky 1998), e.g. the bioremediation of perchlorate-contaminated groundwater in a packed bed biological reactor (Losi et al. 2002) or the treatment of chlorate and perchlorate contaminated water using permeable barriers containing vegetable oil (Hunter 2002). The ability of (per)chlorate-reducing bacteria to produce oxygen can also be applied in other bioremediation processes. The dismutation of chlorite by (per)chlorate-reducing bacteria in anoxic environments can produce extracellular O2. This O2 can for instance be used by bacteria to degrade hydrocarbons. Especially, hydrocarbons that are slowly degraded or persistent under anaerobic conditions could be degraded by this mechanism (Coates et al. 1999a; Mehboob et al. 2010). An example of such a compound is benzene.
Molecular oxygen can be introduced into the anoxic zones of a contaminated environment by injection of compressed air or O2 below the water table (Coates et al. 1999a), but this is a costly and inefficient process due to the low solubility of oxygen. Hydrogen peroxide is often used as an additional soluble O2 source, but this process has some disadvantages, such as toxicity of hydrogen peroxide to many bacteria. Also solid O2-releasing compounds can be used, such as magnesium peroxide (MgO2) or calcium peroxide (CaO2). These compounds consist of powdery material and can also be used as injected slurries for in situ bioremediation. These O2-releasing compounds have some advantages, but the high costs of the O2-releasing compound and the difficulty to evenly distribute the compounds (i.e. the O2) over a large area, for instance a contaminated aquifer, may be major disadvantages. Therefore, chlorite dismutation by (per)chlorate-reducing bacteria offers a good alternative strategy to supply extracellular oxygen to the aerobic hydrocarbon-oxidizing population as was shown by Rikken et al. (1996). Cell suspensions of (per)chlorate reducing cells of strain GR-1 showed formation of (extracellular) oxygen upon the addition of chlorite (Rikken et al. 1996).
The potential use of dissimilatory (per)chlorate reducing bacteria for bioremediation (of soils and sediments) has already been recognized in previous studies (Coates et al. 1999a; Logan and Wu 2002). The amendment of (per)chlorate-reducing bacteria and chlorite to an anoxic soil led to complete degradation of 14C-benzene to 14C-carbon dioxide. Furthermore, the addition of chlorite in anoxic soils samples, inoculated with starved cells of the (per)chlorate reducer Dechloromonas agitata strain CKB, showed that 14C-benzene was rapidly oxidized to 14C-carbon dioxide. This observation further enhances the applicability of these kind or organisms to in situ bioremediation (Coates et al. 1999a). In another study, increased rates of toluene degradation were observed in sand columns inoculated with both (aerobic) toluene-degrading and chlorate-reducing enrichment cultures, which indicated a symbiotic relationship between the toluene-degrading bacteria and chlorate-reducing bacteria (Logan and Wu 2002). Furthermore, the addition of chlorate to a soil column polluted with benzene showed removal of benzene coupled with chlorate reduction (Tan et al. 2006) and the addition of chlorate to groundwater at a site in the Netherlands resulted in the complete removal of benzene (Langenhoff et al. 2009). Recently, the degradation of benzene coupled to (per)chlorate reduction was studied in more detail in enrichments and pure culture studies (Weelink et al. 2007, 2008). A benzene-degrading chlorate-reducing enrichment culture was obtained with reactor material, which was originally inoculated with mixed material from a wastewater treatment plant and soil samples obtained from a location contaminated with benzene (Tan et al. 2004). This stable enrichment culture degraded benzene at rates similar to reported aerobic benzene degradation rates, but 20–1,650 times faster than reported for anaerobic benzene degradation (Weelink et al. 2007). The different partners in the enrichment culture were identified as a bacteria closely related to either Alicycliphilus denitrificans K601T (Mechichi et al. 2003), or Zoogloea resiniphila PIV-3A2w, or Mesorhizobium sp. WG or Stenotrophomonas acidaminiphila (Costa et al. 2000; Assih et al. 2002; Probian et al. 2003). DGGE analysis of cultures grown with different electron donors and acceptors indicated that the bacterium related to Alicycliphilus denitrificans K601 is able to degrade benzene coupled to chlorate reduction. The role of the other bacteria could not be conclusively determined. The bacterium related to Mesorhizobium sp. WG could be enriched with benzene and oxygen, but not with acetate and chlorate, while the bacterium related to Stenotrophomonas acidaminophila grew with acetate and chlorate, but not with benzene and oxygen. As during chlorate reduction oxygen is produced, an aerobic benzene degradation pathway is most likely in this enrichment. Furthermore, as one of these bacteria seems to be able to oxidize benzene aerobically, but not able to reduce chlorate and another bacterium seems able to reduce chlorate but not oxidize benzene, cross-feeding involving interspecies oxygen transfer is a likely mechanism. However, thus far attempts to demonstrate interspecies oxygen transfer between the bacteria in the consortium were not successful.
From the benzene-degrading chlorate-reducing enrichment culture, a bacterium (strain BC) was isolated (Weelink et al. 2008) that is able to degrade benzene coupled to chlorate reduction. Strain BC grows on benzene and some other aromatic compounds with oxygen, or in the absence of oxygen with chlorate as the electron acceptor. Strain BC is a denitrifying bacterium, but it is not able to grow on benzene with nitrate. The closest cultured relative is Alicycliphilus denitrificans K601 (type strain), a cyclohexanol-degrading nitrate-reducing betaproteobacterium. The 16S rRNA gene sequences are 99.7% similar, while genomic DNA was 74.5 ± 3.5% similar as indicated by DNA-DNA hybridization. However, physiological differences are apparent. Strain BC is not able to degrade cyclohexanol, while strain K601T lacks the ability to reduce chlorate.
During chlorate reduction, oxygen is produced by the dismutation of chlorite. Therefore, it seems likely that benzene is degraded via an aerobic degradation pathway in strain BC. An aerobic degradation pathway would require chlorate reductase, chlorite dismutase and oxygenase enzymes. Chlorate reductase and chlorite dismutase activities were determined in cell extracts of strain BC. Subsequently, the total genomic DNA of strain BC was screened for sequence signatures indicating the presence of genes that can potentially encode the key enzymes, i.e. chlorite dismutases, ring activating oxygenases and ring cleavage oxygenases. Chlorite dismutase activity was detected in cell extracts of strain BC, but no signal was obtained using the primers previously reported as designed for the detection of the group of genes assigned to this function (Bender et al. 2004), but the genetic information available for this group of genes is still very scarce (Weelink et al. 2008). Screening of the genomic DNA of strain BC for ring activating oxygenases and ring cleavage oxygenases resulted in the detection of a putative benzene monooxygenase (BC–BMOa) gene and a putative catechol 2,3-dioxygenase (BC–C23O) gene. The sequence features and the physiological data support the hypothesis that these sequences encode functional and active benzene monooxygenases and catechol 2,3-dioxygenases used by strain BC (Weelink et al. 2008).
Based on physiological, genetic and biochemical experiments, a benzene degradation pathway with chlorate as the electron acceptor was proposed in strain BC (Fig. 6). In this pathway, oxygen produced during chlorate reduction is used in oxygenase reactions, i.e. benzene conversion to catechol by two sequential monooxygenase reactions (by benzene monooxygenase, BC-BMOa) and catechol conversion to 2-hydroxymuconic semialdehyde by catechol 2,3-dioxygenase (by catechol-2,3 dioxygenase, BC–C23O). Dihydroxylation of benzene to catechol cannot be ruled out, but genes encoding benzene dioxygenases were not detected with the primers used in this study. Oxygen formed in the dismutation of chlorite may not only be used as terminal electron acceptor in strain BC but is also used to attack molecules by means of oxygenases. This demonstrates the existence of aerobic benzene bacterial biodegradation pathways under essentially anaerobic conditions by the concerted action of chlorite dismutases, providing the metabolic oxygen needed by aromatic activating and cleaving oxygenases. Thus, it was demonstrated that aerobic pathways can be employed under conditions where no external oxygen is supplied (Weelink et al. 2008).

Proposed benzene degradation pathway with chlorate as the electron acceptor in strain BC. A proposed stoichiometric reactions involved in benzene degradation with chlorate as the electron acceptor in strain BC. [H] stands for reducing equivalents. Benzene metabolism involves the hydroxylation of benzene to phenol, phenol hydroxylation to catechol, extradiol (meta-) cleavage of catechol to 2-hydroxymuconic semialdehyde (2-hms) and complete oxidation of 2-hms to carbon dioxide and reducing equivalents. Chlorate metabolism involves the reduction of chlorate to chlorite, dismutation of chlorite into chloride and oxygen and subsequent reduction of oxygen to water
Biodegradation of benzene by Alicycliphilus denitrificans strain BC under both oxic and chlorate-reducing conditions was also included in the study by Fischer et al. (2008) concerning carbon and hydrogen fractionation during benzene biodegradation (Table 4). The carbon enrichment factors for strain BC were higher than reported for dihydroxylation activation of benzene, but corresponded more to reported range of carbon enrichment factors for monohydroxylation. The hydrogen enrichment factors observed for strain BC were in the same range as that observed for the monooxygenase-catalyzed reactions (Table 4). The observed carbon and hydrogen enrichment factors strongly indicate that benzene is attacked by a monooxygenase under aerobic and chlorate-reducing conditions in strain BC. These results confirmed the previously mentioned results of the molecular biology experiments with strain BC in which also evidence for benzene degradation by monooxygenases was obtained.
The genome of strain BC has been sequenced by the DOE Joint Genome Institute (JGI) (http://www.jgi.doe.gov/genome-projects/). With the available genome sequences, insight into the molecular mechanism of intracellular oxygen transfer and the regulation of chlorate-dependent and chlorate-independent growth can be obtained and the suggested pathway for intracellular oxygen transfer can be confirmed. Besides intracellular oxygen transfer, the concept of interspecies oxygen transfer deserves further study. Insight into interspecies oxygen transfer may broaden the degradation possibilities of microbial communities in polluted soils. Introduction of chlorate-reducing strains in a polluted soil may lead to growth of non-chlorate-reducing aerobic bacteria, with the capacity to metabolize a broad spectrum of substrates.
In situ bioremediation by using naturally occurring microorganisms or introduced microorganisms is often for several reasons (e.g. financial reasons) a very attractive option. (Per)chlorate-enhanced bioremediation may become an important and attractive process for stimulated in situ anaerobic soil remediation. The advantage of (per)chlorate-enhanced bioremediation compared to oxygen injection is that (per)chlorate has a higher solubility than oxygen. Unlike oxygen, which is soluble in the mM-range, (per)chlorate is soluble in the M-range. Furthermore, (per)chlorate can be considered as a slow-oxygen release compound. The advantage of using (per)chlorate over other oxygen-releasing chemicals is that oxygen production is biologically controlled. The conversion of (per)chlorate to chlorite requires reducing equivalents that are formed in the oxidation of an organic compound. Without an electron donor (per)chlorate is chemically stable. This is not the case for other oxygen-releasing chemicals, like MgO2 and CaO2. (Per)chlorate reduction and subsequent oxygen formation can also be beneficial for other compounds that are anaerobically difficult to degrade but that are readily degraded aerobically. Compounds of interest are naphthalene, monochlorobenzene (MCB), vinylchloride (Vc), methyl tertiary butylether (MTBE), long and short chain alkanes and a variety of specific compounds like aromatic amines.
When (per)chlorate addition to a contaminated aquifer will be applied as bioremediation technique, it has to be taken into account that (per)chlorate itself is a pollutant as well. Perchlorate is a solid rocket propellant, and chlorate is applied as herbicide and defoliant and is formed during paper bleaching (Versteegh et al. 1993; Coates and Achenbach 2004). However, (per)chlorate is also naturally present in Chilean nitrate deposits and other mineral deposits. It was found recently that the electric discharge in chloride-containing aerosols may be a major source of naturally occurring perchlorate (Dasgupta et al. 2005). Biological (per)chlorate reduction may therefore be a rather ancient process. (Per)chlorate-reducing bacteria are known to use a wide variety of electron donors for growth, which include organic acids (such as acetate and lactate) as well as inorganic compounds like hydrogen and sulfide (Logan 1998; Wolterink et al. 2002; Coates and Achenbach 2004). Because (per)chlorate is a pollutant itself, the application of (per)chlorate addition to contaminated aquifers has to be performed in close consultation with the competent authorities. Remaining (per)chlorate after the organic pollutants have been degraded, could be reduced by the introduction of easily degradable organic compounds to the remediated site. As a result, no environmentally harmful compounds are left after the bioremediation process.
4 Future perspectives on anaerobic benzene degradation
Although anaerobic alkylbenzene (toluene, ethylbenzen and xylene) degradation has been studied in some detail, understanding of the anaerobic degradation of benzene is still in its infancy. This is mostly due to the lack of available pure or enrichment cultures that can degrade benzene anaerobically and the extremely slow growth of the microorganisms in the available enrichment cultures. Apart from the environmental significance of benzene as a contaminant with carcinogenic potential and high toxicity, anaerobic activation of benzene is probably one of the most intriguing reactions in microbial degradation today because the activation of a non-substituted aromatic ring in the absence of molecular oxygen constitutes an unprecedented biochemical reaction (Kunapuli et al. 2008). Furthermore, the evaluation of in situ BTEX biodegradation is essential for the implementation of anaerobic BTEX bioremediation strategies. Traditional methods used to confirm bioremediation in the field include monitoring the contaminant concentrations and electron acceptors, and possible microbial byproducts. However, the challenge is to prove biodegradation in the field, since other processes such as volatilization, dispersion, and sorption can cause contaminant attenuation, and accurate mass balances are difficult to obtain (Mancini et al. 2003). In recent years, stable isotope fractionation analysis has gained attention as a tool for characterizing and assessing in situ biodegradation of organic pollutants in contaminated aquifers. Thus, future studies on anaerobic benzene degradation should focus on:
-
The identification of the bacteria responsible for anaerobic benzene degradation.
-
The elucidation of the anaerobic benzene degradation pathway.
-
The prove and quantification of anaerobic benzene degradation in the field (in particular stable isotope fractionation).
References
Achenbach LA, Michaelidou U, Bruce RA, Fryman J, Coates JD (2001) Dechloromonas agitata gen. nov., sp. nov. and Dechlorosoma suillum gen. nov., sp nov., two novel environmentally dominant (per)chlorate-reducing bacteria and their phylogenetic position. Int J Syst Evol Microbiol 51:527–533
Aihara J (1992) Why aromatic compounds are stable. Sci Am 266:62–68
Anders HJ, Kaetzke A, Kämpfer P, Ludwig W, Fuchs G (1995) Taxonomic position of aromatic-degrading denitrifying Pseudomonad strains K 172 and Kb 740 and their description as new members of the genera Thauera, as Thauera aromatica sp. nov., and Azoarcus, as Azoarcus evansii sp. nov., respectively, members of the beta subclass of the Proteobacteria. Int J Syst Bacteriol 45:327–333
Anderson RT, Lovley DR (1999) Napthtalene and benzene degradation under Fe(III)-reducing conditions in petroleum-contaminated aquifers. Bioremed J 3:121–135
Anderson R, Lovley D (2000) Anaerobic bioremediation of benzene under sulfate-reducing conditions in a petroleum-contaminated aquifer. Environ Sci Technol 34:2261–2266
Anderson RT, Rooney-Varga JN, Gaw CV, Lovley DR (1998) Anaerobic benzene oxidation in the Fe(III) reduction zone of petroleum-contaminated aquifers. Environ Sci Technol 32:1222–1229
Aronson D, Howard PH (1997) Anaerobic biodegradation of organic chemicals in groundwater: a summary of field and laboratory studies. In: North Syracuse, NY Evironmental Science Center, Syracuse Research Corporation, pp 1–268
Assih EA, Ouattara AS, Thierry S, Cayol JL, Labat M, Macarie H (2002) Stenotrophomonas acidaminiphila sp. nov., a strictly aerobic bacterium isolated from an upflow anaerobic sludge blanket (UASB) reactor. Int J Syst Evol Microbiol 52:559–568
Badham HJ, Winn LM (2007) Investigating the role of the aryl hydrocarbon receptor in benzene-initiated toxcicity in vitro. Toxicology 229:177–185
Ball HA, Johnson HA, Reinhard M, Spormann AM (1996) Initial reactions in anaerobic ethylbenzene oxidation by a denitrifying bacterium, strain EB1. J Bacteriol 178:5755–5761
Beller HR, Spormann AM (1997a) Benzylsuccinate formation as a means of anaerobic toluene activation by sulfate-reducing strain PRTOL1. Appl Environ Microbiol 63:3729–3731
Beller HR, Spormann AM (1997b) Anaerobic activation of toluene and o-xylene by addition to fumarate in denitrifying strain T. J Bacteriol 179:670–676
Beller HR, Spormann AM (1998) Analysis of the novel benzylsuccinate synthase reaction for anaerobic toluene activation based on structural studies of the product. J Bacteriol 180:5454–5457
Beller HR, Spormann AM (1999) Substrate range of benzylsuccinate synthase from Azoarcus sp strain T. FEMS Microbiol Lett 178:147–153
Beller HR, Ding W, Reinhard M (1995) Byproducts of anaerobic alkylbenzene metabolism useful as indicators of in situ bioremediation. Environ Sci Technol 29:2864–2870
Beller HR, Spormann AM, Sharma PK, Cole JR, Reinhard M (1996) Isolation and characterization of a novel toluene-degrading, sulfate-reducing bacterium. Appl Environ Microbiol 62:1188–1196
Bender KS, Rice MR, Fugate WH, Coates JD, Achenbach LA (2004) Metabolic primers for detection of (per)chlorate-reducing bacteria in the environment and phylogenetic analysis of cld gene sequences. Appl Environ Microbiol 70:5651–5658
Biegert T, Fuchs G, Heider F (1996) Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur J Biochem 238:661–668
Botton S, Parsons JR (2006) Degradation of BTEX compounds under iron-reducing conditions in contaminated aquifer microcosms. Environ Toxicol 25:2630–2638
Botton S, Parsons JR (2007) Degradation of BTX by dissimilatory iron-reducing cultures. Biodegradation 18:373–381
Botton S, van Harmelen M, Braster M, Parsons JR, Roling WFM (2007) Dominance of Geobacteraceae in BTX-degrading enrichments from an iron-reducing aquifer. FEMS Microbiol Ecol 62:118–130
Burland SM, Edwards EA (1999) Anaerobic benzene biodegradation linked to nitrate reduction. Appl Environ Microbiol 65:529–533
Caldwell ME, Suflita JM (2000) Detection of phenol and benzoate as intermediates of anaerobic benzene biodegradation under different terminal electron-accepting conditions. Environ Sci Technol 34:1216–1220
Carmona M, Zamarro M, Blázquez B, Durante-Rodríguez G, Juárez J, Valderrama J et al (2009) Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol Mol Biol Rev 73:71–133
Cervantes FJ, Dijksma W, Duong Dac T, Ivanova A, Lettinga G, Field JA (2001) Anaerobic mineralization of toluene by enriched sediments with quinones and humus as terminal electron acceptors. Appl Environ Microbiol 67:4471–4478
Chakraborty R, Coates JD (2004) Anaerobic degradation of monoaromatic hydrocarbons. Appl Microbiol Biotechnol 64:437–446
Chakraborty R, Coates JD (2005) Hydroxylation and carboxylation-two crucial steps of anaerobic benzene degradation by Dechloromonas strain RCB. Appl Environ Microbiol 71:5427–5432
Chakraborty R, O’Connor SM, Chan E, Coates JD (2005) Anaerobic degradation of benzene, toluene, ethylbenzene and xylene by Dechloromonas strain RCB. Appl Environ Microbiol 71:8649–8655
Chang W, Um Y, Holoman TRP (2005) Molecular characterization of anaerobic microbial communities from benzene-degrading sediments under methanogenic conditions. Biotechnol Prog 21:1789–1794
Chaudhuri BK, Wiesmann U (1995) Enhanced anaerobic degradation of benzene by enrichment of mixed microbial culture and optimization of the culture medium. Appl Microbiol Biotechnol 43:178–187
Christensen TH, Kjeldsen P, Bjerg PL, Jensen DL, Christensen JB, Baun A et al (2001) Biogeochemistry of landfill leachate plumes. Appl Geochem 16:659–718
Coates JD, Achenbach LA (2004) Microbial perchlorate reduction: rocket-fuelled metabolism. Nat Rev Microbiol 2:569–580
Coates JD, Anderson RT, Lovley DR (1996a) Oxidation of polycyclic aromatic hydrocarbons under sulfate-reducing conditions. Appl Environ Microbiol 62:1099–1101
Coates JD, Anderson RT, Woodward JC, Phillips EJP, Lovley DR (1996b) Anaerobic hydrocarbon degradation in petroleum-contaminated harbor sediments under sulfate-reducing and artificially imposed iron-reducing conditions. Environ Sci Technol 30:2784–2789
Coates JD, Bruce RA, Patrik J, Achenbach LA (1999a) Hydrocarbon bioremediative potential of perchlorate-reducing bacteria. Bioremed J 3:323–334
Coates JD, Michaelidou U, Bruce RA, O’Connor SM, Crespi JN, Achenbach LA (1999b) Ubiquity and diversity of dissimilatory (per)chlorate-reducing bacteria. Appl Environ Microbiol 65:5234–5241
Coates JD, Bhupathiraju VK, Achenbach LA, McInerney MJ, Lovley DR (2001a) Geobacter hydrogenophilus, Geobacter chapellei and Geobacter grbiciae, three new, strictly anaerobic, dissimilatory Fe(III)-reducers. Int J Syst Evol Microbiol 51:581–588
Coates JD, Chakraborty R, Lack JG, O’Connor SM, Cole KA, Bender KS, Achenbach LA (2001b) Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature 411:1039–1043
Coates JD, Chakraborty R, McInerney MJ (2002) Anaerobic benzene biodegradation—a new era. Res Microbiol 153:621–628
Coschigano PW, Wehrman TS, Young LY (1998) Identification and analysis of genes involved in anaerobic toluene metabolism by strain T1: Putative role of a glycine free radical. Appl Environ Microbiol 64:1650–1656
Costa C, Dijkema C, Friedrich M, Garcia-Encina P, Fernandez-Polanco F, Stams AJM (2000) Denitrification with methane as electron donor in oxygen-limited bioreactors. Appl Microbiol Biotechnol 53:754–762
Da Silva MLB, Alvarez PJJ (2007) Assessment of anaerobic benzene degradation potential using 16S rRNA gene-targeted real-time PCR. Environ Microbiol 9:72–80
Dagley S (1985) Microbial metabolism of aromatic compounds. In: Moo-Young M (ed) The principles of biotechnology. Pergamon Press, Oxford, vol I, pp 483–505
Dagley S (1986) Biochemistry of aromatic hydrocarbon degradation in Pseudomonads. In: Sokatch JR (ed) The bacteria. Academic Press, New York, vol 10, pp 527–555
Dasgupta PK, Martinelango PK, Jackson WA, Anderson TA, Tian K, Tock RW, Rajagopalan S (2005) The origin of naturally occurring perchlorate: the role of atmospheric processes. Environ Sci Technol 39:1569–1575
Dean BJ (1978) Genetic toxicology of benzene, toluene, xylenes and phenols. Mutat Res 47:75–97
Dolfing J, Zeyer J, Bindereicher P, Schwarzenbach RP (1990) Isolation and characterization of a bacterium that mineralizes toluene in the absence of molecular-oxygen. Arch Microbiol 154:336–341
Edwards EA, Grbic-Galic D (1992) Complete mineralization of benzene by aquifer microorganisms under strictly anaerobic conditions. Appl Environ Microbiol 58:2663–2666
Edwards EA, Wills LE, Reinhard M, Grbic-Galic D (1992) Anaerobic degradation of toluene and xylene by aquifer microorganisms under sulfate-reducing conditions. Appl Environ Microbiol 58:794–800
Ettwig KF, Butler MK, Le Paslier D, Pelletier E, Mangenot S, Kuypers MMM et al (2010) Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464:543–548
Evans PJ, Mang DT, Kim KS, Young LY (1991) Anaerobic degradation of toluene by a denitrifying bacterium. Appl Environ Microbiol 57:1139–1145
Fischer A, Theuerkorn K, Stelzer N, Gehre M, Thullner M, Richnow HH (2007) Applicability of stable isotope fractionation analysis for the characterization of benzene biodegradation in a BTEX-contaminated aquifer. Environ Sci Technol 41:3689–3696
Fischer A, Herklotz I, Herrmann S, Thullner M, Weelink SAB, Stams AJM et al (2008) Combined carbon and hydrogen isotope fractionation investigations for elucidating benzene biodegradation pathways. Environ Sci Technol 42:4356–4363
Fischer A, Gehre M, Breitfeld J, Richnow HH, Vogt C (2009) Carbon and hydrogen isotope fractionation of benzene during biodegradation under sulfate-reducing conditions: a laboratory to field site approach. Rapid Commun Mass Spec 23:2439–2447
Flesher JW, Myers SR (1991) Methyl-substitution of benzene and toluene in preparations of human bone-marrow. Life Sci 48:843–850
Foght J (2008) Anaerobic biodegradation of aromatic hydrocarbons: pathways and prospects. J Mol Microbiol Biotechnol 15:93–120
Frazer AC, Coschigano PW, Young LY (1995) Toluene metabolism under anaerobic conditions: a review. Anaerobe 1:293–303
Fries MR, Zhou JH, Chee-Sanford J, Tiedje JM (1994) Isolation, characterization, and distribution of denitrifying toluene degraders from a variety of habitats. Appl Environ Microbiol 60:2802–2810
Fries MR, Forney LJ, Tiedje JM (1997) Phenol- and toluene-degrading microbial populations from an aquifer in which successful trichloroethene cometabolism occurred. Appl Environ Microbiol 63:1523–1530
Fuchs G (2008) Anaerobic metabolism of aromatic compounds. Ann NY Acad Sci 1125:82–99
Gibson DT, Subramanian V (1984) Microbial degradation of aromatic hydrocarbons. In: Gibson DT (ed) Microbial degradation of organic compounds. Marcel Dekker, Inc., New York, pp 181–252
Grbic-Galic D (1986) Anaerobic production and transformation of aromatic-hydrocarbons and substituted phenols by ferulic acid degrading BESA-inhibited methanogenic consortia. FEMS Microbiol Ecol 38:161–169
Grbic-Galic D, Vogel TM (1987) Transformation of toluene and benzene by mixed methanogenic cultures. Appl Environ Microbiol 53:254–260
Häner A, Höhener P, Zeyer J (1995) Degradation of p-xylene by a denitrifying enrichment culture. Appl Environ Microbiol 61:3185–3188
Hanzlik RP, Hogberg K, Judson CM (1984) Microsomal hydroxylation of specifically deuterated monosubstituted benzenes—evidence for direct aromatic hydroxylation. Biochemistry 23:3048–3055
Harms G, Rabus R, Widdel F (1999a) Anaerobic oxidation of the aromatic plant hydrocarbon p-cymene by newly isolated denitrifying bacteria. Arch Microbiol 172:303–312
Harms G, Zengler K, Rabus R, Aeckersberg F, Minz D, Rossello-Mora R, Widdel F (1999b) Anaerobic oxidation of o-xylene, m-xylene, and homologous alkylbenzenes by new types of sulfate-reducing bacteria. Appl Environ Microbiol 65:999–1004
Harwood CS, Burchhardt G, Herrmann H, Fuchs G (1999) Anaerobic metabolism of aromatic compounds via the benzoyl-CoA pathway. FEMS Microbiol Rev 22:439–458
Heider J (2007) Adding handles to unhandy substrates: anaerobic hydrocarbon activation mechanisms. Curr Opin Chem Biol 11:188–194
Heider J, Fuchs G (1997) Anaerobic metabolism of aromatic compounds. Eur J Biochem 243:577–596
Heider J, Boll M, Breese K, Breinig S, Ebenau-Jehle C, Feil U et al (1998) Differential induction of enzymes involved in anaerobic metabolism of aromatic compounds in the denitrifying bacterium Thauera aromatica. Arch Microbiol 170:120–131
Heider J, Spormann AM, Beller HR, Widdel F (1999) Anaerobic bacterial metabolism of hydrocarbons. FEMS Microbiol Rev 22:459–473
Herrmann S, Kleinsteuber S, Neu TR, Richnow HH, Vogt C (2008) Enrichment of anaerobic benzene-degrading microorganisms by in situ microcosms. FEMS Microbiol Ecol 63:94–106
Herrmann S, Vogt C, Fischer A, Kuppardt A, Richnow HH (2009) Characterization of anaerobic xylene biodegradation by two-dimensional isotope fractionation analysis. Environ Microbiol 1:535–544
Herrmann S, Kleinsteuber S, Chatzinotas A, Kuppardt S, Lueders T, Richnow HH, Vogt C (2010) Functional characterization of an anaerobic benzene-degrading enrichment culture by DNA stable isotope probing. Environ Microbiol 12:401–411
Hess A, Zarda B, Hahn D, Haner A, Stax D, Hohener P, Zeyer J (1997) In situ analysis of denitrifying toluene- and m-xylene-degrading bacteria in a diesel fuel-contaminated laboratory aquifer column. Appl Environ Microbiol 63:2136–2141
Hunkeler D, Anderson N, Aravena R, Bernasconi SM, Butler BJ (2001) Hydrogen and carbon isotope fractionation during aerobic biodegradation of benzene. Environ Sci Technol 35:3462–3467
Hunter WJ (2002) Bioremediation of chlorate or perchlorate contaminated water using permeable barriers containing vegetable oil. Curr Microbiol 45:287–292
Jahn MK, Haderlein SB, Meckenstock RU (2005) Anaerobic degradation of benzene, toluene, ethylbenzene, and o-xylene in sediment-free iron-reducing enrichment cultures. Appl Environ Microbiol 71:3355–3358
Johnson HA, Pelletier DA, Spormann AM (2001) Isolation and characterization of anaerobic ethylbenzene dehydrogenase, a novel Mo-Fe-S enzyme. J Bacteriol 183:4536–4542
Kane SR, Beller HR, Legler TC, Anderson RT (2002) Biochemical and genetic evidence of benzylsuccinate synthase in toluene-degrading, ferric iron-reducing Geobacter metallireducens. Biodegradation 13:149–154
Kasai Y, Takahata Y, Manefield M, Watanabe K (2006) RNA-based stable isotope probing and isolation of anaerobic benzene-degrading bacteria from gasoline-contaminated groundwater. Appl Environ Microbiol 72:3586–3592
Kasai Y, Kodama Y, Takahata Y, Hoaki T, Watanabe K (2007) Degradative capacities and bioaugmentation potential of an anaerobic benzene-degrading bacterium strain DN11. Environ Sci Technol 41:6222–6227
Kazumi J, Caldwell ME, Suflita JM, Lovley DR, Young LY (1997) Anaerobic degradation of benzene in diverse anoxic environments. Environ Sci Technol 31:813–818
Kleinsteuber S, Schleinitz KM, Breitfeld J, Harms H, Richnow HH, Vogt C (2008) Molecular characterization of bacterial communities mineralizing benzene under sulfate-reducing conditions. FEMS Microbiol Ecol 66:143–157
Kniemeyer O, Fischer T, Wilkes H, Glöckner FO, Widdel F (2003) Anaerobic degradation of ethylbenzene by a new type of marine sulfate-reducing bacterium. Appl Environ Microbiol 69:760–768
Krieger CJ, Beller HR, Reinhard M, Spormann AM (1999) Initial reactions in anaerobic oxidation of m-xylene by the denitrifying bacterium Azoarcus sp strain T. J Bacteriol 181:6403–6410
Kube M, Heider J, Amann J, Hufnagel P, Kühner S, Beck A et al (2004) Genes involved in the anaerobic degradation of toluene in a denitrifying bacterium, strain EbN1. Arch Microbiol 181:182–194
Kuever J, Rainey FA, Widdel F (2005) Desulfosarcina Widdel 1981, 382VP (Effective publication: Widdel 1980, 382) In: Garrity GM (ed) Bergey’s manual of systematic bacteriology. Springer, Two, The Proteobacteria, Part C The Alpha-, Beta-, Delta-, and Epsilonproteobacteria, pp 981–984
Kuhn EP, Colberg PJ, Schnoor JL, Wanner O, Zehnder AJB, Schwarzenbach RP (1985) Microbial transformations of substituted benzenes during infiltration of river water to groundwater—laboratory column studies. Environ Sci Technol 19:961–968
Kühner S, Wöhlbrand L, Fritz I, Wruck W, Hultschig C, Hufnagel P et al (2005) Substrate-dependent regulation of anaerobic degradation pathways for toluene and ethylbenzene in a denitrifying bacterium, strain EbN1. J Bacteriol 187:1493–1503
Kunapuli U, Lueders T, Meckenstock RU (2007) The use of stable isotope probing to identify key iron-reducing microorganisms involved in anaerobic benzene degradation. ISME J 1:643–653
Kunapuli U, Griebler C, Beller HR, Meckenstock RU (2008) Identification of intermediates formed during anaerobic benzene degradation by an iron-reducing enrichment culture. Environ Microbiol 10:1703–1712
Kunapuli U, Jahn MK, Lueders T, Geyer R, Heipieper HJ, Meckenstock RU (2010) Desulfitobacterium aromaticivorans sp. nov. and Geobacter toluenoxydans sp. nov., iron-reducing bacteria capable of anaerobic degradation of monoaromatic hydrocarbons. Int J Syst Evol Microbiol 60:686–695
Laban NA, Selesi D, Jobelius C, Meckenstock RU (2009) Anaerobic benzene degradation by Gram-positive sulfate-reducing bacteria. FEMS Microbiol Ecol 68:300–311
Langenhoff AAM, Nijenhuis I, Tan NCG, Briglia M, Zehnder AJB, Schraa G (1997a) Characterisation of a manganese-reducing, toluene-degrading enrichment culture. FEMS Microbiol Ecol 24:113–125
Langenhoff AAM, Brouwers-Ceiler DL, Engelberting JHL, Quist JJ, Wolkenfelt J, Zehnder AJB, Schraa G (1997b) Microbial reduction of manganese coupled to toluene oxidation. FEMS Microbiol Ecol 22:119–127
Langenhoff AAM, Richnow HH, Gerritse J (2009) Benzene degradation at a site amended with nitrate or chlorate. Bioremed J 13:180–187
Leuthner B, Heider J (1998) A two-component system involved in regulation of anaerobic toluene metabolism in Thauera aromatica. FEMS Microbiol Lett 166:35–41
Leuthner B, Leutwein C, Schulz H, Horth P, Haehnel W, Schiltz E et al (1998) Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica: a new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Mol Microbiol 28:615–628
Liou JS-C, DeRito CM, Madsen EL (2008) Field-based and laboratory stable isotope probing surveys of the indetities of both aerobic and anaerobic benzene-metabolizing microorganisms in freshwater sediment. Environ Microbiol 10:1964–1977
Liu A, Garcia-Dominguez E, Rhine ED, Young LY (2004) A novel arsenate respiring isolate that can utilize aromatic substrates. FEMS Microbiol Ecol 48:323–332
Logan BE (1998) A review of chlorate- and perchlorate-respiring microorganisms. Bioremed J 2:69–79
Logan BE, Wu J (2002) Enhanced toluene degradation under chlorate-reducing conditions by bioaugmentation of sand columns with chlorate- and toluene-degrading enrichments. Bioremed J 6:87–95
Losi ME, Giblin T, Hosangadi V, Frankenberger WT Jr (2002) Bioremediation of perchlorate-contaminated groundwater using a packed bed biological reactor. Bioremed J 6:97–103
Lovley DR (1997) Potential for anaerobic bioremediation of BTEX in petroleum-contaminated aquifers. J Ind Microbiol Biotechnol 18:75–81
Lovley DR (2000) Anaerobic benzene degradation. Biodegradation 11:107–116
Lovley DR, Lonergan DJ (1990) Anaerobic oxidation of toluene, phenol, and para-cresol by the dissimilatory iron-reducing organism, GS-15. Appl Environ Microbiol 56:1858–1864
Lovley DR, Giovannoni SJ, White DC, Champine JE, Phillips EJP, Gorby YA, Goodwin S (1993) Geobacter metallireducens gen. nov. sp. nov., a microorganism capale of coupling the complete oxidation of organic compounds to the reduction of iron and other metals. Arch Microbiol 159:336–344
Lovley DR, Woodward JC, Chapelle FH (1994) Stimulated anoxic biodegradation of aromatic hydrocarbons using Fe(III) ligands. Nature 370:128–131
Lovley DR, Coates JD, Woodward JC, Phillips EJP (1995) Benzene oxidation coupled to sulfate reduction. Appl Environ Microbiol 61:953–958
Lovley DR, Woodward JC, Chapelle FH (1996a) Rapid anaerobic benzene oxidation with a variety of chelated Fe(III) forms. Appl Environ Microbiol 62:288–291
Lovley DR, Coates JD, Blunt-Harris EL, Phillips EJP, Woodward JC (1996b) Humic substances as electron acceptors for microbial respiration. Nature 382:445–448
Lovley DR, Holmes DE, Nevin KP (2004) Dissimilatory Fe(III) and Mn(IV) reduction. Adv Microb Physiol 49:219–286
Major DW, Mayfield CI, Barker JF (1988) Biotransformation of benzene by denitrification in aquifer sand. Ground Water 26:8–14
Mancini SA, Lacrampe-Couloume G, Jonker H, Van Breukelen BM, Groen J, Volkering F, Lollar BS (2002) Hydrogen isotopic enrichment: An indicator of biodegradation at a petroleum hydrocarbon contaminated field site. Environ Sci Technol 36:2464–2470
Mancini SA, Ulrich AC, Lacrampe-Couloume G, Sleep B, Edwards EA, Lollar BS (2003) Carbon and hydrogen isotopic fractionation during anaerobic biodegradation of benzene. Appl Environ Microbiol 69:191–198
Mancini SA, Devine CE, Elsner M, Nandi ME, Ulrich AC, Edwards EA, Lollar BS (2008) Isotopic evidence suggests different initial reaction mechanisms for anaerobic benzene biodegradation. Environ Sci Technol 42:8290–8296
McCarty PL (1971) Energetics and bacterial growth. In: Faust SD, Hunter JV (eds) Organic compounds in aquatic environments. Marcel Dekker, New York, pp 495–531
Mechichi T, Stackebrandt E, Gad’on N, Fuchs G (2002) Phylogenetic and metabolic diversity of bacteria degrading aromatic compounds under denitrifying conditions, and description of Thauera phenylacetica sp nov., Thauera aminoaromatica sp nov., and Azoarcus buckelii sp nov. Arch Microbiol 178:26–35
Mechichi T, Stackebrandt E, Fuchs G (2003) Alicycliphilus denitrificans gen. nov., sp nov., a cyclohexanol-degrading, nitrate-reducing beta-proteobacterium. Int J Syst Evol Microbiol 53:147–152
Meckenstock RU (1999) Fermentative toluene degradation in anaerobic defined syntrophic cocultures. FEMS Microbiol Lett 177:67–73
Meckenstock RU, Annweiler E, Michaelis W, Richnow HH, Schink B (2000) Anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Appl Environ Microbiol 66:2743–2747
Meckenstock RU, Warthmann RI, Schafer W (2004a) Inhibition of anaerobic microbial o-xylene degradation by toluene in sulfidogenic sediment columns and pure cultures. FEMS Micobiol Ecol 47:381–386
Meckenstock RU, Morasch B, Griebler C, Richnow HH (2004b) Stable isotope fractionation analysis as a tool to monitor biodegradation in contaminated aquifers. J Contam Hydrol 75:215–225
Mehboob F, Weelink SAB, Saia FT, Junca H, Stams AJM, Schraa G (2010) Microbial degradation of aliphatic and aromatic hydrocarbons with (per)chlorate as electron acceptor. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, pp 936–945
Mitchell KH, Rogge CE, Gierahn T, Fox BG (2003) Insight into the mechanism of aromatic hydroxylation by toluene 4-monooxygenase by use of specifically deuterated toluene and p-xylene. Proc Natl Acad Sci USA 100:3784–3789
Morasch B, Meckenstock RU (2005) Anaerobic degradation of p-xylene by a sulfate-reducing enrichment. Curr Microbiol 51:127–130
Morasch B, Schink B, Tebbe CC, Meckenstock RU (2004) Degradation of o-xylene and m-xylene by a novel sulfate-reducer belonging to the genus Desulfotomaculum. Arch Microbiol 181:407–417
Musat F, Widdel F (2007) Anaerobic degradation of benzene by a marine sulfate-reducing enrichment culture, and cell hybridization of the dominant phylotype. Environ Microbiol 10:10–19
Nakagawa T, Sato S, Fukui M (2008) Anaerobic degradation of p-xylene in sediment free sulfate-reducing enrichment culture. Biodegradation 19:909–913
Oka AR, Phelps CD, McGuinness LM, Mumford A, Young LY, Kerkhof LJ (2008) Identification of critical members in a sulfidogenic benzene-degrading consortium by DNA Stable Isotope Probing. Appl Environ Microbiol 74:6480–6776
Ommedal H, Torsvik T (2007) Desulfotignum toluenicmum sp. nov., a novel toluene-degrading, sulphate reducing bacterium isolated from an oil-reservoir model column. Int J Syst Evol Microbiol 57:2865–2869
Phelps CD, Young LY (2001) Biodegradation of BTEX under anaerobic conditions: a review. In: Advances in agronomy, 70, pp 329–357
Phelps CD, Kazumi J, Young LY (1996) Anaerobic degradation of benzene in BTX mixtures dependent on sulfate reduction. FEMS Microbiol Lett 145:433–437
Phelps CD, Kerkhof LJ, Young LY (1998) Molecular characterization of a sulfate-reducing consortium which mineralizes benzene. FEMS Microbiol Ecol 27:269–279
Phelps CD, Zhang ZM, Young LY (2001) Use of stable isotopes to identify benzoate as a metabolite of benzene degradation in a sulphidogenic consortium. Environ Microbiol 3:600–603
Probian C, Wülfing A, Harder J (2003) Anaerobic mineralization of quaternary carbon atoms: Isolation of denitrifying bacteria on pivalic acid (2,2-dimethylpropionic acid). Appl Environ Microbiol 69:1866–1870
Rabus R, Heider J (1998) Initial reactions of anaerobic metabolism of alkylbenzenes in denitrifying and sulfate reducing bacteria. Arch Microbiol 170:377–384
Rabus R, Widdel F (1995) Anaerobic degradation of ethylbenzene and other aromatic hydrocarbons by new denitrifying bacteria. Arch Microbiol 163:96–103
Rabus R, Nordhaus R, Ludwig W, Widdel F (1993) Complete oxidation of toluene under strictly anoxic conditions by a new sulfate-reducing bacterium. Appl Environ Microbiol 59:1444–1451
Rabus R, Kube M, Beck A, Widdel F, Reinhardt R (2002) Genes involved in the anaerobic degradation of ethylbenzene in a denitrifying bacterium, strain EbN1. Arch Microbiol 178:506–516
Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhardt R (2005) The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol 183:27–36
Rikken GB, Kroon AGM, van Ginkel CG (1996) Transformation of (per)chlorate into chloride by a newly isolated bacterium: reduction and dismutation. Appl Microbiol Biotechnol 45:420–426
Rooney-Varga JN, Anderson RT, Fraga JL, Ringelberg D, Lovley DR (1999) Microbial communities associated with anaerobic benzene degradation in a petroleum-contaminated aquifer. Appl Environ Microbiol 65:3056–3063
Rotaru AE, Probian C, Wilkes H, Harder J (2010) Highly enriched Betaproteobacteria growing anaerobically with p-xylene and nitrate. FEMS Microbiol Ecol 71:460–468
Safinowski M, Meckenstock RU (2005) Methylation is the initial reaction in anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Environ Microbiol 8:347–352
Salinero KK, Keller K, Feil WS, Feil H, Trong S, Di Bartolo G, Lapidus A (2009) Metabolic analysis of the soil microbe Dechloromonas aromatica str. RCB: indications of a surprisingly complex life-style and cryptic anaerobic pathways for aromatic degradation. BMC Genom 10:1–23
Schink B, Brune A, Schnell S (1992) Anaerobic degradation of aromatic compounds. In: Winkelmann G (ed) Microbial degradation of natural products. VCH, Weinheim, pp 219–242
Schocher RJ, Seyfried B, Vazquez F, Zeyer J (1991) Anaerobic degradation of toluene by pure cultures of denitrifying bacteria. Arch Microbiol 157:7–12
Schühle K, Fuchs G (2004) Phenylphosphate carboxylase: a new C-C lyase involved in anaerobic phenol metabolism in Thauera aromatica. J Bacteriol 186:4556–4567
Shinoda Y, Sakai Y, Uenishi H, Uchihashi Y, Hiraishi A, Yukawa H et al (2004) Aerobic and anaerobic toluene degradation by a newly isolated denitrifying bacterium, Thauera sp strain DNT-1. Appl Environ Microbiol 70:1385–1392
Shinoda Y, Akagi J, Uchihashi Y, Hiraishi A, Yukawa H, Yurimoto H et al (2005) Anaerobic degradation of aromatic compounds by Magnetospirillum strains: isolation and degradation genes. Biosci Biotechnol Biochem 69:1483–1491
Smith MR (1990) The biodegradation of aromatic hydrocarbons by bacteria. Biodegradation 1:191–206
Spormann AM, Widdel F (2000) Metabolism of alkylbenzenes, alkanes, and other hydrocarbons in anaerobic bacteria. Biodegradation 11:85–105
Stumm W, Morgan JJ (1981) Aquatic chemistry: an introduction emphasizing chemical equilibria in natural waters. Wiley, New York
Tan NCG, Van Doesburg W, Langenhoff AAM, Gerritse J, Stams AJM (2004) Benzene degradation coupled with chlorate reduction. In: Verstraete W (ed) Environmental biotechnology, ESEB 2004. A.A. Balkema publishers, Leiden, pp 803–805
Tan NCG, van Doesburg W, Langenhoff AAM, Stams AJM (2006) Benzene degradation coupled with chlorate reduction in a soil column study. Biodegradation 17:113–119
Thauer RK, Jungermann K, Decker K (1977) Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev 41:100–180
Ulrich AC, Edwards EA (2003) Physiological and molecular characterization of anaerobic benzene-degrading mixed cultures. Environ Microbiol 5:92–102
Ulrich AC, Beller HR, Edwards EA (2005) Metabolites detected during biodegradation of 13C6-benzene in nitrate-reducing and methanogenic enrichment cultures. Environ Sci Technol 39:6681–6691
Urbansky ET (1998) Perchlorate chemistry: implications for analysis and remediation. Bioremed J 2:81–95
Van Agteren MH, Keuning S, Janssen DB (1998) Handbook on biodegradation and biological treatment of hazardous organic compounds. Kluwer, Dordrecht
Versteegh JFM, Neele J, Cleeven RFMJ (1993) Chloriet en chloraat in drinkwater: een desinfectie of een milieuprobleem? H2O 26:680–686
Villatoro-Monzon WR, Mesta-Howard AM, Razo-Flores E (2003) Anaerobic biodegradation of BTEX using Mn(IV) and Fe(III) as alternative electron acceptors. Water Sci Technol 48:125–131
Villatoro-Monzon WR, Morales-Ibarria MG, Velázquez EK, Ramírez-Saad H, Razo-Flores E (2008) Benzene biodegradation under anaerobic conditions coupled with metal oxides reduction. Water Air Soil Pollut 192:165–172
Vogel TM, Grbic-Galic D (1986) Incorporation of oxygen from water into toluene and benzene during anaerobic fermentative transformation. Appl Environ Microbiol 52:200–202
Vogt C, Gödeke S, Treutler HC, Weiß H, Schirmer M, Richnow HH (2007) Benzene oxidation under sulfate-reducing conditions in columns simulating in situ conditions. Biodegradation 18:625–636
Washer CE, Edwards EA (2007) Identification and expression of benzylsuccinate synthase genes in a toluene-degrading methanogenic consortium. Appl Environ Microbiol 73:1367–1369
Weelink SAB, Tan NCG, ten Broeke H, van Doesburg W, Langenhoff AAM, Gerritse J, Stams AJM (2007) Physiological and phylogenetic characterization of a stable benzene-degrading chlorate-reducing microbial community. FEMS Microbiol Ecol 60:312–321
Weelink SAB, Tan NCG, Ten Broeke H, van den Kieboom C, Van Doesburg W, Langenhoff AAM et al (2008) Isolation and characterization of Alicycliphilus denitrificans strain BC, which grows on benzene with chlorate as the electron acceptor. Appl Environ Microbiol 74:6672–6681
Weelink SAB, Van Doesburg W, Saia FT, Rijpstra WIC, Roling WFM, Smidt H, Stams AJM (2009) A strictly anaerobic betaproteobacterium Georgfuchsia toluolica gen. nov., sp. nov. degrades aromatic compounds with Fe(III), Mn(IV) or nitrate as an electron acceptor. FEMS Microbiol Ecol 70:243–253
Weiner JM, Lovley DR (1998) Rapid benzene degradation in methanogenic sediments from a petroleum-contaminated aquifer. Appl Environ Microbiol 64:1937–1939
Weiner JM, Lauck TS, Lovley DR (1998) Enhanced anaerobic benzene degradation with the addition of sulfate. Bioremed J 2:159–173
Widdel F, Rabus R (2001) Anaerobic biodegradation of saturated and aromatic hydrocarbons. Curr Opin Biotechnol 12:259–276
Winderl C, Schaefer S, Lueders T (2007) Detection of anaerobic toluene and hydrocarbon degraders in contaminated aquifers using benzylsuccinate synthase (bssA) genes as a functional marker. Environ Microbiol 9:1035–1046
Wöhlbrand L, Kallerhoff B, Lange D, Hufnagel P, Thiermann J, Reinhardt R, Rabus R (2007) Functional proteomic view of metabolic regulation in Aromatoleum aromaticum strain EbN1. Proteomics 7:2222–2239
Wolterink AFWM, Jonker AB, Kengen SWM, Stams AJM (2002) Pseudomonas chloritidismutans sp. nov., a nondenitrifying, chlorate-reducing bacterium. Int J Syst Evol Microbiol 52:2183–2190
Zengler K, Heider J, Rossello-Mora R, Widdel F (1999) Phototrophic utilization of toluene under anoxic conditions by a new strain of Blastochloris sulfoviridis. Arch Microbiol 172:204–212
Zeyer J, Kuhn EP, Schwarzenbach RP (1986) Rapid microbial mineralization of toluene and 1, 3-dimethylbenzene in the absence of molecular oxygen. Appl Environ Microbiol 52:944–947
Zhang XM, Young LY (1997) Carboxylation as an initial reaction in the anaerobic metabolism of naphthalene and phenanthrene by sulfidogenic consortia. Appl Environ Microbiol 63:4759–4764
Acknowledgments
This research was financed through grants of the graduate school WIMEK (Wageningen Institute for Environment and Climate Research, http://www.sense.nl) and of SKB (Dutch Center for Soil Quality Management and Knowledge Transfer, http://www.skbodem.nl). The research was incorporated in the TRIpartite Approaches toward Soil systems processes (TRIAS) program (http://www.nwo.nl/trias/english/index.html). The authors would like to thank Anko Fischer and Howard Junca for the pleasant collaboration.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Weelink, S.A.B., van Eekert, M.H.A. & Stams, A.J.M. Degradation of BTEX by anaerobic bacteria: physiology and application. Rev Environ Sci Biotechnol 9, 359–385 (2010). https://doi.org/10.1007/s11157-010-9219-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11157-010-9219-2